

Abstract
Background
Decades of intensive tomato breeding using wild-species germplasm have resulted in the genomes of domesticated germplasm (Solanum lycopersicum) being intertwined with introgressions from their wild relatives. Comparative analysis of genomes among cultivated tomatoes and wild species that have contributed genetic variation can help identify desirable genes, such as those conferring disease resistance. The ability to identify introgression position, borders, and contents can reveal ancestral origins and facilitate harnessing of wild variation in crop breeding.
Results
Here we present the whole-genome sequences of two tomato inbreds, Gh13 and BTI-87, both carrying the begomovirus resistance locus Ty-3 introgressed from wild tomato species. Introgressions of different sizes on chromosome 6 of Gh13 and BTI-87, both corresponding to the Ty-3 region, were identified as from a source close to the wild species S. chilense. Other introgressions were identified throughout the genomes of the inbreds and showed major differences in the breeding pedigrees of the two lines. Interestingly, additional large introgressions from the close tomato relative S. pimpinellifolium were identified in both lines. Some of the polymorphic regions were attributed to introgressions in the reference Heinz 1706 genome, indicating wild genome sequences in the reference tomato genome.
Conclusions
The methods developed in this work can be used to delineate genome introgressions, and subsequently contribute to development of molecular markers to aid phenotypic selection, fine mapping and discovery of candidate genes for important phenotypes, and for identification of novel variation for tomato improvement. These universal methods can easily be applied to other crop plants.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-014-0287-2) contains supplementary material, which is available to authorized users.
Keywords: Solanum lycopersicum, Solanum pimpinellifolium, Solanum chilense, Genomic introgressions, Genome sequencing, Disease resistance, Single nucleotide polymorphism, Wild species, Domestication, Phylogenetics
Background
A priority in modern plant breeding is the introduction of novel variation for desirable traits; Biotic and abiotic stresses are the most crucial to increase yield and provide reliable food production. Tomato (Solanum lycopersicum) is an important food crop and a model species for studying processes such as fleshy fruit ripening, fruit development [1], and the molecular basis of disease resistance [2,3].
Tomato originated in the South American Andean mountains, deserts, and coastal plains [4]. During the domestication of tomato from its ancestral wild species, the tomato genome went through a genetic bottleneck, reducing its genetic diversity to less than 5% of the diversity found in its closest wild relatives [5,6]. Moreover, human selection for traits related to yield and fruit qualities, such as size, weight, color, sugar content, and shelf life, has disregarded disease resistance traits. Consequently, tomato heirloom cultivars are susceptible to many pathogens, including bacteria, viruses, fungi, nematodes and insect pests, and resistance alleles are present only in wild tomato relatives [7]. Since these species can be outcrossed with cultivated ones, breeders have introgressed wild genomes into cultivated varieties since 1917 [8,9], a practice that continues today [7]. Most disease resistance genes have been introgressed from wild species such as Solanum chilense [10-12], S. peruvianum [13-15], S. habrochaites [16], S. pennellii [17], and S. pimpinellifolium [7,18].
Begomoviruses cause major diseases affecting tomatoes in tropical and subtropical regions. Symptoms vary, but all involve some level of leaf distortion and reduction of growth and yield [19-21]. Management strategies for control of begomovirus-incited tomato diseases have traditionally focused on the insect vector [22]. For begomovirus resistance, at least four loci have been introgressed into tomato from three accessions of S. chilense and S. habrochaites [11,16,21,23].
The release of the reference tomato genome sequence (variety Heinz 1706) in early 2012 has enabled a multitude of new genetic and genomic approaches [24], such as mapping reads from re-sequenced breeding lines. Using the mapping approach, genome regions that contain a limited number of SNPs can be efficiently aligned to the reference sequence, and using paired-end sequencing, insertions and deletions can be detected. However, large insertions and regions that are highly divergent cannot easily be characterized using this mapping approach. More high quality de novo assemblies of reference genomes, especially of wild germplasm, are required for the analysis of re-sequenced genome regions that cannot be mapped using the existing resources [25].
Since virtually all tomato disease resistance genes originate from wild relatives, further knowledge of these genomes will facilitate introgression of multiple disease resistances into elite cultivars. Also, while all tomato species share largely syntenic genomes and can outcross, the genome content of the reference genome is not completely identical even to other commercial tomato cultivars. For example, the fruit shape gene SUN has been duplicated in some varieties, but its functional copy is not present in Heinz 1706 (H1706) [26]. Another example is the bacterial resistance gene Pto, which was introgressed from the wild tomato species, S. pimpinellifolium, in the 1930’s and later positionally cloned [2,27]. A functional version of this gene is also missing in H1706.
Introgression of wild-species genomic regions into domesticated species is a widely used practice for increasing diversity in tomato as well as other crop species [28]. After several generations of backcrossing and selection, larger introgressions carrying favorable traits, as well as cryptic introgressions, are present throughout the genome. While excellent genetic maps exist for tomato [29], many of the available maps are not very dense and do not allow the precise definition of introgression points. The selection process can be accompanied by linkage-drag, producing genomes with tightly linked detrimental alleles, which require many rounds of backcrossing and fine-mapping to eliminate [30]. Thus, the ability to define the borders and contents of wild-species introgressions can contribute significantly to reducing the number of generations required for selecting favorable alleles while minimizing negative variation. Identification of introgressions can help to identify candidate genes responsible for beneficial traits such as disease resistance [31].
Other crops, such as maize, rice, barley [32], bean [33], and melon [34], exhibit wild introgression patterns similar to those found in tomato. These genomes, and those of tomatoes [35], have been studied recently using high-density SNP chips. However, while these technologies are excellent in detecting traits in populations and revealing population structure [36], they are less informative in defining introgression borders and their content. On the other hand, the whole-genome sequencing approach provides more detailed information on genic content and the origins of the introgressed regions through comparison to genomes of wild species involved in the breeding process [37]. Other work related to re-sequencing tomato genomes was published recently, and demonstrates how SNP calling in lines of domesticated tomatoes can reveal substantial differences between domesticated accessions due to wild introgressions [38]. Re-sequencing of tomato accessions has also been used in genome-wide association studies (GWAS) for associating SNPs with agronomically important traits [39].
For this study, two begomovirus-resistant inbreds were chosen, Gh13 [40] and BTI-87 (D.P. Maxwell, unpublished data), which are presumed to originate from different accessions. Gh13 was developed in Guatemala [41] were it has been tested over multiple seasons and consistently shows very good resistance to high begomovirus pressure. Resistance in Gh13 was, until now, presumably derived from S. habrochaites [42]. BTI-87 was also developed in Guatemala and maintains a high level of resistance derived from the begomovirus-resistant inbred Gc171, which is in turn derived from S. chilense accession LA1932 [43]. Both inbred lines carry a Ty-3 resistance allele, as well as several other resistance genes from several wild accession sources.
We used whole-genome sequencing (WGS) to detect introgressions from wild species in two begomovirus-resistant inbreds. The boundaries of the introgressions were established and the source of several introgressions was determined (Figure 1). The findings provide insight into the genome structure of tomato inbreds derived from a breeding program, and demonstrate how breeding can greatly benefit from WGS, which can diminish time consuming phenotypic screening.
Figure 1.
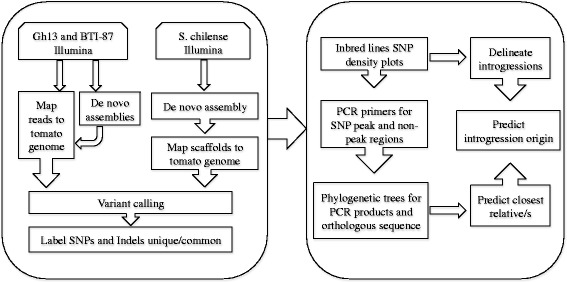
Schematic view of the genome assembly and the introgression detection pipelines.
Results
Sequencing and assembly
Paired-end libraries of the Gh13 and BTI-87 genomes were each sequenced in one Illumina HiSeq lane. Mapping the Gh13 genome to the reference tomato H1706 genome yielded 14.7× coverage of the H1706 genome, after removing low quality reads and duplicates, with 97.6% coverage of the reference genome. Gaps in the Gh13 genome were estimated to span 9.2 Mb, and the total number of SNPs was 288,640 (Table 1). The BTI-87 genome mapping to the reference tomato genome yielded coverage of 32.3×, represented 96.5% of the H1706 genome, with 79.9 Mb of gaps in the assembly, and 702,560 SNPs (Table 1), and 77,652 shared SNPs with Gh13, compared to the reference tomato genome.
Table 1.
