

Abstract
Personalized medicine is rapidly becoming a reality in today's physical medicine. However, as yet this is largely an aspirational goal in psychiatry, despite significant advances in our understanding of the biochemical, genetic and neurobiological processes underlying major mental disorders. Preventive medicine relies on the availability of predictive tools; in psychiatry we still largely lack these. Furthermore, our current diagnostic systems, with their focus on well-established, largely chronic illness, do not support a pre-emptive, let alone a preventive, approach, since it is during the early stages of a disorder that interventions have the potential to offer the greatest benefit. Here, we present a clinical staging model for severe mental disorders and discuss examples of biological markers that have already undergone some systematic evaluation and that could be integrated into such a framework. The advantage of this model is that it explicitly considers the evolution of psychopathology during the development of a mental illness and emphasizes that progression of illness is by no means inevitable, but can be altered by providing appropriate interventions that target individual modifiable risk and protective factors. The specific goals of therapeutic intervention are therefore broadened to include the prevention of illness onset or progression, and to minimize the risk of harm associated with more complex treatment regimens. The staging model also facilitates the integration of new data on the biological, social and environmental factors that influence mental illness into our clinical and diagnostic infrastructure, which will provide a major step forward in the development of a truly pre-emptive psychiatry.
Keywords: Biomarkers, clinical staging, diagnostic reform, early intervention, personalized medicine, pre-emptive psychiatry, youth mental health
As many areas of clinical medicine move towards the introduction of more personalized or stratified treatment selection (1), linked to the stage of illness at which one presents for care, we are still early in the process of evaluation of the relevance of clinical staging for major psychiatric disorders (2-4).
Clinical staging in psychiatry builds on strong epidemiological evidence indicating that what we regard as major mental illnesses evolve over time in terms of clarity and severity. The staging model aims to differentiate earlier and milder clinical phenomena from those that mark illness progression and extension, moving outside the traditional diagnostic boundaries to place strong emphasis on where a person sits within the evolution or resolution of his/her illness.
Advances in research, particularly in the area of early psychosis, have shown that treatment in the very early stages of illness can produce significantly better clinical and functional outcomes for patients (3,5-7). This research has opened the way for a paradigm shift in psychiatry: the movement towards a pre-emptive, rather than largely palliative, psychiatry (8).
Preventive medicine, including pre-emptive psychiatry, requires predictive tools. Predicting who is at risk of developing an illness allows for the possibility of preventive interventions, while predicting an individual's response to the different treatment options available allows for a more personalized therapeutic approach.
While this has become a reality in key areas of physical medicine today, in psychiatry we still largely lack the ideographic tools that would support a profiling strategy to complement the nomothetic staging approach described below (9). At the most basic level, the clinical and trait/state risk factors that form the basis of our current predictive criteria are relatively crude and non-specific.
Despite the lack of detailed knowledge of the risk factors for the onset of mental illness, authors such as Eaton et al (10) have proposed that sub-threshold syndromes may be regarded as risk factors for full-threshold disorders such as schizophrenia and major depression, and could become the relevant targets for preventive interventions.
So far no biomarkers (measurable biological characteristics that index pathogenic processes or treatment responses (11)) or other risk markers are available to create profiles to enhance prediction and therapeutic selection in psychiatry. Stratified or personalized interventions remain aspirational, yet potentially within reach.
The concept of clinical staging is based on the notion of underlying pathophysiology that has, at some point along its course, the capacity to result in persistent or progressive illness. To test the concept, there is a clear need to develop illness markers that are directly linked to pathophysiology and, further, have the capacity to track the extent of the disease process. By examining the relationship between various biomarkers and both syndrome and stage, it may be possible to distinguish between those biomarkers that possess a degree of syndromal specificity from those that do not, and furthermore, to distinguish disease markers, vulnerability markers and risk factors across stages, markers of disease progression, and epiphenomena.
