

Abstract
The generation of excessive amounts of reactive oxygen species (ROS) leads to cellular oxidative stress that underlies a variety of forms of hepatocyte injury and death including that from alcohol. Although ROS can induce cell damage through direct effects on cellular macromolecules, the injurious effects of ROS are mediated largely through changes in signal transduction pathways such as the mitogen-activated protein kinase c-Jun N-terminal kinase (JNK). In response to alcohol, hepatocytes have increased levels of the enzyme cytochrome P450 2E1 (CYP2E1) which generates an oxidant stress that promotes the development of alcoholic steatosis and liver injury. These effects are mediated in large part through overactivation of JNK that alters cell death pathways. Targeting the JNK pathway or its downstream effectors may be a useful therapeutic approach to the oxidative stress generated by CYP2E1 in alcoholic liver disease.
Keywords: Cytochrome P450 2E1, c-Jun N-terminal kinase, Alcoholic liver disease, Oxidative stress, Mitogen-activated protein kinases
Abbreviations: APAP, acetaminophen; CYP2E1, cytochrome P450 2E1; ERK1/2, extracellular signal-regulated kinase 1/2; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MKK, MAPK kinase; MAPKKK, MAPK kinase kinase; NAFLD, nonalcoholic fatty liver disease; PKC, protein kinase C; PKD, protein kinase D; ROS, reactive oxygen species; Sab, SH3 homology associated BTK binding protein; TNF, tumor necrosis factor
Graphical abstract
Highlights
-
•
c-Jun N-terminal kinase (JNK) activation regulates metabolic signaling and cell death pathways in hepatocytes.
-
•
Cytochrome P450 2E1 (CYP2E1) overexpression in fatty liver disease increases levels of reactive oxygen species (ROS) triggering oxidative stress.
-
•
CYP2E1-generated ROS cause JNK overactivation which is a mechanism for the development of alcoholic fatty liver disease.
-
•
The CYP2E1-ROS-JNK injury cascade is a therapeutic target in alcoholic liver disease.
Introduction
Under normal physiological conditions hepatocytes process low levels of reactive oxygen species (ROS) that are constantly generated as a by-product of mitochondrial respiration. Many pathophysiological states of liver injury stimulate increased ROS production, and hepatocytes undergo oxidative stress when ROS generation exceeds the neutralizing capacity of cellular nonenzymatic and enzymatic antioxidants. Potential sources of excessive ROS are increases in the activity of ROS-generating intracellular enzymes including cytochrome P450 2E1 (CYP2E1) and other enzymes such as xanthine oxidase and NADPH oxidase. During liver injury, hepatocytes are also exposed to exogenous ROS produced by macrophages and neutrophils activated as part of the accompanying inflammatory reaction. Oxidative stress is thought to be a critical factor in the development of alcoholic and nonalcoholic fatty liver injury, and CYP2E1 activation is an important source of ROS in chronic alcoholic liver disease [1]. Defining the mechanisms of hepatocyte resistance and injury from CYP2E1-generated ROS is therefore essential to determining the pathophysiological events that underlie alcoholic liver injury. Moreover, delineating the mechanisms by which oxidant stress leads to hepatocyte injury and cell death is important to our understanding of many other forms of liver injury as oxidant stress is thought to be a central mechanism of hepatocyte injury and death in a variety of etiologies of liver injury including that from other toxins [2,3], bile acids [4,5], and ischemia/reperfusion [6].
Oxidative stress can cause hepatocyte injury through direct interactions with critical cellular macromolecules such as DNA, proteins and lipids that destroy their function or trigger their degradation, ultimately leading to cell death. Recent investigations have stressed the importance of oxidant effects on cell signaling pathways which when altered can themselves initiate hepatocellular injury and death [7,8]. Low levels of ROS produced in response to physiological stimuli such as growth factors and hormones function as second messengers to activate cell signal transduction pathways that mediate normal cellular responses to these factors [9]. In contrast, excessive amounts of ROS as occur with liver injury can oxidize the protein kinases and phosphatases that regulate critical cell signals and distort the activation of signaling pathways such as the mitogen-activated protein kinases (MAPKs). Protein modification by oxidants can occur in many ways, but the most important ones involve thiol groups on cysteine residues. Common redox alterations to thiols include oxidation to sulfinic and sulfonic acids, glutathionylation, nitrosylation and the formation of disulfide bonds. These modifications can alter the activity of cell kinases that regulate the process of liver injury and cell death. The mechanisms by which the chronic oxidative stress generated by alcohol-induced CYP2E1 overexpression affects signal transduction pathways and in particular c-Jun N-terminal kinase (JNK) signaling in alcoholic liver disease is the focus of this review.
JNK MAPK pathway
Critical regulators of cellular responses to oxidant stress are the members of the MAPK family of serine/threonine kinases of which the principal members are JNK, extracellular signal-regulated kinase (ERK) 1/2 and p38 [10]. The MAPK signaling pathways modulate cellular responses to a variety of extracellular and intracellular stimuli including oxidant stresses that induce cellular injury and death. In the liver, overactivation of JNK signaling in particular has been identified as a common mechanism underlying hepatocyte death including that from oxidant stress, tumor necrosis factor (TNF), ischemia reperfusion injury and fatty liver disease [11–13].
There are multiple JNK isoforms that are encoded for by three genes. Two of these genes, jnk1 and jnk2, are expressed in all tissues including the liver whereas jnk3 expression is restricted to heart, brain and testes [10]. An examination of jnk gene function in vivo has been possible through studies of jnk1 and jnk2 knockout mice [10,14]. Loss of either one of the two jnk genes leads only to mild phenotypic abnormalities in T cell apoptosis and immune responses [15]. In contrast, the jnk1/jnk2 double knockout is an embryonic lethal because of severe dysregulation of brain apoptosis [16,17]. These findings suggest that there are overlapping functions of jnk1 and jnk2, but recent studies have emphasized distinct cellular roles for the products of the two genes. Distinguishing the specific functions of each gene has been complicated by the fact that both jnk genes undergo alternative splicing to create multiple 46 and 54 kDa protein isoforms that differ by the presence of a COOH-terminal extension [10]. Distinct functions for the products of the two genes have now been established for many cellular processes including that of hepatocyte injury from oxidant stress. However, whether protein isoforms from the same gene have specific functions remains unknown. It has been postulated that the different isoforms may exist to allow interactions with specific substrates but this possibility remains a speculation.
JNK activation results from the sequential activation of a kinase cascade (Fig. 1). Initiating events remain unclear, but G-proteins such as Rac and cdc-42, the TNF receptor associated factor group of adaptor proteins, and death effector domain containing proteins can modulate the activation of JNK [18]. Activation proceeds through a three tier, protein kinase cascade that starts with the activation of any of at least 14 MAPK kinase kinases (MAPKKKs) (Fig. 1), a redundancy that may allow for responses to distinct stimuli [19]. The MAPKKK converge to activate the MAPK kinases (MAPKKs) MKK4 and MKK7, which preferentially phosphorylate JNK on tyrosine 185 and threonine 183, respectively [14]. JNK activation can also be potentiated by kinase interactions with JNK-interacting proteins [20]. Differential activation of either MKK4 or MKK7 can occur with certain stimuli, but dual phosphorylation is required for full JNK activation. Phosphorylation-dependent activation of JNK is counterbalanced by kinase dephosphorylation by phosphatases [21]. An important mechanism for the dysregulation of phosphatases and altered JNK signaling is phosphatase inactivation by oxidant stress. For example, JNK activation is sustained in response to TNF in the absence of NF-κB signaling through TNF-generated ROS which inactive JNK phosphatases through oxidation of a cysteine in the catalytic domain [22]. Whether CYP2E1-generated ROS phosphatase inhibition contributes to JNK activation with alcohol has not been examined. Increased proteasomal degradation of MAPK phosphatase 1 has been demonstrated with ethanol treatment of hepatoma cells, suggesting an alternative mechanism by which phosphatases may be involved in JNK overactivation with alcohol [23]. Additional studies are needed to examine whether alcohol affects the activity of this phosphatase, or other MAPK phosphatases, in ethanol-treated primary hepatocytes or mouse liver and whether CYP2E1-induced oxidant stress regulates phosphatase activity. The level of JNK activity under pathophysiological states such as alcohol-induced liver disease therefore represents a complex balance between the stimulatory actions of upstream kinases and down regulation by phosphatases, both of which may be altered by ROS derived from alcohol metabolism.