Reference-guided assembly metrics
| Heinz 1706^ | LA1589 | Gh13 | BTI-87 | LA1932* | |
|---|---|---|---|---|---|
| Filtered reads in millions | 462.7 | 281.5 | 392.9 | 402.267066 | |
| Mapped reads (% mapped) | 426.1 (92.1%) | 247.7 (88%) | 385.4 (98%) | 380.9 (94.7%) | |
| Coverage depth | 39.3 | 25 | 14.7 | 32.3 | |
| Coverage of tomato genome | 0.992 | 0.95 | 0.976 | 0.965 | |
| Number of gaps (Mb) | 76,276 (5.9) | 209,919 (38.9) | 90,727 (9.2) | 165,894 (79.9) | |
| Gaps >500 bp | 1,660 | 14,396 | 3,058 | 19,479 | |
| Gaps >5000 bp | 247 | 286 | |||
| SNPs | 2,753,307 (0.35%) | 288,640 (0.037%) | 702,560 (0.09%) | 8,123,431 (1%) | |
| Indels | 437,943 | 69,289 | 130,029 | 718,185 |
^Subset of the available libraries for comparison purposes.
*Low coverage reference-based assembly.
LA1589 (S. pimpinellifolium) and LA1932 (S. chilense).
The major difference in coverage depth between lines Gh13 and BTI-87 (14.7× and 32.3×, respectively) was attributed to the quality of the genomic DNA. The DNA library of BTI-87 was of higher quality than the one of Gh13, in that it contained fewer exact-duplicate reads. The difference in coverage did not affect the ability to map the reads to the reference genome and to call SNPs with high confidence using the same criteria. These genomes yielded similar genome coverage levels (97.6% and 96.5%), but the coverage in Gh13 is slightly higher since it has fewer SNPs and gaps than BTI-87, mainly due to fewer regions of introgressions from wild species.
Both Gh13 and BTI-87 genome sequences are available on the Sol Genomics Network (SGN; http://solgenomics.net). Positions of SNPs in both genomes can be found in the Genome Browser track, and can be used for designing new markers.
SNP distribution
The large SNP density peak region on chromosome 6 in Gh13, which spans the position of the Ty-3 region [21] (30.6–34.22 Mb; Figure 2A; Additional file 1: Figure S1), shows that this SNP analysis methodology can effectively identify introgressed genomic regions. Moreover, we identified an introgression in line BTI-87 that has the Ty-3a locus from S. chilense LA1932. BTI-87 has a similar SNP density peak on chromosome 6, spanning a smaller region of 1.33 Mb around the Ty-3 locus region (30.81–32.14 Mb. Additional file 2: Figure S2).
Figure 2.
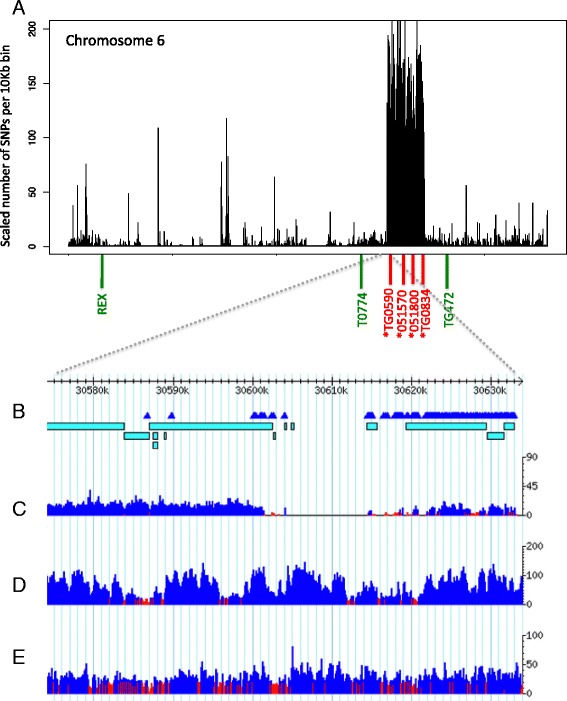
SNP density and coverage plots for chromosome 6. A) SNP density plot of the Gh13 chromosome 6. Peak region on chromosome 6 around 30.6 Mb–34.24 Mb. (*) Denotes PCR markers within the SNP peak region. B) Visualization of the 50-Kb region around the beginning of the SNP peak region (30.58–30.63 Mb). SNP marks are denoted in triangles. Bars represent de novo scaffolds of Gh13. C) Illumina coverage plot of the Gh13 genome mapped to the reference H1706 genome D) coverage of the H1706 genome E) coverage of the S. pimpinellifolium genome. Y axes for plots C-E represent number of Illumina reads mapped in that region.
We also identified a number of other distinct regions of SNP density peaks across the entire Gh13 genome, the most notable of which is apparent on chromosome 11, with two large peak regions spanning 11.76 Mb (23.18–34.94 Mb) and 4.49 Mb (43.18–47.67 Mb) (Figure 3A; Additional file 3: Table S1). Other notable SNP peak regions were identified on chromosome 4 (2.17 Mb and 2.11 Mb), chromosome 7 (1.29 Mb), and chromosome 10 (1.79 Mb). Other candidate SNP peak regions were identified on all chromosomes, ranging in length between 50 Kb to 11.76 Mb (Table 2). We defined a SNP peak as a region having 10 SNPs or more in five or more continuous 10-Kb windows, allowing gaps of up to 40 Kb, to include regions that may have low coverage due to insufficient number of reads or inability to map to the region in the reference genome, while not allowing maximum gap size to exceed the minimum SNP-peak size of 50 Kb. Our goal was to test whether it is possible to reveal relatively small introgressions by defining a minimum window size as small as 50 Kb. Using the criteria of 150 Kb used in the H1706 genome analysis [24], would yield only 32 SNP-peak regions in Gh13 and overlooking many regions of significantly high number of SNPs. To test the cutoff for selecting minimum number of SNPs per 10 Kb window for defining SNP-peak regions we calculated the average number of SNPs per 10 Kb window in the entire genome of Gh13 and compared it to the average number of SNPs in the non-peak regions when calling peak regions using a minimum number of 3, 5, 10, 15, and 20 SNPs per 10 Kb. Our statistical analysis shows the average number SNPs in the entire genome is not significantly different from the non-peak regions when using minimum number of 3 and 5 SNPs (p < 0001, p = 0.0026), but is significantly higher when using 10, 15, and 20 SNPs per 10 Kb window (p = 0.2152, p = 0.4009, p = 0.8383). Therefore we chose a minimum value of 10 SNPs per 10 Kb window, which provides statistical confidence for distinguishing SNP-peak regions from non-peak regions. For testing the reference value of minimum number of SNPs per 10 Kb window in line BTI-87 we have excluded chromosomes 4 and 9, since these have very large SNP peaks covering more than 70% in each of the two chromosomes. The statistical analysis of the remaining 10 chromosomes of BTI-87 shows similar results to the statistical analysis of the Gh13 genome (minimum of 3 and 5 SNPs; p = 0.0003, p = 0.0106. Minimum of 10, 15, and 20 SNPs; p = 0.1793, p = 0.6284, p = 0.6909).
Figure 3.
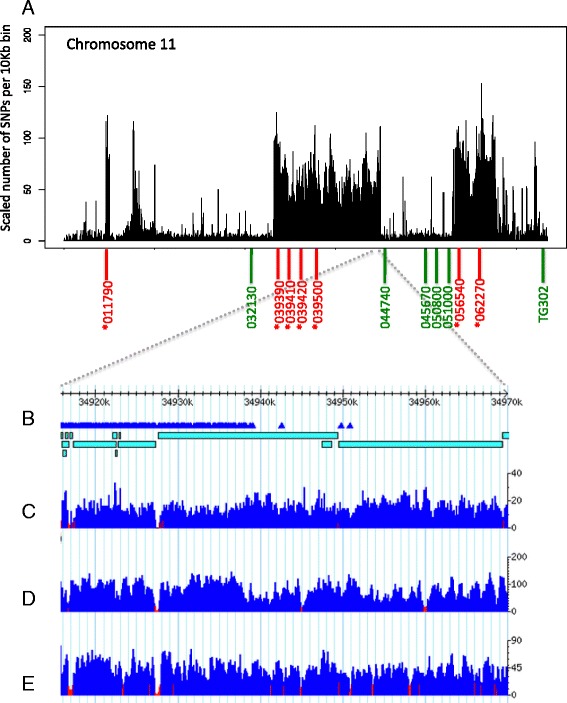
SNP density and coverage plots for chromosome 11. A) SNP density plot of the Gh13 chromosome 11. (*) Denotes PCR markers within the three assayed SNP peak regions (4.58–5.01 Mb, 23.12–34.94 Mb, 42.89–47.79 Mb). B) Visualization of the 50-Kb region around the end of the largest SNP peak region (34.92–34.97 Mb). SNP marks are denoted in triangles. Bars represent de novo scaffolds of Gh13. C) Illumina coverage plot of the Gh13 genome mapped to the reference H1706 genome D) coverage of the H1706 genome E) coverage of the S. pimpinellifolium genome. Y axes for plots C-E represent the number of Illumina reads mapped in that region.