Despite our current lack of detailed knowledge linking clinical phenotypes to specific pathophysiologies, it is possible to outline the framework of a useful staging model based on presenting clinical features (Figure 1). We propose that major mental illnesses develop from an “at-risk” but asymptomatic state (stage 0), through an initial stage of undifferentiated general symptoms such as mild anxiety, depressive and somatic symptoms, followed by a worsening of these existing symptoms and the acquisition of new ones, associated clinically with hints of greater syndromal specificity, and with behavioural and functional decline (stage 1). Personality development and functioning are typically affected in young people, and this dimension should be incorporated in staging definitions rather than split off in another axis. Further progression of illness may then take place, resulting in the occurrence of a first episode of a full-threshold syndrome(s) (stage 2), which may be followed by the development of persistent symptoms, frequent relapses and ongoing impairment (stage 3), or even severe, unremitting illness (stage 4) (2-4). Remission and amelioration is possible at every stage, though it is less likely with advancing stage (Figure 1).
Figure 1.
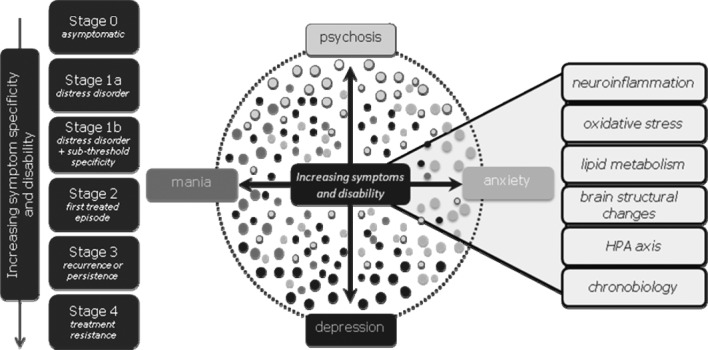
Clinical staging model for mental disorders and putative biomarkers. HPA – hypothalamic-pituitary-adrenal
Within this model, there is an implied “step function”, rather than a simple severity gradient, between the “attenuated syndromes” (of psychosis or severe mood disorder) in stage 1 and the onset and persistence of full-blown syndromes in stage 2 and beyond. The risk of progression and of persistence and recurrence are proposed to increase with each stage. However, this is only part of the justification for what is primarily a heuristic strategy of imposing a categorical framework upon what otherwise is likely dimensional in nature. Hence the notion of a “step” or discontinuity remains a hypothesis, since each stage may merely represent an arbitrary boundary rather than carving nature at a biologically or therapeutically relevant “joint”.
Underlying this framework (at least when applied to youth-onset disorders) is the assumption that the emergence and persistence of mental illness is driven by risk factors, some of which are earlier vulnerability factors or trait markers (e.g., genotype, intrauterine infection, adverse childhood experiences, childhood-onset learning, developmental or behavioural disorders), while others operate later, notably in the peri- or post-pubertal periods (e.g., developmental and life stress, illicit drug use, etc.). Presumably, at least some of these later factors, which may not only affect onset but also the risk of persistence and progression, are potentially modifiable (e.g., alcohol, cannabis or other substance misuse, adverse social environments, social isolation).
Within the model, the actual persistence and progression of illness over time is presumed to be determined by a gene-environment interaction between the underlying pathophysiology, influenced by genetic, epigenetic and other factors, and the balance of other risk or protective factors. Within this framework, these modifiable factors, as well as the underling pathophysiology, become major targets for preventive intervention.
This model has an explicitly preventive stance, emphasizing that progression of illness or “transition” from one stage to the next is by no means inevitable. The specific goals of therapeutic interventions are therefore broadened to include the prevention of illness onset or progression, and to minimize the risk of harm associated with more complex treatment regimens that are typically provided to those with more severe, persistent or recurrent disorders. Ensuring that early stage treatment is demonstrably safer is a key principle underpinning preventive medicine and early intervention. A further advantage of this model is that it facilitates the integration of new data on the biological, social, and environmental factors that influence mental illness into our clinical and diagnostic infrastructure. Ultimately, we seek to replace our prototypical clinical staging model with the development of a more robust clinico-pathological framework.