Fig. 1.
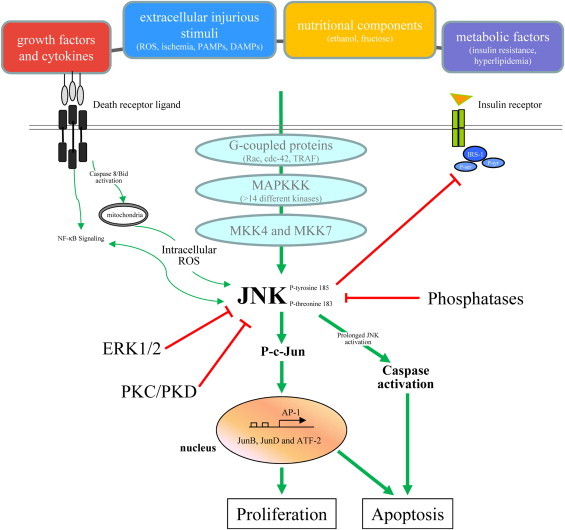
Regulation and effects of JNK signaling in hepatocytes. Activation of JNK in hepatocytes occurs through phosphorylation of tyrosine and threonine residues that is triggered by extracellular stimuli including growth factors and cytokines, injurious mediators including pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPS) and nutritional and metabolic factors. This phosphorylation involves a multi-step protein kinase signaling cascade with MAPKKKs and the downstream mitogen-activated protein kinase kinases MKK4 and MKK7. JNK phosphorylation is negatively regulated by signaling through ERK1/2, PKC and PKD. JNK activity is also down regulated by phosphatases. JNK signaling effects are predominantly mediated through transcriptional activation of the c-Jun/AP-1 signaling pathway, but also by the direct effects of JNK to lead to biological effects such as cell proliferation or death. Red lines show inhibitory pathways.
The primary action of JNK has been thought to be its phosphorylation of c-Jun at serine-63 and −73 which increases the transcriptional activity of this critical AP-1 subunit. However, increasing numbers of JNK substrates have been described subsequently and now number over 50 [24]. Prominent among these factors are other AP-1 transcription factors, Jun B, Jun D and ATF-2 [24]. However, additional substrates include other transcription factors (c-Myc, p53 and nuclear hormone receptors), mediators of protein degradation (E3 ligase Itch), mitochondrial proteins (SH3 homology associated BTK binding protein (Sab)), metabolic regulators (insulin receptor substrate 1), microtubule-associated proteins (stathmin) and cell death pathway proteins (Bcl-2, Bcl-XL, Bid, Bim, Bad and Bax) [24]. This wide array of JNK phosphorylation targets further broadens the possible effects of JNK on cell physiology and pathophysiology.
Regulation of ROS-induced cell death by MAPK signaling
JNK signaling in menadione-induced oxidant stress
Investigations of the effects on cultured hepatocytes of an oxidative stress from sources other than alcohol have demonstrated the importance of MAPKs including JNK in regulating hepatocyte death from ROS. Studies using the model of superoxide-induced oxidant stress generated by the redox recycler menadione in a nontransformed rat hepatocyte cell line have revealed that non-toxic concentrations of menadione induce transient, low-level activation of JNK and ERK1/2, but fail to activate p38 MAPK [25]. In contrast, treatment with higher, toxic menadione concentrations leads to markedly increased and prolonged JNK and ERK1/2 activation [25]. JNK overactivation mediates hepatocyte death from menadione-induced oxidant stress as adenoviral expression of a dominant negative c-Jun, the downstream substrate of JNK, blocks death from menadione [25]. These findings are consistent with the emerging concept that the mechanisms by which MAPK signaling can be induced by many different stimuli, yet mediate very specific and even opposing biological effects, include alterations in either the duration of kinase activation or in the subcellular localization of the MAPK. Prolonged hepatocyte JNK activation has emerged as an important signal that mediates death not only from oxidant stress but also from many other factors such as TNF [26]. Subsequent studies revealed that the two jnk genes have opposing functions with jnk1 promoting menadione-induced cell death and jnk2 blocking cell death [27]. The specific targets that mediate the death and survival signals of jnk1 and jnk2 respectively, remain to be determined, although studies in a nonalcoholic fatty liver disease (NAFLD) model have identified that an important jnk2 protective function in the liver is to down regulate the pro-apoptotic family member Bim [28].
Activation of ERK1/2 MAPK functions in hepatocyte resistance to cell death from menadione through crosstalk with JNK. ERK1/2 inhibition sensitizes hepatocytes to apoptosis from previously nontoxic concentrations of menadione in association with prolonged JNK activation. That ERK1/2’s protective effect is mediated by down regulation of JNK/AP-1 signaling was confirmed by the finding that a c-Jun dominant negative blocked death from ERK1/2 inhibition and low-dose menadione, identical to findings that c-Jun inhibition prevents death from high, toxic concentrations of menadione [25]. Thus, the MAPKs JNK and ERK1/2 perform opposing functions in the regulation of hepatocyte survival after oxidant stress. ERK1/2 effectively curtails JNK activation at low levels of oxidative stress, whereas with higher levels of oxidants, JNK overactivation occurs despite ERK1/2 activity and cell death ensues. ERK1/2 may function to induce phosphatases that effectively inactivate JNK at low levels of oxidative stress. At higher levels of oxidant stress these phosphatases may be inactivated by oxidant-induced protein modifications, leading to JNK overactivation despite ERK1/2 activity. The pro-death effect of JNK activation and protective function of ERK1/2 signaling in menadione-induced oxidative stress have been confirmed in similar studies performed in primary rat hepatocytes [29]. Hepatic ERK1/2 activation occurs in rodent alcohol models [30], but whether it alters JNK signaling induced by alcohol has not been examined.
Given the critical importance of the regulation of JNK activity in determining hepatocyte sensitivity to death from oxidant stress, a more complex network of checks on JNK signaling is needed in addition to the ERK1/2 pathway. One identified negative regulator is menadione-induced activation of protein kinase C (PKC) which when activated phosphorylates protein kinase D (PKD) causing its nuclear translocation [31]. PKC inhibition leads to increased cell death from menadione by the mechanism of JNK overactivation and can be prevented by the expression of a constitutively active PKD indicating that PKD is the downstream effector of PKC signaling. Co-inhibition of ERK1/2 and PKC/PKD signaling results in significant increases in levels of phosphorylated JNK/c-Jun and cell death when compared to inhibition of either ERK1/2 or PKC alone. These findings indicate that the ERK1/2 and PKC/PKD signaling pathways act independently and in parallel to down regulate JNK/c-Jun signaling and protect against hepatocyte death from oxidative stress [31]. Factors other than kinase-dependent signaling pathways are also likely involved in JNK regulation and need to be more carefully examined for a role in JNK overactivation. For example, menadione has been shown to induce heme oxygenase in hepatocytes, and the carbon monoxide produced by the actions of this enzyme protects against menadione-induced cell death [32]. This effect occurs in association with p54 JNK down regulation [32], although decreased JNK has not been proven to account for the protective effects of carbon monoxide.