Table 2.
Introgression metrics for Gh13 and BTI-87
| Gh13 | BTI-87 | |
|---|---|---|
| Number of introgressions | 144 | 146 |
| Introgressions in Heinz 1706 | 60 | 37 |
| Total size (Mb) | 49.42 | 150.16 |
| SNPs in introgressions | 171,711 | 641,454 |
| Gene models in introgressions | 2,326 | 5,633 |
| Smallest introgression (Kb) | 50 | 50 |
| Largest introgression (Kb) | 11,760 | 42,870 |
| Average introgression size (Kb) | 343 | 1,028 |
| Median introgression size (Kb) | 130 | 200 |
The total number of SNP-peak regions identified using these criteria was 144, spanning 49.42 Mb with a total of 171,711 SNPs, of which 94 regions were 100 Kb or larger (Table 2; Additional file 3: Table S1). Using the same criteria for calling SNP peaks in BTI-87, we also detected 146 regions in its genome, spanning 150.16 Mb with a total of 641,454 SNPs (Table 2; Additional file 4: Table S2). The SNP peak flanking the Ty-3 locus region on chromosome 6 is 1.33 Mb. A striking difference between SNP-distribution in the two genomes is the large introgressions detected in chromosomes 4, 6, and 9 of BTI-87 (total of 48.89 Mb in 11 regions in chromosome 4, 18.51 Mb in 47 regions in chromosome 6, and 53.39 Mb in 10 regions in chromosome 9).
Detection of putative introgressions
To identify potential introgressions, we identified SNPs between Gh13 and the reference genome, and discovered regions that were significantly different from the reference genome (tomato SL2.40 genome build, http://solgenomics.net/organism/Solanum_lycopersicum/genome). These regions could indicate introgressions in either the analyzed genome or in the reference genome. By plotting the number of SNPs in the Gh13 and BTI-87 genomes in windows of 10 Kb, a number of regions across the genome that could be potential introgressions from wild species were identified (Additional file 1: Figure S1, Additional file 2: Figure S2).
To test the hypothesis that regions with high SNP density correspond to introgressions from wild species, the SNPs between each of the inbred lines, Gh13 and BTI-87, and the reference tomato genome were compared to SNPs in the genomes of S. pimpinellifolium LA1589 [24], and the heirloom line Yellow Pear (YP). S. pimpinellifolium is a close relative of the domesticated tomato species, S. lycopersicum [4], and the reference tomato genome, H1706, has a S. pimpinellifolium parent in its background [24,44]. Therefore, we expected to find regions of introgressions from S. pimpinellifolium in the reference tomato genome, and perhaps from other wild species. YP does not show any traces of introgressions from wild species [37]. Thus any regions displaying a high density of SNPs between YP and H1706 could indicate regions in H1706 that did not originate from S. lycopersicum, and were likely introgressed during the breeding of this line [24,44]. The SNP density plots of both Gh13 and BTI-87 display regions with major differences between each genome and the reference tomato genome, but it is impossible to determine from this information alone whether the SNP peak represents an introgression in the inbred line or in the H1706 genome. By determining SNPs shared between Gh13 and S. pimpinellifolium, it is possible to predict which introgressions in Gh13 are most likely from S. pimpinellifolium. SNP peak regions that are shared between Gh13 and YP (Gh13 X YP) but different in H1706 (H1706 X Gh13 and H1706 X YP) most likely represent wild introgressions in the H1706 genome.
The SNP peak regions in Gh13 that do not correspond to peaks in the YP or to the S. pimpinellifolium genome, can be designated as introgressions in Gh13 originating from a different wild species (Additional file 3: Table S1). H1706 is not introgression-free, containing introgressions from S. pimpinellifolium [24,44] and possibly other wild accessions. We have detected in Gh13 SNP-peak regions that share SNPs with YP (60 out of the 144 detected candidate introgression regions). Since YP has no wild introgressions and is considered to have 100% S. lycopersicum genome [37] we can conclude these regions in the inbred Gh13 correspond to the introgression-free S. lycopersicum genome (Additional file 3: Table S1; Table 2). For example, on chromosome 10 of Gh13, 5.18 Mb in 15 SNP peak regions are shared with YP and not shared with S. pimpinellifolium, indicating all these regions are introgressions from unknown wild species in H1706 which were not recorded in its pedigree [44]. Pedigree origins are also not always reliable, as we have demonstrated with the Ty-3 gene in line Gh13, which was reported to have S. habrochaites as the source of resistance, but the Ty-3 locus was introduced from S. chilense, which is not recorded in the line’s pedigree.
The SNP peak detected in chromosome 6 of Gh13 (Figure 2A) and BTI-87 (Additional file 2: Figure S2) shows no significant overlap either with SNPs of S. pimpinellifolium or with those of YP, indicating these are introgressions of a wild species other than S. pimpinellifolium (Figure 4A; Additional file 3: Table S1). Chromosome 11 of line Gh13 shows three distinct regions which we conclude are introgressed from S. pimpinellifolium, because the majority of the SNPs are shared between the two (Figure 5A). In contrast, the SNP introgressions in chromosome 11 of BTI-87 are different than those in Gh13 (Additional file 2: Figure S2; Additional file 3: Table S1, Additional file 4: Table S2).
Figure 4.
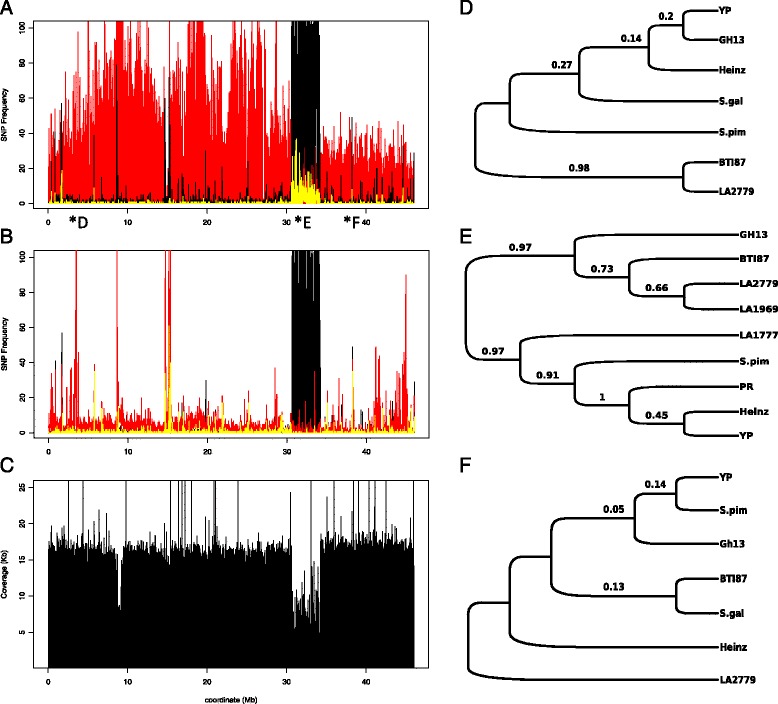
Chromosome 6 SNPs and gene trees of line Gh13 compared to selected tomato wild species and accessions. A) Chromosome 6 SNP plots of inbred line Gh13 (black) and S. pimpinellifolium (red) compared to H1706. Shared SNPs are denoted in yellow. B) Chromososme 6 SNP plots of inbred line Gh13 (black) and heirloom line YP (red) compared to H1706. Shared SNPs are denoted in yellow. C) Coverage plot of chromosome 6 of Gh13. D) Gene tree of non-peak region (marker REX). E) Gene tree of SNP peak region (marker TG590). F) Gene tree of non-peak region (marker TG472).
Figure 5.