Clearly, the identification of cross-sectional and longitudinal biomarkers that provide accurate assessments of risk and illness progression is of crucial importance. Ideally, these markers should link to relevant pathophysiology and enhance our understanding of the biological mechanisms that underlie the onset and progression of illness. Here, we discuss examples of how certain biological markers that have already undergone some systematic evaluation could be integrated into a clinical staging framework. We review some of the well-replicated risk factors for the early stages of psychiatric disorders within the neurobiological and other biomarker domains. These biomarkers may reflect causal mechanisms (such as genetic and oxidative stress markers) or consequences of the pathophysiology (such as cognitive, structural and physiological alterations). Some may reflect both (e.g., inflammation). Some biomarkers (e.g., imaging findings) may have potential predictive and/or diagnostic value (12), while others may suggest novel therapeutic interventions (e.g., N-acetylcysteine and omega-3 fatty acids for oxidative stress and anti-inflammatory agents for inflammation (13,14)).
COGNITIVE MARKERS
Examination of cognitive factors in major psychiatric disorders has primarily focused on neurocognition – mental operations underlying goal-directed behaviour such as attention, working memory, processing speed, learning and memory, executive functions, and global intellectual functions, including IQ. However, social cognition, a related but independent construct involving the perception, interpretation and processing of social information, has also received increasing investigation. It includes the domains of emotion recognition, theory of mind, social perception/knowledge and attributional style.
Neurocognition and social cognition are potentially highly valuable markers within the staging model. They may provide clues regarding underlying pathophysiology and/or genetic etiology (i.e., endophenotypes). They are also strongly related to functioning and disability across a range of psychiatric disorders (which form a key element of defining the stage of illness) independently of symptoms (15,16), can be relatively easily assessed within the clinical setting, and are amenable to intervention (17).
There is extensive evidence of neurocognitive, and increasingly, social cognitive deficits across all putative clinical stages of psychosis relative to healthy controls. Mild significant premorbid neurocognitive deficits are found in asymptomatic at-risk and ultra-high risk individuals. Deficits around 0.5 standard deviations below healthy controls are seen in global indices such as IQ (18-20), as well as specific accentuated deficits, particularly in verbal and visual memory, working memory, olfactory identification and social cognition (18,21,22). A recent meta-analysis found that deficits are greater in high-risk individuals who later develop full-threshold schizophrenia-spectrum disorders relative to those who do not (21).
In patients with first-episode psychosis, significant medium to large impairments (0.64-1.20 SDs below healthy controls) are present across all neurocognitive domains, including IQ and social cognition, with the most severe deficits observed in immediate verbal memory and information processing speed (23). The pattern and magnitude of these deficits are remarkably similar to those reported in meta-analyses of older, more chronic patients (0.46-1.57 SDs below healthy controls) (24). Furthermore, longitudinal studies indicate that the profile and severity of neurocognitive impairments tend to be stable for up to the first 10 years of illness (25) and over periods of up to 6 years in older people with chronic schizophrenia (24-26). Preliminary findings also point to relative stability in social cognition from early through to later stages of psychosis (27).
A cautionary note regarding the heterogeneity of psychotic disorders is important, as there is evidence that specific subgroups (i.e., those with unremitting symptoms and poorer functioning) may experience a more deteriorating cognitive course (24). Collectively, however, findings indicate that both cognitive lag (failure to develop normally) and progressive decline occurring primarily somewhere between premorbid and first-episode psychosis phases may be present, and followed by relative stability of deficits.
Medium to large neurocognitive impairments are also well documented in stable euthymic bipolar I disorder (28,29). Verbal learning and memory, executive functioning, attention and processing speed are most consistently impaired, with relative preservation of verbal abilities and intelligence (28). Similar milder impairments are also observed in bipolar II disorder (30). These neurocognitive deficits are evident early in the course of bipolar disorder (stage 2), but are less severe than in later illness stages (31-33). Specifically, cross-sectional studies have demonstrated a relationship between the degree of neurocognitive dysfunction and number of mood episodes, as well as illness duration (31,32). In general, neurocognitive deficits in bipolar disorder are not as severe as those observed in psychotic disorders (29,34).
Contrary to psychotic disorders, the presence of neurocognitive deficits prior to the onset of first-episode mania (stages 0-1) remains less clear, mostly because few studies have been conducted. Relatively intact or even higher global intellectual function (e.g., IQ) was found in children and adolescents who later developed bipolar disorder (19,35). However, there is preliminary evidence for specific neurocognitive deficits compared to healthy controls in at-risk young people who later developed a bipolar spectrum disorder (36,37). Milder neurocognitive deficits are found in healthy first-degree relatives, although the specific domains implicated have been inconsistent (28,29,38). Together these findings suggest that neurocognitive deficits are present after the onset of full-threshold bipolar illness and may progressively worsen with illness chronicity, but their role as a specific early risk marker remains undetermined, since few studies have focused on the premorbid and sub-threshold stages.