MAPK signaling in hepatocyte death from hydrogen peroxide
Additional confirmation of the role of ERK1/2 signaling in resistance to oxidant stress has come from studies of hydrogen peroxide-treated primary rat hepatocytes. Although one study indicated that MAPK signaling is not affected by hydrogen peroxide-induced oxidant stress [29], investigations by Rosseland et al. [33], demonstrated that hydrogen peroxide induces ERK1/2 activation that mediates hepatocyte survival from hydrogen peroxide toxicity. In contrast to the proliferative stimulus of epithelial growth factor receptor-dependent activation of ERK1/2 that induces its nuclear translocation, hydrogen peroxide-induced activation is epithelial growth factor receptor independent and results in ERK1/2 retention in the cytoplasm. This difference in subcellular localization may determine whether hepatocyte ERK1/2 activation triggers a proliferative or cytoprotective cellular response after an oxidative stress. Whether the protective effects of ERK1/2 in hydrogen peroxide-induced hepatocyte death are mediated through JNK down regulation has not been determined and needs to be examined.
MAPK involvement in acetaminophen-induced oxidant stress
Another well-studied model of hepatocyte injury that results from acute CYP2E1-mediated oxidant stress is that induced by acetaminophen (APAP). Hepatocellular injury from APAP results from its CYP2E1-dependent metabolism to the electrophile N-acetyl-p-benzoquinone imine which binds glutathione, and APAP at sufficient concentrations depletes the cell of this critical nonenzymatic antioxidant [34]. When glutathione depletion ensues, free electrophiles bind to other thiol-containing proteins and this process or the resultant oxidative stress induces hepatocellular injury and death. Similar to findings in the menadione and hydrogen peroxide models of oxidant injury, liver injury from APAP is not the result of direct oxidant damage but rather is actively mediated by sustained JNK activation [35]. This conclusion is derived from comprehensive studies by Kaplowitz and colleagues in which APAP injury was prevented when JNK function was inhibited by a pharmacological agent, genetic knockout or antisense oligonucleotides [35]. In contrast to findings in the menadione model, the death effect of JNK is principally mediated by JNK2 isoforms as inhibition of JNK2 but not JNK1 decreases liver injury, although in the absence of JNK2 the loss of JNK1 promotes additional resistance to toxicity. In response to APAP-generated ROS from the mitochondria, activated JNK translocates to mitochondria and compromises mitochondrial bioenergetics by inhibiting mitochondrial respiration which leads to cell death [36]. Recruitment of JNK to the mitochondria is dependent on its binding to the mitochondrial outer membrane protein Sab [37]. These studies further support the critical involvement of JNK overactivation as a common mechanism of hepatocyte injury and death from a variety of forms of oxidative stress, and suggest that Sab may be an appropriate therapeutic target in any ROS-mediated hepatic disease that leads to JNK overactivation, including that induced by alcohol.
Effects of CYP2E1 on JNK signaling and cell death from chronic oxidative stress
CYP2E1-induced JNK overactivation is a mechanism for the development of alcohol-induced steatohepatitis
CYP2E1 belongs to a family of heme-containing proteins that regulate the hepatic metabolism of a variety of endogenous and exogenous compounds. Among the various CYP family members, CYP2E1 is characterized by its broad spectrum of substrates and generation of large amounts of ROS when enzymatically active [38]. The role of CYP2E1 in alcoholic liver disease has been well demonstrated through a series of studies in models of hepatoma cell and hepatocyte and mouse CYP2E1 overexpression or pharmacological or genetic inhibition largely by the studies of Cederbaum and coworkers (summarized in Table 1). Ethanol is a well-known inducer of CYP2E1 [38], and increased hepatic CYP2E1 expression has been mechanistically linked to both the excessive lipid accumulation and hepatocellular injury that underlie the pathophysiology of alcoholic liver disease [39]. CYP2E1 knockout mice exhibit significantly reduced hepatic steatosis and liver injury from chronic ethanol exposure [40]. A possible mechanism by which CYP2E1 increases liver injury is through the sensitization of hepatocytes to injury from lipopolysaccharide-stimulated TNF through activation of JNK signaling [41]. This injury is secondary to oxidative stress as it can be inhibited by antioxidants such as N-acetylcysteine and triggers apoptosis that can be blocked by cyclosporine [42,43]. Restoration of redox equivalents using a-den-o-syl-methionine also prevents injury in ethanol-treated mice and apoptosis that is accompanied by mitochondrial dysfunction and swelling [44]. In all of these studies, oxidative stress was found to over activate JNK which then promoted liver injury. As with other types of hepatocyte injury-associated oxidative stress, the JNK isoforms contribute differentially to alcohol-induced liver injury. Deletion of jnk1, but not jnk2 reduces alcohol-induced liver injury [45]. JNK1 has a comparable role in nonalcoholic liver disease [46], clearly implicating JNK1 overactivation as a central mechanism of liver injury in a fatty liver.
Table 1.
Summary of the major studies of CYP2E1 and JNK in alcohol-induced oxidative stress.
| Experimental system | Model | Read out | Mechanism | Effect | Reference |
|---|---|---|---|---|---|
| CYP2E1 knockout mice | Chronic ethanol | Liver injury, oxidative stress, steatosis | Increased oxidative stress | Increased steatosis and injury | [40] |
| C57BL/6 mice | Pyrazole±LPS | Liver injury and oxidative stress | MAPK activation | Increased injury | [41] |
| C57BL/6 mice | Ethanol±Jo2 antibody | Liver injury and oxidative stress | Mitochondrial dysfunction | Increased injury | [44] |
| JNK1 and JNK2 knockout mice | Pyrazole±TNF | Liver injury, oxidative stress, mitochondrial dysfunction | JNK1 activation | Decreased injury in jnk1−/− mice | [45] |
| C57BL/6 mice | Pyrazole±TNF | Liver injury, oxidative stress, MAPK activation | ASK-1 activation | Increased injury | [41] |
| CYP2E1 knockout mice | Ethanol±Jo2 antibody | Liver injury, MAPK activation | Increased death receptor signaling | increased injury | [68] |
| C57BL/6 mice and CYP2E1 overexpressing HepG2 cells | Ethanol gavage in mice and ethanol treatment in cells | Cellular injury, oxidative stress, hepatic steatosis | Increased autophagy | Decreased steatosis | [58] |
| CYP2E1 overexpressing HepG2 cells | siRNA knockdown of thioredoxin | MAPK activation, cell survival | ASK-1 and JNK activation | Increased cell death | [48] |
| CYP2E1 overexpressing HepG2 cells | 3-Methyladenine or Atg 7 siRNA | Oxidative stress, MAPK activation, cell survival | Decreased autophagy | Increased injury | [57] |
In vitro studies in a transgenic, CYP2E1-overexpressing hepatoma cell line have similarly demonstrated that CYP2E1 overexpression sensitizes to injury from cofactors such as TNF [47]. That CYP2E1 alters the antioxidant capacities of hepatocytes was confirmed by studies of thioredoxin, a redox protein and cytosolic sensor of oxidative stress. siRNA-induced knockdown of cytosolic and mitochondrial thioredoxin in hepatoma cells augments CYP2E1-induced activation of JNK signaling and the resultant hepatocyte cell death [48].