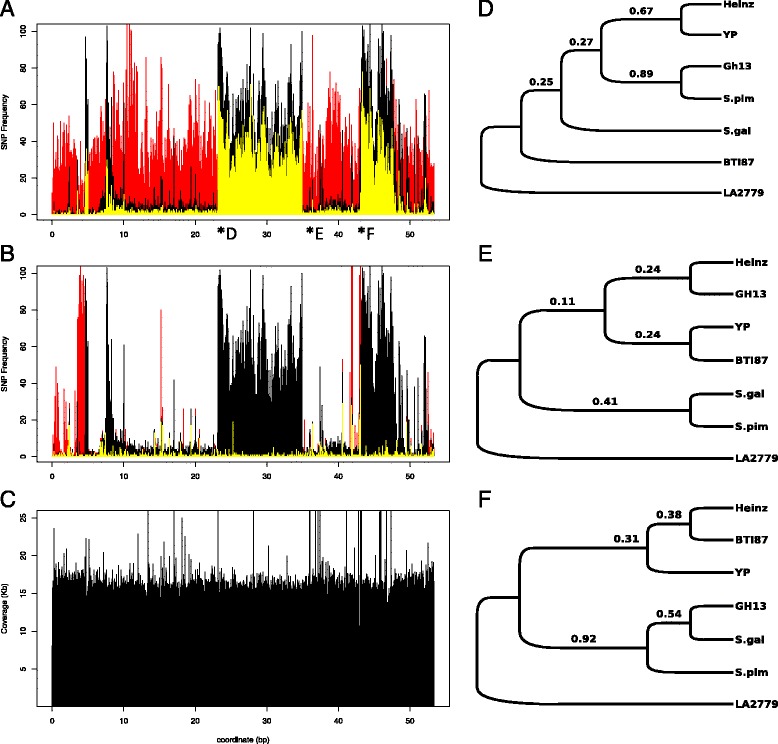
Chromosome 11 SNPs and gene trees of line Gh13 compared to selected tomato wild species and accessions. A) Chromosome 11 SNP plots of inbred line Gh13 (black) and S. pimpinellifolium (red) compared to H1706. Shared SNPs are denoted in yellow. B) Chromosome 11 SNP plots of inbred line Gh13 (black) and heirloom line YP (red) compared to H1706. Shared SNPs are denoted in yellow. C) Coverage plot of chromosome 11 of Gh13. Gene trees of three regions from chromosome 11. D) Gene tree of SNP peak region (marker P11-039390). E) Gene tree of nonpeak region (marker P11-050800). F) Gene tree of SNP-peak region (marker P11-062270).
On chromosome 4 of Gh13 we detected a large 2.17-Mb introgression (from 53.35 Mb to 55.52 Mb), which is closest to S. pimpinellifolium. However, this introgression includes a few fragments that range in size between 10 and 200 Kb for which YP has a significant number of matching SNPs (more than 10 SNPs in 10 Kb). The second largest SNP peak in chromosome 4 shows similarity to S. pimpinellifolium from 57.53 Mb to 57.91 Mb, immediately followed by 1.73-Mb region (57.91 Mb to 59.64 Mb) that most likely corresponds to an introgression in H1706 due to the high SNP density shared between Gh13 and YP (Additional file 3: Table S1). In some of those regions of high SNP density in YP, it is unclear as to the origin of introgression in Gh13 (Additional file 3: Table S1). Further phylogenetic analysis is required for each of those regions to clarify its origins.
PCR sequencing and gene trees
To investigate the origin of each detected SNP peak region on chromosomes 6 and 11 of Gh13, PCR primers were designed for amplifying fragments outside and inside the selected SNP peak regions (Figures 2A, 3A). PCR sequences were aligned, analyzed for SNPs (Table 3) and indels, and used for building phylogenetic gene trees including sequences from H1706, the heirloom lines YP and Purple Russian (PR), the inbred lines Gh13 and BTI-87, and the wild species S. pimpinellifolium, S. galapagense, S. chilense, and S. habrochaites.
Table 3.
PCR primers and fragment sequencing results
| Marker | GenBank number | Position | SNP region^ | Heinz* | YP* | PR | Gh13 | LA1589* | LA2779 | LA1969 | LA1777 | LA0386 | BTI-87 | S. gal * |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| REX | KF887310, KF887311 | 2,633,235 | Chr6 NP | a | a | - | a | a | b | - | - | - | b | a |
| T0774 | KF887301, KF887302 | 30,027,677 | Chr6 NP | a | ac | - | a | ac | d | - | - | - | ad | ab |
| TG590 | KF887295–KF887300 | 31,166,442 | Peak Chr6 | a | a | ab | be | ac | b | b | ad | d | bf | - |
| P6-051570 | KF887303–KF887307 | 31,568,208 | Peak Chr6 | a | a | a | b | a | b | b | c | c | b | a |
| T0834 | KF887312–KF887316 | 33,353,915 | Peak Chr6 | a | a | a | c | ab | cd | c | e | - | a | a |
| TG472 | KF887308, KF887309 | 37,982,169 | Chr6 NP | a | a | - | a | a | c | - | - | - | a | ab |
| P11-011790 | KF887317, KF887318 | 4,777,374 | Peak Chr11 | a | a | - | b | b | c | - | - | - | b | a |
| P11-032130 | KF887319, KF887320 | 21,629,704 | Chr11 NP | a | a | - | a | ab | c | - | - | - | a | ab |
| P11-039390 | KF887321, KF887322 | 23,182,355 | Peak Chr11 | a | a | - | c | c | d | - | - | - | a | ab |
| P11-039410 | KF887323, KF887324 | 23,342,156 | Peak Chr11 | a | a | - | b | b | - | - | d | - | a | bc |
| P11-039420 | KF887325 | 23,390,919 | Peak Chr11 | a | a | - | b | b | - | - | - | - | a | bc |
| P11-039500 | KF887326 | 24,113,034 | Peak Chr11 | a | a | - | b | b | - | - | - | - | a | c |
| P11-044740 | KF887327, KF887328 | 36,050,109 | Chr11 NP | a | a | - | a | a | b | - | - | - | a | a |
| P11-045670 | KF887329, KF887330 | 40,368,253 | Chr11 NP | a | a | - | a | a | b | - | - | - | a | a |
| P11-050800 | KF887331, KF887332 | 41,218,579 | Chr11 NP | a | a | - | a | b | c | - | - | - | a | b |
| P11-051000 | KF887333, KF887334 | 42,147,976 | Chr11 NP | a | a | - | a | ab | c | - | - | - | a | ac |
| P11-056540 | KF887335 | 43,330,076 | Peak Chr11 | a | a | - | b | b | - | - | - | - | a | bc |
| P11-062270 | KF887336, KF887337 | 46,239,133 | Peak Chr11 | a | a | - | b | b | c | - | - | - | a | b |
| TG0302 | KF887338–KF887341 | 51,878,967 | Chr11 NP | a | a | - | a | b | c | - | d | d | a | b |
^NP - Non SNP-peak.
*Heinz 1706, Yellow Pear, S. galapagense, and S. pimpinellifolium (LA1589) sequences were extracted from their genome assemblies.
On chromosome 6, the three selected regions outside the SNP peak (markers REX, T0774, TG472; Figure 2A) showed, as expected, that the Gh13 sequence was identical to the sequences from the two S. lycopersicum genomes, H1706, and YP, and very different from the wild species S. chilense and S. galapagense. Non-peak sequences of Gh13 were also nearly identical to S. pimpinellifolium sequences (REX fragments had 1 SNP, while the other two markers were identical) (Figures 4A, D, and E). The three markers tested in the SNP peak region, TG590, T0834, P6_051570 (Figure 2A), showed that the Gh13 sequence is different from the S. lycopersicum genomes, H1706, YP, and Purple Russian for TG590 and T0834 as well as for S. pimpinellifolium and S. galapagense. Other wild species tested for the chromosome 6 SNP peak region were two of the reported Gh13 pedigree parental lines of S. habrochaites (accessions LA1777 and LA0386) [42], and two other Solanum chilense accessions (LA2779 and LA1969) known to be sources of alleles of the Ty-3 locus [21]. Phylogenetic analyses of the sequences for all three markers showed that Gh13 sequence was always closest to the two S. chilense accessions (Figure 4E) rather than the expected wild species S. habrochaites.
A similar approach was applied for chromosome 11, where we detected three candidate introgressed regions in the Gh13 genome (Figure 3A). The SNP plot of Gh13, S. pimpinellifolium, and the H1706 genome showed the Gh13 introgression regions overlap mostly with S. pimpinellifolium SNPs (Figure 5A). As expected, the seven markers tested in the three SNP peak regions showed that the Gh13 sequences had highest identity to S. pimpinellifolium (Figures 5D, and F). The six markers tested in the non-SNP-peak flanking regions all showed that Gh13 sequences were identical to the S. lycopersicum genomes H1706 and YP (Table 3, Figure 5E). Sequences for all thirteen markers on chromosome 11 were compared with those of two other wild tomato species. S. chilense sequences were mostly different than all the other genome sequences for all markers, and the S. galapagense sequence was intermediate between S. lycopersicum and S. pimpinellifolium (Figures 5D, E, and F; Table 3).
SNP chip genotyping
The SolCAP SNP chip array containing 7,720 SNP markers [45] was used for genotyping Gh13 and HUJ-VF, a begomovirus-susceptible inbred. We defined regions having three or more polymorphic SNPs in 100 Kb as candidate introgressions, and found a total of 49 regions spanning 96.76 Mb with 968 polymorphic SNPs (Additional file 5: Table S3), compared with 171,711 SNPs spanning 49.42 Mb predicted with WGS. Of the 49 introgression-regions detected by the SolCAP chip, 25 have at least partial overlap with the Gh13 introgressions including, as expected, a full overlap with the predicted chromosome-6 introgression containing the Ty-3 locus. The SolCAP introgressions that were not detected by WGS could be attributed to the comparison with two different susceptible lines (H1706 and HUJ-VF) that have different genome contents.