While neurocognitive deficits are also common in unipolar depression, the evidence for staging is less clear, largely due to limited pre-diagnosis and longitudinal studies. In adolescents, young adults and older adults with depression, evidence exists for deficits in several neurocognitive domains, with executive functioning abilities (particularly in set-shifting), psychomotor speed and verbal and visual explicit memory most consistently impaired (39-45). Deficits are observed in mild, moderate and severe depression, with an apparent relationship between depression severity and neurocognitive impairment (39,43,45,46). Moreover, endogenous (melancholic) relative to non-endogenous depression seems to be associated with greater neurocognitive decrement (39,43,46).
Together, these findings tend to suggest that neurocognitive impairment is state-dependent. Nevertheless, it remains undetermined whether the neurocognitive deficits in depression may also reflect trait-based phenomena (41,47). Two studies of offspring at genetic risk for depression found no significant impairment in neurocognitive function relative to healthy controls (48,49). Mild premorbid deficits in psychomotor speed and attention were reported in one population study of children who later developed unipolar depression (50). Furthermore, mild residual deficits have been reported in older remitted patients, particularly in executive functions (41,42), but studies of remitted patients are sparse and the relationship of neurocognitive dysfunction with factors such as age, age of onset, duration of illness and number of episodes is equivocal (43,46,51). One study reported a relationship between neurocognitive dysfunction and recurrent episodes (52), suggesting a possible progressive course, while others have found no relationship (43). Much more work is needed to characterize cognitive dysfunction in terms of trait, state and progressive manifestations of depressive illness.
In summary, there has been a paucity of longitudinal studies examining cognition from premorbid to first-episode and more chronic illness phases, particularly in unipolar and bipolar illness. Nevertheless, the pattern of cognition according to stages of illness suggests that neurocognitive impairment may reflect both neurodevelopmental trait vulnerabilities and progressive illness-related deficits in major psychiatric illness. The evidence for social cognition is less advanced, but is beginning to parallel the neurocognitive findings, at least in psychotic disorders.
Points of difference between psychotic and bipolar disorder appear to be in severity and pattern of progression. Cognitive deficits are more broad and severe across all stages of illness in psychotic relative to bipolar disorder. In psychosis the greatest progression of cognitive dysfunction occurs between the sub-threshold and full-threshold stages, whereas cognitive decline is more evident after the onset of bipolar disorder and with increasing episodes. Although state-related neurocognitive deficits are clearly evident in depression, research is much less advanced, precluding any firm assertions regards to vulnerability and progression.
To exactly determine the timing and pattern of cognitive deterioration, more longitudinal studies are required to serially compare cognitive functioning of the same at-risk samples before and after development of full-threshold psychiatric disorders.
BRAIN STRUCTURAL MARKERS
Neuroimaging evidence for clinical staging in psychotic disorders has been reviewed recently (53), and space permits merely a summary here. A substantive body of literature has accumulated showing subtle structural brain changes in non-symptomatic relatives at risk for schizophrenia. A meta-analysis by Boos et al (54) showed modest reductions in gray matter volume, notably in the hippocampus, but also more widely spread. Such changes may tend towards normalization in relatives that do not show emergent psychopathology (55), but seem to be more prominent in those who already manifest schizotypal (56), cognitive (57) or early prodromal symptoms (58).
The brain region most commonly shown to be abnormal in chronic schizophrenia, the lateral ventricles, is less affected in first-episode patients (59), and unaffected in ultra-high risk individuals (60). A recent study has shown that gray matter decreases, predominantly in the frontal cortex, are far more pronounced in chronic schizophrenia than in first-episode psychosis patients (61), while other work has extended this evidence to ultra-high risk subjects (62). We have found similar patterns in the superior temporal gyri (63) and insular cortex (64,65), with clear evidence for increasing abnormality across the stages of illness (66). Similarly, marked reductions in hippocampal volume have been noted in chronic schizophrenia patients, while this is less clear in first-episode patients (67), and non-significant in ultra-high risk subjects (68-70). Meta-analyses confirm progression of ventricular enlargement and gray and white matter reductions (71), and these changes have been associated with poor outcome (72).