CYP2E1-mediated JNK-dependent effects on autophagy in alcoholic liver disease
Another possible mechanism by which CYP2E1-induced JNK signaling may regulate the development of alcohol-induced liver disease is through effects on the lysosomal degradative pathway of macroautophagy [49]. Autophagy modulates both hepatocyte lipid metabolism and cell death suggesting that the activity of this pathway can be an important determinant of the development and progression of fatty liver disease [49,50]. The effects of alcohol on levels of autophagic function are controversial with initial studies in rodent binge alcohol models demonstrating an increase in autophagy with ethanol [51], whereas subsequently the effects of chronic alcohol have been reported to decrease autophagy [52]. Autophagy is critical for hepatocyte resistance to death from menadione-induced oxidative stress [53], suggesting that au-toph-a-gy may be protective against CYP2E1-generated oxidant injury. Liver injury from alcohol is blocked by agents that augment autophagy [51,54], indicating a protective function for this degradative pathway in this disease.
Some evidence suggests that JNK may mediate the effects of CYP2E1 on autophagy. CYP2E1 has been reported to decrease autophagy in an acute in vivo alcohol model by a JNK-dependent mechanism, however the studies did not include actual measures of autophagic function [55]. This result runs counter to findings in non-hepatic cell types in which JNK acts as an inducer of autophagy [56]. In vitro inhibition of autophagy in CYP2E1-expressing HepG2 cells increases levels of active JNK that promote liver injury, suggesting that autophagy may also regulate the effects of CYP2E1 on JNK activation [57]. With regards to the regulation of lipid metabolism, acute alcohol exposure decreases autophagic function with a resulting increase in lipogenic sterol regulatory element-binding protein and subsequent hepatic steatosis [55]. These findings were supported by both in vitro and in vivo studies using cannabidiol, a cannabinoid receptor agonist. Pretreatment of mice and hepatoma cells with cannabidiol decreased hepatic steatosis following acute ethanol exposure by blocking autophagy [58]. The clinical applicability of these observations is limited by the nervous system side effects of cannabinoids, but peripherally acting cannabinoid agonists are being developed. Likewise, pharmacological inhibition of autophagy in a short-term in vivo ethanol model worsens steatosis and liver injury in CYP2E1-overexpressing mice but not in knockout mice [59]. Thus, overall the data suggest that CYP2E1-dependent JNK activation may impair autophagic function that is necessary to limit alcohol-induced steatosis and injury thereby providing a novel mechanism by which JNK activation promotes alcoholic liver disease.
Alcohol-induced CYP2E1 expression in cells other than hepatocytes
This review has focused on the relationship between CYP2E1 overexpression and JNK signaling in hepatocytes. Recent studies have documented that alcohol feeding increases CYP2E1 expression in cells other than hepatocytes including adipocytes, lymphocytes and monocytes. Adipocyte CYP2E1 overexpression promotes adipose tissue inflammation and reduces adiponectin secretion although these effects have not been linked to MAPKs [60,61]. With acute or chronic ethanol exposure CYP2E1 is increased in peripheral lymphocytes resulting in increased lipid peroxidation and activation of JNK and c-Jun in these cells [62]. This finding suggests that blood lymphocytes could serve as a more readily available source to determine human CYP2E1 and JNK activity and predict an individual’s likelihood to develop liver injury. In vitro ethanol treatment increases CYP2E1 expression in monocytes by a JNK-dependent mechanism [63]. In nonalcoholic liver disease controversy exists over whether it is JNK overactivation in hepatocytes and/or inflammatory macrophages that mediates the development of steatosis and hepatic injury [13]. The specific liver cell types that undergo JNK activation and mediate the various manifestations of alcoholic liver disease have not been established and require further study.
CYP2E1/JNK overexpression alters other forms of hepatocyte injury
Changes in cellular signaling pathways such as JNK MAPK that occur in response to chronic oxidant stress generated by alcohol-induced CYP2E1 expression can precondition the hepatocyte and alter the outcome of injury from other sources of acute oxidative stress. CYP2E1 overexpression in a non-transformed hepatocyte cell line sensitizes to APAP injury but protects against hydrogen peroxide- and superoxide-induced injury [64]. These findings demonstrate that alcohol-induced increases in CYP2E1 may have differential effects on cellular injury from other sources of oxidative stress in the liver. Low levels of oxidative stress from CYP2E1 overexpression induce protective ERK1/2 MAPK overactivation that decreases menadione-induced hepatocyte injury but sensitizes to cell death from fatty acids and 4-hydroxynonenal [65,66]. Likewise, increased expression of CYP2E1 in vitro and in vivo sensitizes hepatocytes to death from TNF [67,68], a central mediator of many forms of liver injury other than that from alcohol. In both studies the increase in injury was shown to be JNK mediated, indicating that synergistic effects of oxidant stress from CYP2E1 overexpression and other injurious stimuli on JNK can potentiate liver injury.
ROS-JNK signaling as a therapeutic target in fatty liver disease
The presumed central involvement of oxidative stress in many types of human pathophysiology has led to a number of clinical trials of antioxidant therapies. In general these investigations have shown no benefit, or even detrimental effects, on the disease process being treated [69]. This failure may be the result of the fact that low, physiological levels of ROS have critical beneficial effects through the modulation of cell signaling pathways. In hepatocytes, basal ROS production may mediate antioxidant responses that precondition these cells to block subsequent injury from higher, injurious levels of ROS [70]. The failure of antioxidant therapies has suggested that what may be needed is more directed therapy against downstream effectors of ROS-stimulated death pathways. In alcoholic liver disease, JNK in particular may be one target since JNK activation is central to the death effects of both CYP2E1 and other injurious factors such as proinflammatory cytokines. With excessive alcohol intake hepatic inflammation develops from disruption of the intestinal barrier, and translocation of lipopolysaccharide into the hepatic circulation causes Kupffer cell JNK activation and release of pro-inflammatory cytokines [71]. These cytokines - among which TNF is most prominent - in turn cause hepatocyte activation of JNK. Thus, JNK could serve as a more specific and central target in alcohol-induced chronic liver disease. However, despite the development of small molecule JNK inhibitors with nanomolar potency, their use has been tempered by the complexity of JNK’s biological effects including the ability of JNK isoforms to have different protective and pro-death effects [27]. It may therefore be necessary to target factors even more downstream of JNK such as Sab.