Discussion
In this study, introgressions were detected and their origins inferred using whole-genome sequence analysis (re-sequencing), SNP calling, PCR sequencing, and phylogenetics. Two tomato inbreds (Gh13 and BTI-87) with alleles at the begomovirus resistance locus Ty-3 were used to demonstrate that a known introgression for the Ty-3 locus on chromosome 6 could be detected and boundaries determined (Figure 6A, and B). This re-sequencing strategy provides a wealth of polymorphism data (SNPs) between the reference genome and the re-sequenced lines Gh13 and BTI-87. To assess SNP regions, the chromosomes were divided into contiguous windows of 10 Kb. Plotting of the SNP frequency in each window, along the reference sequence, revealed regions of higher SNP density. These regions were tentatively labeled as introgressions. However, there were many smaller regions, from 40 Kb to a few hundred Kb in length, which showed high SNP density. These regions could represent smaller, ‘cryptic’ introgressions, or could be regions of high divergence due to other factors, such as transposon sequences. A total of 144 heretofore unknown putative introgressions, ranging in size from 50 Kb to more than 11 Mb, from different wild species were detected across the entire Gh13 genome, and 146 predicted introgressions in BTI-87 (ranging from 50 Kb to 42.87 Mb).
Figure 6.
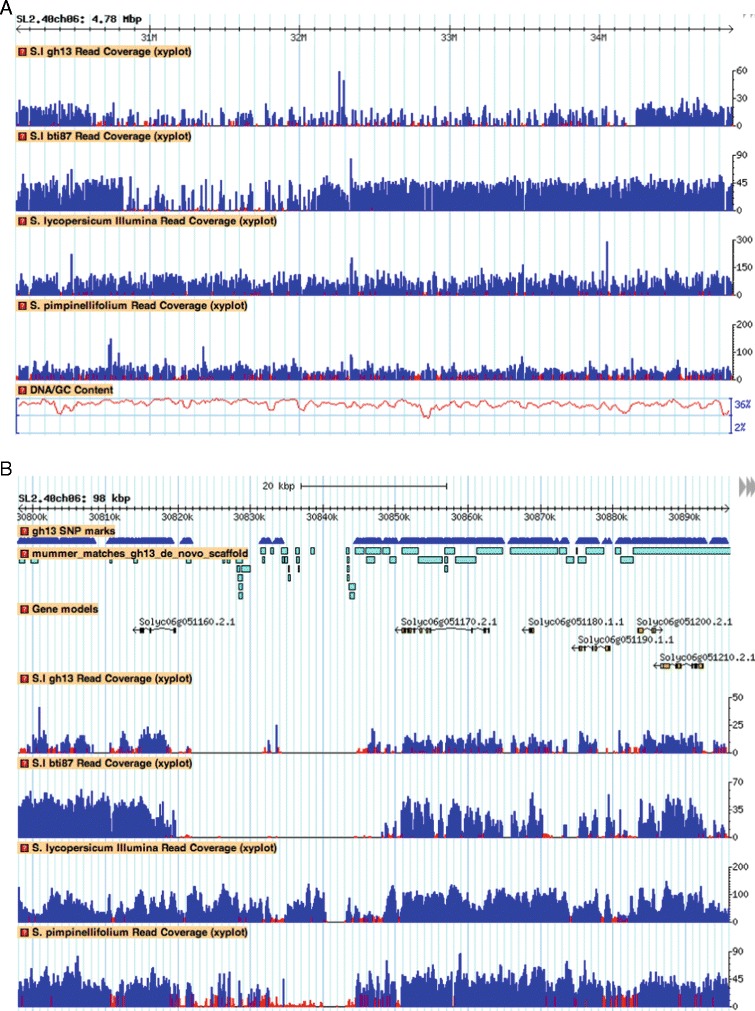
Genome regions of the Ty-3 introgression in lines Gh13 and BTI-87. A) Genome coverage plot of the chromosome 6 introgression (Gh13 and BTI-87). B) Zooming in an 80-Kb region from Figure 5A, spanning the Ty-3 region.
We detected, in both inbreds, chromosome-6 introgressions encompassing the Ty-3 locus. As the breeding pedigrees of these begomovirus-resistant lines are mostly unknown, yet both originate from a number of wild tomato species, we determined the origins of the introgressions by constructing phylogenetic trees based on sequencing of PCR fragments. Our results show that the introgressed regions in BTI-87 and in Gh13 cluster closely with S. chilense, identifying this wild species as the source for the Ty-3 locus. Other notable introgressions were detected on chromosomes 4 and 11, where their origin is most likely S. pimpinellifolium. SNP peak regions that show high similarity between Gh13 and YP indicate introgressed region in H1706 from an unknown source, or from a different S. pimpinellifolium accession. The more than double the number of BTI-87 SNPs compared to Gh13 (Table 1; Additional file 2: Figure S2) is attributed to the large introgressions in chromosomes 4, 6 and 9. These results demonstrate that tomato breeding has resulted in numerous cryptic introgressions from various wild species. Current genome sequencing technologies, coupled with the available genomic resources, permit fast discovery of such candidate introgressions, could further assist in breeding programs, and facilitate the discovery of novel genetic variation and the study of gene function.
An important property of introgression detection is the ability to determine its boundaries accurately. The ability to detect the starting and ending nucleotide of the S. chilense introgression in chromosome 6 of Gh13 was tested by extracting the unique SNPs of S. chilense in the Gh13 genome by selecting only unique SNPs that do not occur in the other tested genomes, having a coverage greater than 10× and allele frequency greater than 90%. This analysis yielded 4,931 unique S. chilense SNP positions in the Gh13 genome, with 148 SNPs in the 30.6- to 34.22-Mb chromosome 6 region of the predicted S. chilense introgression. The first SNP position within this region is at nucleotide 30,620,481, and the last is at nucleotide 34,051,365. This analysis should be repeated with the fully sequenced reference genome of S. chilense and other wild parental lines for delineating the accurate introgressions throughout the genome. The SolCAP SNP chip gave similar results for the Ty-3 introgression (30,623,784 to 33,972,992 nucleotides); however, only 29 SNPs were polymorphic, compared to more than 35,000 SNPs detected with WGS, thereby providing a greater breadth of data related to the introgression content.
The Ty-1 and Ty-3 loci were recently mapped to the same region of chromosome 6 [21], which is within the introgression for chromosome 6 for both Gh13 and BTI-87. Mapping the Ty-1 and Ty-3 loci was time-consuming and required large mapping populations over many generations of selection [21]. With re-sequencing and SNP analysis, it is possible to facilitate fine-mapping and eventually cloning of a target gene, since putative introgressions from wild species can be easily detected and possibly narrow the genomic region to be screened.
Conclusions
We utilized the H1706 reference genome and other genome sequences from S. pimpinellifolium, S. chilense, and YP, to detect introgressions in two begomovirus-resistant inbreds and identify the origin of some of these introgressions. The discovered introgressions vary greatly in size, location, and content, and our analysis with the heirloom line YP shows many of the introgressions are in the H1706 genome, which is known to have S. pimpinellifolium in its pedigree. These findings emphasize the need for additional genomic sequences of tomato wild species, which can be used to identify the origin of tomato introgressions, and study genome sequences that may not exist in the H1706 genome [46]. In addition, approaches outlined here can be used to develop SNP markers for specific regions and to determine the boundaries for introgressions. Our approach, in this report, represents a proof of concept that can readily be applied to other species with available reference genomes.
Methods
Plant material
Solanum lycopersicum inbred Gh13 was derived from the TYLCV-resistant germplasm FAVI 9 [42] by multiple generation selection of single begomovirus-resistant plants in the field in Sanarate, Guatemala [41,46]. Disease resistance genes in Gh13 were detected by SNP analysis by AgBiotech, Inc. and results were: homozygous for the begomovirus-resistance locus Ty-3 on chromosome 6; homozygous for Ve on chromosome 9; heterozygous for I2 on chromosome 11, susceptible for Mi, Sw5, Ty2, Ph3, Tm2a, and Pto. Molecular scanning by sequencing PCR fragments showed that Gh13 had an introgression on chromosome 6 from 20 to 32 cM (C. Martin and D.P. Maxwell, personal communication), which corresponds to the location of the Ty-3 locus [47,48]. Gh13 was used in several research projects to determine the effectiveness of the Ty-3 locus in conferring resistance to begomoviruses [40,49].
The proprietary begomovirus-resistant S. lycopersicum inbred, BTI-87, was obtained from the commercial seed company Semillas Tropicales, S.A. The source of begomovirus resistance in BTI-87 was from the inbred line Gc171, which is known to have the Ty-3a and Ty-4 resistance loci on chromosome 6 and chromosome 3, respectively [47,50]. These resistant loci were introgressed from S. chilense LA1932 [43]. Disease resistance genes in BTI-87 were detected by SNP analysis by AgBiotech, Inc. and results were: homozygous for the begomovirus-resistance locus Ty-3 or Ty-3a on chromosome 6; heterozygous for Mi on chromosome 6; homozygous for the gene Tm2a on chromosome 9; and susceptible for I2 and Sw5.