A key issue that has arisen recently is the role of antipsychotic medication in these progressive brain changes, given that illness duration and treatment intensity have been shown to be predictive of brain tissue loss, with illness severity having a smaller effect (73,74). Relapse duration and antipsychotic treatment intensity have both been shown to contribute to this tissue loss, although their effects are relatively small (74). These findings have important implications and indicate that, while relapse prevention is a key therapeutic strategy, the lowest possible doses of antipsychotic medications should be used.
Structural brain abnormalities have also been widely investigated in patients with both unipolar and bipolar affective disorders (75-77). Typically, the degree of gray matter loss reported appears to be correlated with the duration of illness and lack of exposure to antidepressant therapies. White matter changes have also been documented, particularly among those who develop depression for the first time in later life, which appear to be secondary to micro-vascular pathology (78). For depressive disorders, therefore, it is particularly important to differentiate early-onset disorders (typically post-puberty) from those that develop in mid- or later life.
For depressive disorders, it has been assumed that the observed structural abnormalities – even in young people – are a consequence rather than a risk factor for illness. This assumption is now being challenged by studies focusing on young people, particularly those who go on to develop more severe bipolar or psychotic disorders. An accelerated loss in ventral and rostral prefrontal cortex volume in adolescents/young adults with bipolar disorder has been found in a longitudinal study (79). To date, there has been a major lack of longitudinal studies comparing findings prior to onset, early or later in the course of illness, or the effects of exposure to the range of treatments employed in those with early-onset disorders.
Affective symptoms, in particular depression, are common in the early phases of schizophrenia, and the emergence of psychosis is often the harbinger of what might later evolve as a non-affective psychosis. Of relevance in this context are imaging studies across the psychotic spectrum which can inform whether patients with psychotic versus non-psychotic affective disorders might have qualitative and quantitative differences in brain structure. There is evidence that, compared to psychotic depression, non-psychotic depression may be associated with preserved ventricular and white matter volumes (80). Brain structural differences are also seen between psychotic and non-psychotic bipolar disorder, supporting the categorical distinction between these entities (81). Strasser et al (82) have also observed smaller ventricular volumes in non-psychotic versus psychotic bipolar subjects. Recent meta-analyses have shown that gray matter reductions and ventricular enlargement may be more common in schizophrenia, while white matter volume reductions (83) and amygdala volume increases (75) may be more likely in bipolar disorder. In general, structural changes appear to be more confined to regions involving affect regulation in bipolar disorder, while the alterations are more pervasive in schizophrenia (84). Thus, it is likely that differential structural brain changes might become more prominent as syndromal specificity increases, such as may be seen with the emergence of psychotic or affective symptoms.
Taken together, these observations support the view that progressive brain structural changes in schizophrenia clearly involve a linear gradient of change, though a non-linear step function is also possible. Thus, it is likely that hippocampal alterations are subtle or absent in the early stages of psychotic disorders, but may appear and progress during the prodromal phase before reaching a plateau. Gray and white matter losses might, however, progress over time, and prominent ventriculomegaly might characterize chronic, poor outcome cases of schizophrenia, while more focal alterations in brain regions involved in affect regulation might signify affective psychoses. Such progressive evolution and differentiation of neuroanatomical changes may be paralleled by changes in function, connectivity and neurochemical integrity in these circuits, and will need systematic research.
A key issue is to clarify the role of antipsychotic medication in progressive brain changes. There is an association that could prove to be causal or turn out to be merely an epiphenomenon, but this requires urgent investigation (73). For now, structural magnetic resonance imaging (MRI) measures are a valuable first step towards the neurobiological characterization of staging across clinical phenotypes and syndromes.
MISMATCH NEGATIVITY AS A MARKER OF BRAIN FUNCTION
Several electroencephalographic properties have been studied in psychotic and mood disorders. Mismatch negativity (MMN) is a change in the activity of the brain induced by the occurrence of novel stimuli, leading to a switch of attention in the subject. MMN amplitude is thought to reflect the functioning of N-methyl-D-aspartate (NMDA) receptors, and impaired MMN generation has been associated with poor social and global functioning. Classically, this event-related potential is induced by the occurrence of a deviant sound in an otherwise contiguous stream of events, and it can be measured with electroencephalography.