NAFLD bears many similarities to alcohol-induced fatty liver, and oxidant stress has been implicated as a critical mechanism of liver injury and inflammation in this disease as well [72]. Toxic lipids are thought to promote mitochondrial dysfunction, endoplasmic reticulum stress and resultant ROS generation that are in part responsible for disease progression to liver injury [73]. Interestingly CYP2E1 overexpression is seen in nonalcoholic fatty liver disease. Increased CYP2E1 expression occurs in the nutritional mouse models of NAFLD induced by a methionine- choline-deficient or high fat diet [74,75]. CYP2E1 overexpression also occurs in human NAFLD [76,77]. In high fat diet-induced murine NAFLD, CYP2E1 promotes the development of fat accumulation, oxidative stress, insulin resistance and liver injury as demonstrated by a reduction in all of these events in CYP2E1 knockout mice [74]. JNK overexpression is also mechanistically linked to both hepatic steatosis and injury in nonalcoholic fatty liver injury [28,46], but whether CYP2E1 overexpression contributes to JNK activation in this disease is unknown. CYP2E1 null mice do have decreased JNK activation in response to HFD-feeding, but it is has not been determined whether this effect is a mechanism of the decreased liver injury in these mice, or merely an effect secondary to the prevention of liver damage.
Increased expression of CYP2E1 may also affect hepatocyte insulin sensitivity in NAFLD. CYP2E1-induced oxidant stress decreases insulin-mediated reductions in hepatic gluconeogenesis through increased inhibitory serine phosphorylation of insulin receptor substrate 1 in hepatocytes [78]. The role of CYP2E1 in insulin resistance is further supported by studies in which hepatocyte-specific overexpression of human CYP2E1 in mice led to increased insulin resistance with impaired insulin signaling and decreased activation of the downstream transcription factor FoxO1a even on a standard diet with only 20% of calories derived from fat [79]. Insulin itself decreases CYP2E1 expression by a regulatory feedback loop dependent on phosphatidylinositol 3-kinase signaling [80]. Conversely, ketone bodies, a catabolic product of metabolism in the absence of insulin, elevate CYP2E1 protein levels by posttranslational mechanisms to increase insulin resistance [81]. In human NAFLD other complex interactions may regulate CYP2E1 in the context of insulin resistance such as hypoxia from the associated syndrome of obstructive sleep apnea [82]. In mice with a hepatocyte knockout of the phosphatase and tensin homolog deleted on chromosome 10 gene, hypoxia aggravated steatohepatitis in concert with increased expression of CYP2E1 [83].
Although antioxidant administration has not been shown to have beneficial therapeutic effects in alcoholic liver disease in the clinical trials conducted to date, a multicenter, placebo-controlled trial has demonstrated beneficial effects of vitamin E in nonalcoholic steatohepatitis [84]. In this trial inflammation, signs of hepatocellular injury (ballooning) and hepatic steatosis, but not hepatic fibrosis, improved in vitamin E-treated patients. Only 43% of nonalcoholic steatohepatitis patients on vitamin E responded, and subsequent analysis revealed that a positive response was associated with decreased activation of the hedgehog signaling pathway [85]. Responsiveness was dependent on genetic polymorphisms of the vitamin E metabolizing cytochrome CYP4F2 [86]. In alcoholic liver disease, vitamin E exerts antioxidant effects in animal models of ethanol exposure [87], however these effects have been not successfully transferred into the clinic. Further studies that aim at identifying selective and targetable mediators of the CYP2E1-ROS-JNK injury cascade may yield new therapies for alcoholic liver disease, but may have to account for genetic differences towards antioxidant treatment.
Conclusions
Oxidant stress from excessive ROS generation regulates physiological and pathophysiological processes in the liver and contributes to a variety of liver diseases. The generation of ROS by CYP2E1 is centrally involved in alcoholic, and perhaps nonalcoholic liver disease as well, through the regulation of hepatic steatosis, hepatocellular death and insulin sensitivity. Critical to the ability of CYP2E1 overexpression to promote alcoholic liver disease is the overactivation of JNK MAPK. The mechanisms by which CYP2E1-induced JNK overactivation modulates alcoholic liver injury remain to be determined but may be through additive effects on JNK activation together with other injurious factors such as the proinflammatory cytokine TNF. Alternatively CYP2E1-induced JNK activation may alter cellular processes such as autophagy. A better understanding of the downstream effects of the CYP2E1-ROS-JNK cascade may lead to the development of more targeted and therefore effective therapies for alcoholic liver disease.
Acknowledgements
JMS received support from the Deutsche Forschungsgemeinschaft (DFG) (Grant no. schu1239/3-1) and the Deutsche Krebshilfe (Grant no. 110994), MJC received support from the National Institutes of Health Grants R01DK044234, R01DK061498 and R01AA022601.
References
- 1.Cederbaum A.I. Role of CYP2E1 in ethanol-induced oxidant stress, fatty liver and hepatotoxicity. Digestive Diseases (Basel, Switzerland) 2010;28:802–811. doi: 10.1159/000324289. 21525766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Osawa Y., Nagaki M., Banno Y., Yamada Y., Imose M., Nozawa Y., Moriwaki H., Nakashima S. Possible involvement of reactive oxygen species in D-galactosamine-induced sensitization against tumor necrosis factor-alpha-induced hepatocyte apoptosis. Journal of Cellular Physiology. 2001;187:374–385. doi: 10.1002/jcp.1088. 11319761 [DOI] [PubMed] [Google Scholar]
- 3.Ramachandran A., Lebofsky M., Baines C.P., Lemasters J.J., Jaeschke H. Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free Radical Research. 2011;45:156–164. doi: 10.3109/10715762.2010.520319. 20942566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lesage G., Glaser S., Ueno Y., Alvaro D., Baiocchi L., Kanno N., Phinizy J.L., Francis H., Alpini G. Regression of cholangiocyte proliferation after cessation of ANIT feeding is coupled with increased apoptosis. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2001;281:G182–G190. doi: 10.1152/ajpgi.2001.281.1.G182. 11408271 [DOI] [PubMed] [Google Scholar]
- 5.Yerushalmi B., Dahl R., Devereaux M.W., Gumpricht E., Sokol R.J. Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology (Baltimore, MD) 2001;33:616–626. doi: 10.1053/jhep.2001.22702. 11230742 [DOI] [PubMed] [Google Scholar]
- 6.Zhou W., Zhang Y., Hosch M.S., Lang A., Zwacka R.M., Engelhardt J.F. Subcellular site of superoxide dismutase expression differentially controls AP-1 activity and injury in mouse liver following ischemia/reperfusion. Hepatology (Baltimore, MD) 2001;33:902–914. doi: 10.1053/jhep.2001.23073. 11283855 [DOI] [PubMed] [Google Scholar]
- 7.Czaja M.J. Cell signaling in oxidative stress-induced liver injury. Seminars in Liver Disease. 2007;27:378–389. doi: 10.1055/s-2007-991514. 17979074 [DOI] [PubMed] [Google Scholar]