Seeds of accessions S. habrochaites LA0386 and LA1777, S. chilense LA1932, LA1969, and LA2779, and S. galapagense LA0436 were obtained from the Tomato Genetics Resource Center at UC Davis (http://tgrc.ucdavis.edu).
Seeds of S. lycopersicum H1706 (LA4345) and YP were provided by Gregory Martin, Boyce Thompson Institute for Plant Research (BTI). S. lycopersicum Purple Russian seeds were available from the laboratory of Douglas Maxwell, University of Wisconsin-Madison. The SNP assay for resistance loci by AgBiotech, Inc. showed that the S. lycopersicum lines, H1706, YP, and Purple Russian, had susceptible loci for Ty-3, Mi, I2, Sw5, and Tm2a.
DNA extraction
Gh13 seedlings were grown at the University of Wisconsin-Madison. DNA was extracted using CTAB method [51], yielding about 500 ng/ul of genomic DNA for whole-genome sequencing.
About 20 seedlings of tomato line BTI-87 were grown in a greenhouse under standard conditions (22°C, 14 h light) at Boyce Thompson Institute for Plant Research. Young leaves of 4- week-old seedlings were collected for DNA extraction using CsCl gradient as described previously [52]. Plants of Purple Russian, LA0386, LA1777, LA1932, LA1969, LA2779, and H1706 (LA4345) were grown under the same conditions as BTI-87 and young leaf tissue was collected and DNA extracted with CTAB protocol.
Genome sequencing
Paired-end (PE) libraries of Gh13, BTI-87, and S. chilense LA1932 were generated and sequenced on Illumina HiSeq 2000 machine at the Weill-Cornell Genomics Core Facility, New York, NY. Each PE library had an insert size of 300 bp. The reference genome for S. lycopersicum H1706 used is from the international tomato genome project, version SL2.40 (http://solgenomics.net/organism/Solanum_lycopersicum/genome). Dr. Zach Lippman, at the Cold Spring Harbor Laboratory, sequenced the S. pimpinellifolium accession, LA1589, [24]. S. galapagense accession LA0436 and the S. lycopersicum heirloom line YP sequences were obtained from a previous study at BTI [37].
Genome assembly
Illumina reads were inspected for quality using FastQC and rechecked after cleaning. Cleaning was performed with fastq-mcf (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were mapped to the S. lycopersicum H1706 reference assembly version 2.40 using BWA [53] with default parameters. Duplicate reads as well as reads with a mapping quality less than 30 were removed for variation analysis with Picard (http://picard.sourceforge.net) and Samtools (http://samtools.sourceforge.net/) [54], respectively. SNPs and indels were detected using Samtools mpileup (http://samtools.sourceforge.net/mpileup.shtml).
Whole genome de novo assemblies of Gh13 and BTI-87 were created using SOAPdenovo version 1.05 (http://soap.genomics.org.cn/) [55]. Assemblies were produced using a kmer range between 25 and 63. Scripts supplied with the SOAPdenovo package were used for error correction and gap filling of the scaffolds. De novo reads were mapped to the reference H1706 genome to increase coverage in regions with poor mapping from the BWA-aligned sequences.
For determining exact S. chilense introgression breakpoints in Gh13, variants of accession LA1932 were called using VarScan2 [56] and unique LA1932 SNPs in the Gh13 genomes were extracted using custom Perl scripts (https://github.com/nmenda/GenomeTools).
SNP plots
SNPs of S. pimpinellifolium, Gh13, and BTI87 that were called in reference to H1706 were compared to each other, and labeled ‘unique’ or ‘common’. SNPs for each group were then aggregated into bins of 10 Kb using a custom Perl script (https://github.com/nmenda/GenomeTools). SNP density for each comparison was plotted along every S. lycopersicum ‘Heinz’ chromosome using R statistics (http://www.R-project.org).
Introgression detection
Introgressions were defined as SNP-peaks having at least 10 SNPs per 10 Kb window, with minimum size of 50 Kb, and up to 40 Kb of continuous gaps. Minimum size was chosen for capturing small introgressions, and the gaps were introduced to offset the significant decrease in genome coverage in introgressed regions due to the difficulty to map those regions to the reference H1706 genome. The minimum number of SNPs per window was selected based on the hypothesis that having no introgressions means the average number of SNPs per 10 Kb window in the entire genome will be similar to this number in non-peak regions. If introgressions can be defined as having significantly higher number of SNPs in peak-regions and lower number of SNPs in non-peak regions, then the average number of SNPs per window in the entire genome should be higher than the number of SNPs in the non-peak regions. We tested introgressions using minimum number of 3, 5, 10, 15, or 20 SNPs per 10 Kb, extracting for each condition the SNP-peak and non-peak regions, and comparing the average number of SNPs in 10 Kb windows in the non-peak regions to that number in the entire genome of Gh13, and comparing each pair using Student’s t-test [57,58].
PCR and Sanger sequencing
PCR primers were developed for regions of interest based on previous markers and genic regions. PCR products were generated from S. chilense, S. habrochaites, and S. lycopersicum (lines Gh13, and Purple Russian). PCR was performed at 55 degrees Celsius, 32 amplification cycles, 60 seconds extension step. All designed primers are listed in Table 3. PCR products were cleaned with Qiagen QIAquick PCR Purification Kit, and sent for Sanger sequencing to the Life Science Core Laboratory Center at Cornell University (Ithaca, NY) or to the University of Wisconsin-Madison Biotechnology Center. Sequences from S. lycopersicum H1706 and YP, the inbred BTI-87, S. pimpinellifolium, and S. galapagense were extracted from their genome assemblies by best BLAST match of primer pairs.
Phylogenetic trees
Putative orthologous sequences for regions of interest were obtained from draft genome assemblies by using S. lycopersicum H1706 sequence selecting the top BLAST hit followed by reciprocal BLAST back to S. lycopersicum H1706. Sequences from Gh13, BTI-87, S. lycopersicum H1706, YP and Purple Russian, S. pimpinellifolium, S. galapagense, S. chilense, and S. habrochaites when available, were aligned using ClustalW [59] with default settings. Alignments were inspected to ensure accuracy. Mega5 was used to construct maximum likelihood trees using 500 bootstrap replicates and the Tamura-Nei substitution model [60]. FigTree (http://tree.bio.ed.ac.uk/software/figtree/) was used for drawing the gene tree figures. All trees were submitted to TreeBase http://purl.org/phylo/treebase/phylows/study/TB2:S16453.
SNP array genotyping
Lines Gh13 and a begomovirus-susceptible inbred, HUJ-VF that lacked the Ty-3 locus, were genotyped using a tomato array with 7,720 SNPs as implemented in the Infinium assay (Illumina Inc., San Diego, CA, USA). HUJ-VF, a processing type tomato, was provided by Dr. Favi Vidavsky, Hebrew University of Jerusalem. For each accession, genomic DNA was isolated from fresh, young leaf tissue using a Qiagen DNeasy kit (Qiagen, USA) at the University of Wisconsin-Madison. Double-stranded DNA concentrations were quantified using the PicoGreen assay (Life Technologies Corp., Grand Island, NY, USA) and normalized to 50 ng/ul with 10 mM Tris–HCl pH 8.0, 1 mM EDTA. Genotyping was conducted with 250 ng of DNA per accession following the manufacturer’s protocol for the Infinium assay. For SNP calls, the resulting intensity data was loaded in GenomeStudio version 1.7.4 (Illumina Inc., San Diego, CA, USA). In order to determine SNP genotype, the automated cluster algorithm was first used to generate initial SNP calls. Clustering for every SNP was determined using the SolCAP cluster file [45].
Availability of supporting data
The genomes of lines Gh13 and BTI-87 are available to browse, BLAST, and download at the Sol Genomics Network website (http://solgenomics.net/organism/Solanum_lycopersicum/inbred_genomes). Sequences of PCR products and primers designed and sequences in this work are available from the NCBI GenBank nucleotide database, accession numbers KF887310–KF887341.
Custom perl scripts are available from GitHub https://github.com/nmenda/GenomeTools.
Acknowledgments
We thank Dr. Mark Massoudi, AgBiotech Inc. (San Juan Bautista, California) for the KASP SNP marker assays for resistance loci; Dr. Allen Van Deynze, University of California-Davis and the SolCAP project (USDA NIFA AFRI Plant Breeding, Genetics and Genome grant 2009-85606-05673) for the SNP array genotyping; and TGRC for the seeds of wild tomato accessions.
We thank Martha Maxwell and Monica Franciscus for proofreading the manuscript and Sarah Refi-Hind for critical reading of the manuscript. This work was supported by BTI startup funds to the Mueller lab (NM, SRS, JDE, AB), and by National Science Foundation grant IOS-1025642 (GBM).