A meta-analysis of 32 studies reports that a decrease in the amplitude of the MMN has been consistently replicated in schizophrenia (85). Very few data on the amplitude of MMN in people suffering from major depressive disorder are available, and the studies on this topic have all been conducted in stages 3 or 4 of the illness. One study showed no difference in the MMN amplitude between patients with major depression and controls (86), while others showed either an increase (87,88) or a decrease (89,90). Of the two studies that showed a decrease, one used a visual paradigm and the other measured MMN with magnetoencephalography, making it difficult to compare the results to those obtained with schizophrenia patients. Research into the MMN deficit in individuals with bipolar disorder has also been conducted during the later stages of the illness. Only two of six studies have shown impairment in MMN generation, with an MMN amplitude lying between that of controls and schizophrenia patients (see 91). Thus, the impairment in the generation of MMN appears to have some specificity for schizophrenia.
Umbricht and Krljes' meta-analysis (85) also revealed that the effect sizes of MMN induced by pitch-deviant sounds were significantly correlated with duration of illness, indicating that the pitch-deviant MMN amplitude attenuation could reflect disease progression. Results of MMN studies in people at ultra-high risk for psychosis are in line with this proposition. Indeed, the generation of pitch-deviant MMN was shown to be intact in first-degree relatives (92,93) and in individuals with sub-threshold psychotic symptoms (94). Furthermore, the amplitude of the pitch-deviant MMN was intact in patients with first-episode psychosis (93,95-100), with a subsequent significant decrease in the same patients in the post-acute phase (95), or one year after their first psychotic experience (97). In the later follow-up study, deterioration in MMN generation was correlated with gray matter volume reduction in the primary auditory cortex, a correlation that was also observed in chronic schizophrenia (101). Two studies showed contradictory results on pitch-deviant MMN in first-episode psychosis, but both groups recruited individuals presenting with a first episode of schizophrenia specifically, rather than a first episode of psychosis more broadly (94,102).
On the other hand, MMN induced by a duration-deviant sound seems to behave like a trait marker of psychosis. Indeed, in all four studies conducted in ultra-high risk individuals, i.e., those with sub-threshold psychotic symptoms and/or genetic vulnerability, a decrease in duration-deviant MMN amplitude was observed compared to controls (91,94,103,104). Most interestingly, this decrease in amplitude was significant only in ultra-high risk participants who later transitioned to psychosis, suggesting that this impairment in duration-deviant MMN could be used as a predictor of illness (94,103). The amplitude of the MMN for a change in the duration of the sound was equally attenuated in schizophrenia and first-episode psychosis (94,102,105), but this was not reported in all studies (93,99).
In summary, while the attenuation of frequency-deviant MMN seems to follow the staging model, the duration-deviant MMN deficit appears to be more closely related to the genetic disposition of the patient.
SLEEP AND CHRONOBIOLOGICAL MARKERS
A ubiquitous characteristic of the onset period of most major psychiatric disorders is disruption of sleep, which is often accompanied by specific shifts in the sleep-wake cycle. While there is a normal tendency in adolescence for later onset of sleep, prolonged sleep and later daytime rising compared with younger children, those experiencing their first major onset of depression are particularly prone to further changes in this key homeostatic and developmental function. Shortened sleep duration appears to be a risk factor to the onset of common forms of psychological distress, and their persistence over 12 months, in late adolescence (106).
Among those with first-onset depressive syndromes, a significant sub-population develop “atypical” syndromes characterized by over-sleeping (and over-eating) and reduced daytime activity. In more severe cases, this phenotype may be characterized by distinct phase-delay in circadian timing, with shifting to later times of sleep onset and sleep offset, as well as increased total sleep time (107). In the most extreme forms, there may be a loss of normal circadian synchronization of mood, energy, cognitive performance and neurohormonal parameters, as well as the development of more somatically-focused “prolonged” or “chronic fatigue” syndromes. Those with more severe somatic syndromes, accompanied by persistent mood disturbance, appear to be at increased risk of later development of bipolar spectrum disorders (108).