- 8.Singh R., Czaja M.J. Regulation of hepatocyte apoptosis by oxidative stress. Journal of Gastroenterology and Hepatology. 2007;22(Suppl. 1):S45–S48. doi: 10.1111/j.1440-1746.2006.04646.x. 17567464 [DOI] [PubMed] [Google Scholar]
- 9.Holmström K.M., Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nature Reviews. Molecular Cell Biology. 2014;15:411–421. doi: 10.1038/nrm3801. 24854789 [DOI] [PubMed] [Google Scholar]
- 10.Davis R.J. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. 11057897 [DOI] [PubMed] [Google Scholar]
- 11.Czaja M.J. The future of GI and liver research: Editorial perspectives. III. JNK/AP-1 regulation of hepatocyte death. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2003;284:G875–G879. doi: 10.1152/ajpgi.00549.2002. 12736142 [DOI] [PubMed] [Google Scholar]
- 12.Czaja M.J. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends in Endocrinology and Metabolism: TEM. 2010;21:707–713. doi: 10.1016/j.tem.2010.08.010. 20888782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seki E., Brenner D.A., Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–320. doi: 10.1053/j.gastro.2012.06.004. 22705006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weston C.R., Davis R.J. The JNK signal transduction pathway. Current Opinion in Genetics and Development. 2002;12:14–21. doi: 10.1016/s0959-437x(01)00258-1. 11790549 [DOI] [PubMed] [Google Scholar]
- 15.Constant S.L., Dong C., Yang D.D., Wysk M., Davis R.J., Flavell R.A. JNK1 is required for T cell-mediated immunity against Leishmania major infection. Journal of Immunology (Baltimore, MD: 1950) 2000;165:2671–2676. doi: 10.4049/jimmunol.165.5.2671. 10946297 [DOI] [PubMed] [Google Scholar]
- 16.Kuan C.Y., Yang D.D., Samanta Roy D.R., Davis R.J., Rakic P., Flavell R.A. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. 10230788 [DOI] [PubMed] [Google Scholar]
- 17.Sabapathy K., Jochum W., Hochedlinger K., Chang L., Karin M., Wagner E.F. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mechanisms of Development. 1999;89:115–124. doi: 10.1016/s0925-4773(99)00213-0. 10559486 [DOI] [PubMed] [Google Scholar]
- 18.Schattenberg J.M., Wörns M.A., Zimmermann T., He Y.W., Galle P.R., Schuchmann M. The role of death effector domain-containing proteins in acute oxidative cell injury in hepatocytes. Free Radical Biology and Medicine. 2012;52:1911–1917. doi: 10.1016/j.freeradbiomed.2012.02.049. 22406316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen W., White M.A., Cobb M.H. Stimulus-specific requirements for MAP3 kinases in activating the JNK pathway. Journal of Biological Chemistry. 2002;277:49105–49110. doi: 10.1074/jbc.M204934200. 12351623 [DOI] [PubMed] [Google Scholar]
- 20.Morrison D.K., Davis R.J. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annual Review of Cell and Developmental Biology. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 21.Keyse S.M. Protein phosphatases and the regulation of MAP kinase activity. Seminars in Cell and Developmental Biology. 1998;9:143–152. doi: 10.1006/scdb.1997.0219. 9599409 [DOI] [PubMed] [Google Scholar]
- 22.Kamata H., Honda S., Maeda S., Chang L., Hirata H., Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. 15766528 [DOI] [PubMed] [Google Scholar]
- 23.Venugopal S.K., Chen J., Zhang Y., Clemens D., Follenzi A., Zern M.A. Role of MAPK phosphatase-1 in sustained activation of JNK during ethanol-induced apoptosis in hepatocyte-like VL-17A cells. Journal of Biological Chemistry. 2007;282:31900–31908. doi: 10.1074/jbc.M703729200. 17848570 [DOI] [PubMed] [Google Scholar]
- 24.Bogoyevitch M.A., Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiology and Molecular Biology Reviews: MMBR. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. 17158707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Czaja M.J., Liu H., Wang Y. Oxidant-induced hepatocyte injury from menadione is regulated by ERK and AP-1 signaling. Hepatology (Baltimore, MD) 2003;37:1405–1413. doi: 10.1053/jhep.2003.50233. 12774020 [DOI] [PubMed] [Google Scholar]
- 26.Liu H., Lo C.R., Czaja M.J. NF-kappaB inhibition sensitizes hepatocytes to TNF-induced apoptosis through a sustained activation of JNK and c-Jun. Hepatology (Baltimore, MD) 2002;35:772–778. doi: 10.1053/jhep.2002.32534. 11915022 [DOI] [PubMed] [Google Scholar]
- 27.Amir M., Liu K., Zhao E., Czaja M.J. Distinct functions of JNK and c-Jun in oxidant-induced hepatocyte death. Journal of Cellular Biochemistry. 2012;113:3254–3265. doi: 10.1002/jcb.24203. 22644775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh R., Wang Y., Xiang Y., Tanaka K.E., Gaarde W.A., Czaja M.J. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology (Baltimore, MD) 2009;49:87–96. doi: 10.1002/hep.22578. 19053047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conde de la Rosa L., Schoemaker M.H., Vrenken T.E., Buist-Homan M., Havinga R., Jansen P.L., Moshage H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. Journal of Hepatology. 2006;44:918–929. doi: 10.1016/j.jhep.2005.07.034. 16310883 [DOI] [PubMed] [Google Scholar]
- 30.Aroor A.R., Jackson D.E., Shukla S.D. Elevated activation of ERK1 and ERK2 accompany enhanced liver injury following alcohol binge in chronically ethanol-fed rats. Alcoholism, Clinical and Experimental Research. 2011;35:2128–2138. doi: 10.1111/j.1530-0277.2011.01577.x. 21790671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y., Schattenberg J.M., Rigoli R.M., Storz P., Czaja M.J. Hepatocyte resistance to oxidative stress is dependent on protein kinase C-mediated down-regulation of c-Jun/AP-1. Journal of Biological Chemistry. 2004;279:31089–31097. doi: 10.1074/jbc.M404170200. 15145937 [DOI] [PubMed] [Google Scholar]
- 32.Conde de la Rosa L., Vrenken T.E., Hannivoort R.A., Buist-Homan M., Havinga R., Slebos D.J., Kauffman H.F., Faber K.N., Jansen P.L., Moshage H. Carbon monoxide blocks oxidative stress-induced hepatocyte apoptosis via inhibition of the p54 JNK isoform. Free Radical Biology Medicine. 2008;44:1323–1333. doi: 10.1016/j.freeradbiomed.2007.12.011. 18206660 [DOI] [PubMed] [Google Scholar]
- 33.Rosseland C.M., Wierød L., Oksvold M.P., Werner H., Ostvold A.C., Thoresen G.H., Paulsen R.E., Huitfeldt H.S., Skarpen E. Cytoplasmic retention of peroxide-activated ERK provides survival in primary cultures of rat hepatocytes. Hepatology (Baltimore, MD) 2005;42:200–207. doi: 10.1002/hep.20762. 15962331 [DOI] [PubMed] [Google Scholar]
- 34.David Josephy P. The molecular toxicology of acetaminophen. Drug Metabolism Reviews. 2005;37:581–594. doi: 10.1080/03602530500205200. 16393886 [DOI] [PubMed] [Google Scholar]
- 35.Gunawan B.K., Liu Z.X., Han D., Hanawa N., Gaarde W.A., Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–178. doi: 10.1053/j.gastro.2006.03.045. 16831600 [DOI] [PubMed] [Google Scholar]
- 36.Hanawa N., Shinohara M., Saberi B., Gaarde W.A., Han D., Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. Journal of Biological Chemistry. 2008;283:13565–13577. doi: 10.1074/jbc.M708916200. 18337250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Win S., Than T.A., Han D., Petrovic L.M., Kaplowitz N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. Journal of Biological Chemistry. 2011;286:35071–35078. doi: 10.1074/jbc.M111.276089. 21844199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leung T.M., Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. Journal of Hepatology. 2013;58:395–398. doi: 10.1016/j.jhep.2012.08.018. 22940046 [DOI] [PubMed] [Google Scholar]