Abbreviations
- SNP
Single nucleotide polymorphism
- WGS
Whole genome sequencing
- YP
Yellow pear
- H1706
Heinz 1706
- PR
Purple Russian
Additional files
Gh13 SNP density and coverage plots. X axes are positions in bp, Y axes are number of SNPs, and negative Y axes are genome coverage. Introgression regions are highlighted in red.
BTI-87 SNP density and coverage plots. X axes are positions in bp, Y axes are number of SNPs, and negative Y axes are genome coverage. Introgression regions are highlighted in red.
Introgressions in Gh13 by 10 Kb windows. Overlapping SNPs of YP, S. pimpinellifolium, BTI-87, S. chilense LA1932, and their overlapping SNPs with Gh13.
Summary of introgressions in BTI-87 by 10 Kb windows.
Gh13 introgressions summary and SolCAP introgression regions.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NM performed the genome assemblies of Gh13 and BTI-87, wrote scripts for the bioinformatics analysis, performed the PCR sequencing, and drafted the manuscript. SR performed the phylogenetic analysis, PCR sequencing, wrote scripts for the bioinformatics analysis. JE contributed to the bioinformatics analysis tools. AB wrote scripts for the bioinformatics analysis of the genomes. DD grew the plants and extracted DNA, and contributed to the PCR sequencing. GM contributed to the PCR sequencing and to the analysis of the introgressions. LM developed, phenotyped and genotyped the inbred lines. SH contributed to the analysis of the Ty-3 introgressions. MH extracted genomic DNA, and contributed to the analysis of the introgressions. DM performed PCR sequencing, developed, genotyped and phenotyped the inbred lines, and contributed to the analysis of the inbred genomes, the introgressions, and the phylogenetic trees. LAM contributed to the bioinformatics analysis of the genomes and to the introgressions analysis. All authors read and approved the final manuscript.
Contributor Information
Naama Menda, Email: nm249@cornell.edu.
Susan R Strickler, Email: srs57@cornell.edu.
Jeremy D Edwards, Email: jde22@cornell.edu.
Aureliano Bombarely, Email: ab782@cornell.edu.
Diane M Dunham, Email: dmd248@cornell.edu.
Gregory B Martin, Email: gbm7@cornell.edu.
Luis Mejia, Email: lmejia.gt@gmail.com.
Samuel F Hutton, Email: sfhutton@ufl.edu.
Michael J Havey, Email: mjhavey@wisc.edu.
Douglas P Maxwell, Email: douglas.maxwell08@gmail.com.
Lukas A Mueller, Email: lam87@cornell.edu.
References
- 1.Giovannoni JJ. Fruit ripening mutants yield insights into ripening control. Curr Opin Plant Biol. 2007;10(3):283–289. doi: 10.1016/j.pbi.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Pedley KF, Martin GB. Molecular basis of Pto-mediated resistance to bacterial speck disease in tomato. Annu Rev Phytopathol. 2003;41:215–243. doi: 10.1146/annurev.phyto.41.121602.143032. [DOI] [PubMed] [Google Scholar]
- 3.Scofield SR, Tobias CM, Rathjen JP, Chang JH, Lavelle DT, Michelmore RW, Staskawicz BJ. Molecular basis of gene-for-gene specificity in bacterial speck disease of tomato. Science. 1996;274(5295):2063–2065. doi: 10.1126/science.274.5295.2063. [DOI] [PubMed] [Google Scholar]
- 4.Blanca J, Canizares J, Cordero L, Pascual L, Jose Diez M, Nuez F. Variation revealed by SNP genotyping and morphology provides insight into the origin of the tomato. PLoS One. 2012;7(10):e48198. doi: 10.1371/journal.pone.0048198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sim S, Robbins M, Van Deynze A, Michel A, Francis D. Population structure and genetic differentiation associated with breeding history and selection in tomato (Solanum lycopersicum L.) Heredity. 2010;106(6):927–935. doi: 10.1038/hdy.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanksley SD, McCouch SR. Seed banks and molecular maps: unlocking genetic potential from the wild. Science. 1997;277(5329):1063–1066. doi: 10.1126/science.277.5329.1063. [DOI] [PubMed] [Google Scholar]
- 7.Foolad MR: Genome mapping and molecular breeding of tomato.Int J Plant Genomics 2007, doi:10.1155/2007/64358. [DOI] [PMC free article] [PubMed]
- 8.Alexander LJ: Leaf mold resistance in the tomato.Ohio Agr Exp Sta Bul 1934, 539.
- 9.Allan EW. United States Department of Agriculture: States Relations Service Office of Experiment Stations Experiment Station Record. 1919. p. 41. [Google Scholar]
- 10.Grandillo S, Chetelat R, Knapp S, Spooner D, Peralta I, Cammareri M, Perez O, Termolino P, Tripodi P, Chiusano ML, Ercolano MR, Frusciante L, Monti L, Pignone D. Solanum sect. Lycopersicon. In: Chittaranjan K, editor. Wild Crop Relatives: Genomic and Breeding Resources. Heidelberg/Dordrecht/London/New York: Springer; 2011. pp. 129–215. [Google Scholar]
- 11.Ji Y, Scott JW, Hanson P, Graham E, Maxwell DP. Sources of resistance, inheritance, and location of genetic loci conferring resistance to members of the tomato-infecting begomoviruses. In: Czosnek H, editor. Tomato Yellow Leaf Curl Virus Disease. Netherlands: Springer; 2007. pp. 343–362. [Google Scholar]
- 12.Zamir D, Ekstein-Michelson I, Zakay Y, Navot N, Zeidan M, Sarfatti M, Eshed Y, Harel E, Pleban T, van-Oss H, Kedar N, Rabinowitch HD, Czosnek H. Mapping and introgression of a tomato yellow leaf curl virus tolerance gene, TY-1. Theor Appl Genet. 1994;88(2):141–146. doi: 10.1007/BF00225889. [DOI] [PubMed] [Google Scholar]
- 13.Barham WS, Winstead NN. Inheritance of resistance to root-knot nematodes in tomatoes. Proc Am Soc of Horticultural Sci. 1957;69:372–377. [Google Scholar]
- 14.Lanfermeijer FC, Warmink J, Hille J. The products of the broken Tm-2 and the durable Tm-22 resistance genes from tomato differ in four amino acids. J Exp Bot. 2005;56(421):2925–2933. doi: 10.1093/jxb/eri288. [DOI] [PubMed] [Google Scholar]
- 15.Seah S, Yaghoobi J, Rossi M, Gleason C, Williamson V. The nematode-resistance gene, Mi-1, is associated with an inverted chromosomal segment in susceptible compared to resistant tomato. Theor Appl Genet. 2004;108(8):1635–1642. doi: 10.1007/s00122-004-1594-z. [DOI] [PubMed] [Google Scholar]
- 16.Hanson P, Green S, Kuo G. Ty-2, a gene on chromosome 11 conditioning geminivirus resistance in tomato. Tomato Genet Coop Rep. 2006;56:17–18. [Google Scholar]
- 17.Parniske M, Wulff BB, Bonnema G, Thomas CM, Jones DA, Jones JD. Homologues of the Cf-9 disease resistance gene (Hcr9s) are present at multiple loci on the short arm of tomato chromosome 1. Mol Plant-Microbe Interact. 1999;12(2):93–102. doi: 10.1094/MPMI.1999.12.2.93. [DOI] [PubMed] [Google Scholar]
- 18.Chunwongse J, Chunwongse C, Black L, Hanson P. Molecular mapping of the Ph-3 gene for late blight resistance in tomato. J Horticultural Sci Biotechnol. 2002;77(3):281–286. [Google Scholar]
- 19.Anbinder I, Reuveni M, Azari R, Paran I, Nahon S, Shlomo H, Chen L, Lapidot M, Levin I. Molecular dissection of Tomato leaf curl virus resistance in tomato line TY172 derived from Solanum peruvianum. Theor Appl Genet. 2009;119(3):519–530. doi: 10.1007/s00122-009-1060-z. [DOI] [PubMed] [Google Scholar]
- 20.Moriones E, Navas-Castillo J. Tomato yellow leaf curl virus, an emerging virus complex causing epidemics worldwide. Virus Res. 2000;71(1):123–134. doi: 10.1016/S0168-1702(00)00193-3. [DOI] [PubMed] [Google Scholar]
- 21.Verlaan MG, Hutton SF, Ibrahem RM, Kormelink R, Visser RG, Scott JW, Edwards JD, Bai Y. The Tomato Yellow Leaf Curl Virus resistance genes Ty-1 and Ty-3 are allelic and code for DFDGD-class RNA–dependent RNA polymerases. PLoS Genet. 2013;9(3):e1003399. doi: 10.1371/journal.pgen.1003399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polston JE, Lapidot M: Management of tomato yellow leaf curl virus: US and Israel perspectives. In Tomato Yellow Leaf Curl Virus Disease. Edited by Czosnek H. Springer; 2007:251–262
- 23.Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Res. 2011;39(suppl 1):D19–D21. doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomato Genome Consortium The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485(7400):635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang X, Lu T, Han B. Resequencing rice genomes: an emerging new era of rice genomics. Trends Genet. 2013;29(4):225–232. doi: 10.1016/j.tig.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Xiao H, Jiang N, Schaffner E, Stockinger EJ, van der Knaap E. A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science. 2008;319(5869):1527–1530. doi: 10.1126/science.1153040. [DOI] [PubMed] [Google Scholar]
- 27.Martin GB, Brommonschenkel SH, Chunwongse J, Frary A, Ganal MW, Spivey R, Wu T, Earle ED, Tanksley SD. Map-based cloning of a protein kinase gene conferring disease resistance in tomato. Science. 1993;262(5138):1432–1436. doi: 10.1126/science.7902614. [DOI] [PubMed] [Google Scholar]
- 28.Hajjar R, Hodgkin T. The use of wild relatives in crop improvement: a survey of developments over the last 20 years. Euphytica. 2007;156(1–2):1–13. doi: 10.1007/s10681-007-9363-0. [DOI] [Google Scholar]
- 29.Tanksley SD, Ganal MW, Prince JP, de Vicente MC, Bonierbale MW, Broun P, Fulton TM, Giovannoni JJ, Grandillo S, Martin GB. High density molecular linkage maps of the tomato and potato genomes. Genetics. 1992;132(4):1141–1160. doi: 10.1093/genetics/132.4.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Labate JA, Robertson LD. Evidence of cryptic introgression in tomato (Solanum lycopersicum L.) based on wild tomato species alleles. BMC Plant Biol. 2012;12(1):133. doi: 10.1186/1471-2229-12-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viquez-Zamora M, Vosman B, van de Geest H, Bovy A, Visser RG, Finkers R, van Heusden AW. Tomato breeding in the genomics era: insights from a SNP array. BMC Genomics. 2013;14:354. doi: 10.1186/1471-2164-14-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henry RJ. Next-generation sequencing for understanding and accelerating crop domestication. Brief Funct Genomics. 2012;11(1):51–56. doi: 10.1093/bfgp/elr032. [DOI] [PubMed] [Google Scholar]
- 33.Blair MW, Cortés AJ, Penmetsa RV, Farmer A, Carrasquilla-Garcia N, Cook DR. A high-throughput SNP marker system for parental polymorphism screening, and diversity analysis in common bean (Phaseolus vulgaris L.) Theor Appl Genet. 2013;126(2):535–548. doi: 10.1007/s00122-012-1999-z. [DOI] [PubMed] [Google Scholar]
- 34.Esteras C, Formisano G, Roig C, Díaz A, Blanca J, Garcia-Mas J, Gómez-Guillamón ML, López-Sesé AI, Lázaro A, Monforte AJ. SNP genotyping in melons: genetic variation, population structure, and linkage disequilibrium. Theor Appl Genet. 2013;126:1285–1303. doi: 10.1007/s00122-013-2053-5. [DOI] [PubMed] [Google Scholar]
- 35.Robbins MD, Sim S, Yang W, Van Deynze A, van der Knaap E, Joobeur T, Francis DM. Mapping and linkage disequilibrium analysis with a genome-wide collection of SNPs that detect polymorphism in cultivated tomato. J Exp Bot. 2011;62(6):1831–1845. doi: 10.1093/jxb/erq367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sim SC, Van Deynze A, Stoffel K, Douches DS, Zarka D, Ganal MW, Chetelat RT, Hutton SF, Scott JW, Gardner RG, Panthee DR, Mutschler M, Myers JR, Francis DM. High-density SNP genotyping of tomato (Solanum lycopersicum L.) reveals patterns of genetic variation due to breeding. PLoS One. 2012;7(9):e45520. doi: 10.1371/journal.pone.0045520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strickler SR, Bombarely A, Munkvold JD, Menda N, Martin GB, Mueller LA. Comparative genomics and phylogenetic discordance of cultivated tomato and close wild relatives. Peer J Pre Prints. 2014;2:e377v1. doi: 10.7717/peerj.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Causse M, Desplat N, Pascual L, Le Paslier MC, Sauvage C, Bauchet G, Berard A, Bounon R, Tchoumakov M, Brunel D, Bouchet JP. Whole genome resequencing in tomato reveals variation associated with introgression and breeding events. BMC Genomics. 2013;14(1):791. doi: 10.1186/1471-2164-14-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shirasawa K, Fukuoka H, Matsunaga H, Kobayashi Y, Kobayashi I, Hirakawa H, Isobe S, Tabata S. Genome-wide association studies using single nucleotide polymorphism markers developed by re-sequencing of the genomes of cultivated tomato. DNA Res. 2013;20(6):593–603. doi: 10.1093/dnares/dst033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mejía L, Garcia BE, Fulladolsa AC, Sánchez-Pérz A, Havey MJ, Teni R, Maxwell DP. Effetiveness of the Ty-3 introgression for conferring resistance in recombinant inbred lines of tomato to bipartite begomoviruses in Guatemala. Tomato Genet Coop Rep. 2009;59:42–47. [Google Scholar]
- 41.Mejía L, Teni R, Vidavski F, Czosnek H, Lapidot M, Nakhla M, Maxwell D. Evaluation of tomato germplasm and selection of breeding lines for resistance to begomoviruses in Guatemala. Acta Hort. 2004;695:251–256. [Google Scholar]
- 42.Vidavsky F, Czosnek H. Tomato breeding lines resistant and tolerant to tomato yellow leaf curl virus issued from Lycopersicon hirsutum. Phytopathology. 1998;88(9):910–914. doi: 10.1094/PHYTO.1998.88.9.910. [DOI] [PubMed] [Google Scholar]
- 43.Scott JW, Schuster DJ. Gc9, Gc171, and Gc173 begomovirus resistant inbreds. Tom Gen Coop Rept. 2007;57:45–46. [Google Scholar]
- 44.Ozminkowski R. Pedigree of variety Heinz 1706. Report Tomato Genet Cooper. 2004;54:26. [Google Scholar]
- 45.Sim S, Durstewitz G, Plieske J, Wieseke R, Ganal MW, Van Deynze A, Hamilton JP, Buell CR, Causse M, Wijeratne S, Francis DM. Development of a large SNP genotyping array and generation of high-density genetic maps in tomato. PLoS One. 2012;7(7):e40563. doi: 10.1371/journal.pone.0040563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finkers R, van Heusden S. The 150+ tomato genome (re-)sequence project; lessons learned and potential applications. Chaing Mai, Thailand: Tomato Breeder’s Roundtable; 2013. [Google Scholar]
- 47.Ji Y, Salus M, Van Betteray B, Smeets J, Jensen K, Martin C, Mejia L, Scott J, Havey M, Maxwell D. Co-dominant SCAR markers for detection of the Ty-3 and Ty-3a loci from Solanum chilense at 25 cM of chromosome 6 of tomato. Tomato Genet Cooper. 2008;57:25–29. [Google Scholar]
- 48.Ji Y, Schuster DJ, Scott JW. Ty-3, a begomovirus resistance locus near the Tomato yellow leaf curl virus resistance locus Ty-1 on chromosome 6 of tomato. Mol Breed. 2007;20(3):271–284. doi: 10.1007/s11032-007-9089-7. [DOI] [Google Scholar]
- 49.Garcia BE, Mejia L, Melgar S, Teni R, Sanchez-Perez A, Barillas AC, Montes L, Keuler NS, Salus MS, Havey MJ, Maxwell DP. Effectiveness of the Ty-3 introgression for conferring resistance in F3 families of tomato to bipartite begomoviruses in Guatemala. Tomato Genet Coop Rep. 2008;58:22–28. [Google Scholar]
- 50.Ji Y, Scott JW, Maxwell DP, Schuster DJ. Ty-4, a tomato yellow leaf curl virus resistance gene on chromosome 3. Tomato Genet Coop Rep. 2008;58:29–31. [Google Scholar]
- 51.Doyle JJ. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15. [Google Scholar]
- 52.Bombarely A, Rosli HG, Vrebalov J, Moffett P, Mueller LA, Martin GB. A draft genome sequence of Nicotiana benthamiana to enhance molecular plant-microbe biology research. Mol Plant Microbe Interact. 2012;25(12):1523–1530. doi: 10.1094/MPMI-06-12-0148-TA. [DOI] [PubMed] [Google Scholar]
- 53.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24(5):713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 56.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.JMP®, Version 11.2. SAS Institute Inc., Cary, NC, 1989–2007.
- 58.RStudio Team: RStudio: Integrated Development for R. Boston, MA: RStudio, Inc.; 2012 [http://www.RStudio.com/ide]
- 59.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 60.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]