- 39.Cederbaum A.I., Lu Y., Wu D. Role of oxidative stress in alcohol-induced liver injury. Archives of Toxicology. 2009;83:519–548. doi: 10.1007/s00204-009-0432-0. 19448996 [DOI] [PubMed] [Google Scholar]
- 40.Lu Y., Wu D., Wang X., Ward S.C., Cederbaum A.I. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free Radical Biology and Medicine. 2010;49:1406–1416. doi: 10.1016/j.freeradbiomed.2010.07.026. 20692331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu D., Cederbaum A. Activation of ASK-1 and downstream MAP kinases in cytochrome P4502E1 potentiated tumor necrosis factor alpha liver injury. Free Radical Biology and Medicine. 2010;49:348–360. doi: 10.1016/j.freeradbiomed.2010.04.021. 20438834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cederbaum A.I., Yang L., Wang X., Wu D. CYP2E1 sensitizes the liver to LPS- and TNF α-induced toxicity via elevated oxidative and nitrosative stress and activation of ASK-1 and JNK mitogen-activated kinases. International Journal of Hepatology. 2012;2012:582790. doi: 10.1155/2012/582790. 22028977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhuge J., Cederbaum A.I. Inhibition of the mitochondrial permeability transition by cyclosporin A prevents pyrazole plus lipopolysaccharide-induced liver injury in mice. Free Radical Biology and Medicine. 2009;46:406–413. doi: 10.1016/j.freeradbiomed.2008.10.037. 19026739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X., Cederbaum A.I. S-adenosyl-l-methionine decreases the elevated hepatotoxicity induced by Fas agonistic antibody plus acute ethanol pretreatment in mice. Archives of Biochemistry and Biophysics. 2008;477:1–11. doi: 10.1016/j.abb.2008.04.033. 18482574 [DOI] [PubMed] [Google Scholar]
- 45.Wang X., Wu D., Yang L., Cederbaum A.I. Hepatotoxicity mediated by pyrazole (cytochrome P450 2E1) plus tumor necrosis factor alpha treatment occurs in c-Jun N-terminal kinase 2−/− but not in c-Jun N-terminal kinase 1−/− mice. Hepatology (Baltimore, MD) 2011;54:1753–1766. doi: 10.1002/hep.24540. 21748763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schattenberg J.M., Singh R., Wang Y., Lefkowitch J.H., Rigoli R.M., Scherer P.E., Czaja M.J. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology (Baltimore, MD) 2006;43:163–172. doi: 10.1002/hep.20999. 16374858 [DOI] [PubMed] [Google Scholar]
- 47.Jimenez-Lopez J.M., Wu D., Cederbaum A.I. Synergistic toxicity induced by prolonged glutathione depletion and inhibition of nuclear factor-kappaB signaling in liver cells. Toxicology in Vitro: An International Journal Published in Association with BIBRA. 2008;22:106–115. doi: 10.1016/j.tiv.2007.08.012. 17920235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang L., Wu D., Wang X., Cederbaum A.I. Depletion of cytosolic or mitochondrial thioredoxin increases CYP2E1-induced oxidative stress via an ASK-1-JNK1 pathway in HepG2 cells. Free Radical Biology and Medicine. 2011;51:185–196. doi: 10.1016/j.freeradbiomed.2011.04.030. 21557999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Czaja M.J. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology. 2011;140:1895–1908. doi: 10.1053/j.gastro.2011.04.038. 21530520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Czaja M.J. Autophagy in health and disease. 2. Regulation of lipid metabolism and storage by autophagy: pathophysiological implications. American Journal of Physiology. Cell Physiology. 2010;298:C973–C978. doi: 10.1152/ajpcell.00527.2009. 20089934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding W.X., Li M., Chen X., Ni H.M., Lin C.W., Gao W., Lu B., Stolz D.B., Clemens D.L., Yin X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. 20659474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dolganiuc A., Thomes P.G., Ding W.X., Lemasters J.J., Donohue T.M., Jr. Autophagy in alcohol-induced liver diseases. Alcoholism, Clinical and Experimental Research. 2012;36:1301–1308. doi: 10.1111/j.1530-0277.2012.01742.x. 22551004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y., Singh R., Xiang Y., Czaja M.J. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology (Baltimore, MD) 2010;52:266–277. doi: 10.1002/hep.23645. 20578144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin C.W., Zhang H., Li M., Xiong X., Chen X., Chen X., Dong X.C., Yin X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. Journal of Hepatology. 2013;58:993–999. doi: 10.1016/j.jhep.2013.01.011. 23339953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang L., Wu D., Wang X., Cederbaum A.I. Cytochrome P4502E1, oxidative stress, JNK, and autophagy in acute alcohol-induced fatty liver. Free Radical Biology Medicine. 2012;53:1170–1180. doi: 10.1016/j.freeradbiomed.2012.06.029. 22749809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei Y., Pattingre S., Sinha S., Bassik M., Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Molecular Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. 18570871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu D., Cederbaum A.I. Inhibition of autophagy promotes CYP2E1-dependent toxicity in HepG2 cells via elevated oxidative stress, mitochondria dysfunction and activation of p38 and JNK MAPK. Redox Biology. 2013;1:552–565. doi: 10.1016/j.redox.2013.10.008. 24273738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang L., Rozenfeld R., Wu D., Devi L.A., Zhang Z., Cederbaum A. Cannabidiol protects liver from binge alcohol-induced steatosis by mechanisms including inhibition of oxidative stress and increase in autophagy. Free Radical Biology andMedicine. 2014;68:260–267. doi: 10.1016/j.freeradbiomed.2013.12.026. 24398069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu D., Wang X., Zhou R., Yang L., Cederbaum A.I. Alcohol steatosis and cytotoxicity: The role of cytochrome P4502E1 and autophagy. Free Radical Biology and Medicine. 2012;53:1346–1357. doi: 10.1016/j.freeradbiomed.2012.07.005. 22819980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang H., Sebastian B.M., Axhemi A., Chen X., Hillian A.D., Jacobsen D.W., Nagy L.E. Ethanol-induced oxidative stress via the CYP2E1 pathway disrupts adiponectin secretion from adipocytes. Alcoholism, Clinical and Experimental Research. 2012;36:214–222. doi: 10.1111/j.1530-0277.2011.01607.x. 21895711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sebastian B.M., Roychowdhury S., Tang H., Hillian A.D., Feldstein A.E., Stahl G.L., Takahashi K., Nagy L.E. Identification of a cytochrome P4502E1/Bid/C1q-dependent axis mediating inflammation in adipose tissue after chronic ethanol feeding to mice. Journal of Biological Chemistry. 2011;286:35989–35997. doi: 10.1074/jbc.M111.254201. 21856753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma A., Saurabh K., Yadav S., Jain S.K., Parmar D. Ethanol induced induction of cytochrome P450 2E1 and activation of mitogen activated protein kinases in peripheral blood lymphocytes. Xenobiotica; The Fate of Foreign Compounds in Biological Systems. 2012;42:317–326. doi: 10.3109/00498254.2011.624648. 21999510 [DOI] [PubMed] [Google Scholar]
- 63.Jin M., Ande A., Kumar A., Kumar S. Regulation of cytochrome P450 2e1 expression by ethanol: role of oxidative stress-mediated pkc/jnk/sp1 pathway. Cell Death and Disease. 2013;4:e554. doi: 10.1038/cddis.2013.78. 23519123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jones B.E., Liu H., Lo C.R., Koop D.R., Czaja M.J. Cytochrome P450 2E1 expression induces hepatocyte resistance to cell death from oxidative stress. Antioxidants a Redox Signaling. 2002;4:701–709. doi: 10.1089/152308602760598846. 12470497 [DOI] [PubMed] [Google Scholar]
- 65.Schattenberg J.M., Wang Y., Rigoli R.M., Koop D.R., Czaja M.J. CYP2E1 overexpression alters hepatocyte death from menadione and fatty acids by activation of ERK1/2 signaling. Hepatology (Baltimore, MD) 2004;39:444–455. doi: 10.1002/hep.20067. 14767997 [DOI] [PubMed] [Google Scholar]
- 66.Singh R., Wang Y., Schattenberg J.M., Xiang Y., Czaja M.J. Chronic oxidative stress sensitizes hepatocytes to death from 4-hydroxynonenal by JNK/c-Jun overactivation. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2009;297:G907–G917. doi: 10.1152/ajpgi.00151.2009. 20501438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu H., Jones B.E., Bradham C., Czaja M.J. Increased cytochrome P-450 2E1 expression sensitizes hepatocytes to c-Jun-mediated cell death from TNF-alpha. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2002;282:G257–G266. doi: 10.1152/ajpgi.00304.2001. 11804847 [DOI] [PubMed] [Google Scholar]
- 68.Wang X., Lu Y., Xie B., Cederbaum A.I. Chronic ethanol feeding potentiates Fas Jo2-induced hepatotoxicity: Role of CYP2E1 and TNF-alpha and activation of JNK and P38 MAP kinase. Free Radical Biology and Medicine. 2009;47:518–528. doi: 10.1016/j.freeradbiomed.2009.05.021. 19477265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ristow M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nature Medicine. 2014;20:709–711. doi: 10.1038/nm.3624. 24999941 [DOI] [PubMed] [Google Scholar]
- 70.Kawagishi H., Finkel T. Unraveling the truth about antioxidants: ROS and disease: finding the right balance. Nature Medicine. 2014;20:711–713. doi: 10.1038/nm.3625. 24999942 [DOI] [PubMed] [Google Scholar]
- 71.Stickel F., Seitz H.K. Update on the management of alcoholic steatohepatitis. Journal of Gastrointestinal and Liver Diseases: JGLD. 2013;22:189–197. 23799218 [PubMed] [Google Scholar]
- 72.Tariq Z., Green C.J., Hodson L. Are oxidative stress mechanisms the common denominator in the progression from hepatic steatosis towards non-alcoholic steatohepatitis (Nash)? Liver International: Official Journal of the International Association for the Study of the Liver. 2014;34:e180–e190. doi: 10.1111/liv.12523. 24621397 [DOI] [PubMed] [Google Scholar]
- 73.Neuschwander-Tetri B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology (Baltimore, MD) 2010;52:774–788. doi: 10.1002/hep.23719. 20683968 [DOI] [PubMed] [Google Scholar]
- 74.Abdelmegeed M.A., Banerjee A., Yoo S.H., Jang S., Gonzalez F.J., Song B.J. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. Journal of Hepatology. 2012;57:860–866. doi: 10.1016/j.jhep.2012.05.019. 22668639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weltman M.D., Farrell G.C., Liddle C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology. 1996;111:1645–1653. doi: 10.1016/s0016-5085(96)70028-8. 8942745 [DOI] [PubMed] [Google Scholar]
- 76.Chalasani N., Gorski J.C., Asghar M.S., Asghar A., Foresman B., Hall S.D., Crabb D.W. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology (Baltimore, MD) 2003;37:544–550. doi: 10.1053/jhep.2003.50095. 12601351 [DOI] [PubMed] [Google Scholar]
- 77.Weltman M.D., Farrell G.C., Hall P., Ingelman-Sundberg M., Liddle C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology (Baltimore, MD) 1998;27:128–133. doi: 10.1002/hep.510270121. 9425928 [DOI] [PubMed] [Google Scholar]
- 78.Schattenberg J.M., Wang Y., Singh R., Rigoli R.M., Czaja M.J. Hepatocyte CYP2E1 overexpression and steatohepatitis lead to impaired hepatic insulin signaling. Journal of Biological Chemistry. 2005;280:9887–9894. doi: 10.1074/jbc.M410310200. 15632182 [DOI] [PubMed] [Google Scholar]
- 79.Kathirvel E., Morgan K., French S.W., Morgan T.R. Overexpression of liver-specific cytochrome P4502E1 impairs hepatic insulin signaling in a transgenic mouse model of nonalcoholic fatty liver disease. European Journal of Gastroenterology and Hepatology. 2009;21:973–983. doi: 10.1097/MEG.0b013e328328f461. 19307976 [DOI] [PubMed] [Google Scholar]
- 80.Woodcroft K.J., Hafner M.S., Novak R.F. Insulin signaling in the transcriptional and posttranscriptional regulation of CYP2E1 expression. Hepatology (Baltimore, MD) 2002;35:263–273. doi: 10.1053/jhep.2002.30691. 11826398 [DOI] [PubMed] [Google Scholar]
- 81.Abdelmegeed M.A., Carruthers N.J., Woodcroft K.J., Kim S.K., Novak R.F. Acetoacetate induces CYP2E1 protein and suppresses CYP2E1 mRNA in primary cultured rat hepatocytes. Journal of Pharmacology and Experimental Therapeutics. 2005;315:203–213. doi: 10.1124/jpet.105.084608. 15980059 [DOI] [PubMed] [Google Scholar]
- 82.Musso G., Cassader M., Olivetti C., Rosina F., Carbone G., Gambino R. Association of obstructive sleep apnoea with the presence and severity of non-alcoholic fatty liver disease. A systematic review and meta-analysis. Obesity Reviews: An Official Journal of the International Association for the Study of Obesity. 2013;14:417–431. doi: 10.1111/obr.12020. 23387384 [DOI] [PubMed] [Google Scholar]
- 83.Piguet A.C., Stroka D., Zimmermann A., Dufour J.F. Hypoxia aggravates non-alcoholic steatohepatitis in mice lacking hepatocellular PTEN. Clinical Science (London, England: 1979) 2010;118:401–410. doi: 10.1042/CS20090313. 19832698 [DOI] [PubMed] [Google Scholar]
- 84.Sanyal A.J., Chalasani N., Kowdley K.V., McCullough A., Diehl A.M., Bass N.M., Neuschwander-Tetri B.A., Lavine J.E., Tonascia J., Unalp A., Van Natta M., Clark J., Brunt E.M., Kleiner D.E., Hoofnagle J.H., Robuck P.R. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. New England Journal of Medicine. 2010;362:1675–1685. doi: 10.1056/NEJMoa0907929. 20427778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guy C.D., Suzuki A., Abdelmalek M.F., Burchette J.L., Diehl A.M. Treatment response in the PIVENS Trial is associated with decreased hedgehog pathway activity. Hepatology (Baltimore, MD) 2014 doi: 10.1002/hep.27235. 24849310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Athinarayanan S., Wei R., Zhang M., Bai S., Traber M.G., Yates K., Cummings O.W., Molleston J., Liu W., Chalasani N. Genetic polymorphism of cytochrome P450 4F2, vitamin E level and histological response in adults and children with nonalcoholic fatty liver disease who participated in PIVENS and TONIC clinical trials. PloS One. 2014;9:e95366. doi: 10.1371/journal.pone.0095366. 24759732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McDonough K.H. Antioxidant nutrients and alcohol. Toxicology. 2003;189:89–97. doi: 10.1016/s0300-483x(03)00155-0. 12821285 [DOI] [PubMed] [Google Scholar]