A large body of data have accumulated since the 1980s on alterations in sleep architecture. Polysomnographic studies have received relatively little attention in recent years, perhaps because of lack of diagnostic specificity (109). However, there appear to be distinct sleep “signatures”, with consistent reductions in slow wave sleep (SWS) in schizophrenia, and shortened rapid eye movement (REM) latency and increases in REM density in depression. These alterations may precede the emergence of full-blown disorder; for example, shortened REM latency can predict emergence of depression in at-risk adolescents (110). On the other hand, SWS reductions are seen in asymptomatic relatives of schizophrenia patients (111,112). Sleep disturbances are also predictive of psychosis in prodromal cases at clinical high risk for schizophrenia (113). Sleep alterations may possibly be stage-specific and vary with the course of illness, with acute symptoms being associated with REM density and latency (114), while chronicity, poor outcome and cognitive deficits are associated with SWS alterations (115,116).
Sleep disturbances are treatable, though it is not known whether such interventions can potentially prevent the emergence of psychopathology in individuals at high risk. Clearly, sleep studies may offer an early marker that is treatable, with likely preventive value.
NEUROENDOCRINE MARKERS
An impaired ability to cope with stress, both at the psychological and biological level, is thought to play a key role in the development and maintenance of psychiatric disorders (117). The hypothalamic-pituitary-adrenal (HPA) axis is the main biological mediator of the stress response, and its function is often altered in psychiatric disorders, with early life stress or trauma being an important moderating factor in determining the level of stress reactivity later in life (118).
Hyperactivity of the HPA axis, characterized by elevated cortisol secretion, an enlarged pituitary gland volume and impaired negative feedback of the HPA system, has been observed in affective (119) and psychotic disorders (120,121). Patients with psychosis also exhibit increased reactivity to daily minor stressors and this in turn correlates with greater negative affect and psychotic symptoms (122).
It is hypothesized that HPA axis abnormalities are involved in the genesis of psychopathology and cognitive deficits via the neurotoxic effects of elevated cortisol in brain regions such as the hippocampus and prefrontal cortex (117), and via interactions with the dopaminergic and other neurotransmitter systems (117). Elevated cortisol secretion is linked with greater severity of positive and negative symptoms in patients with schizophrenia (123) and first-episode psychosis (124). Furthermore, decreases in cortisol and the cortisol/dehydroepiandrosterone sulfate (DHEAS) ratio during the initial three months of treatment in first-episode psychosis were directly related to an improvement in depression, negative and psychotic symptoms (124). Cortisol hypersecretion has also been associated with smaller left hippocampal volume in patients with first-episode psychosis (125), and an abnormal cortisol awakening response has been associated with greater cognitive deficits (126).
Atypical antipsychotics have been shown to dampen HPA axis activity (127) and reduce pituitary volume in a dose-dependent manner (128), providing further support for the stress-vulnerability model. These medication effects may also partly explain the discrepancies in HPA axis findings in patients with differing length of illness. Further longitudinal studies are urgently needed to better understand the role of the HPA axis in progressive brain change and cognitive decline associated with illness progression.
A recent body of literature has demonstrated that HPA axis abnormalities are evident prior to illness onset and in unaffected relatives of patients. An enlarged pituitary (a marker of HPA activation) was found to predict the subsequent transition to psychosis in ultra-high risk individuals (129). Consistent with this, a later study conducted in young at-risk individuals found that increased cortisol secretion was predictive of later transition to psychosis (130). Circulating cortisol was also positively associated with depressive and anxiety symptoms in ultra-high risk individuals (131). Healthy first-degree relatives of patients with a psychotic disorder have been shown to have increased stress reactivity, elevated cortisol levels (132) and an enlarged pituitary (133). The study by Collip et al (132) also reported an association between cortisol secretion and intensity of psychotic-like experiences and negative emotions, similar to that observed in patients with psychosis.
Taken together, these findings suggest that stress/HPA axis activity may be an important biological marker for vulnerability to develop psychopathology. It is important to note that distinct patterns of HPA axis dysfunction may occur within and across psychiatric diagnoses, and HPA activity may also change as a function of illness duration.
INFLAMMATORY AND OXIDATIVE STRESS MARKERS
There is increasing evidence to implicate inflammatory processes in the pathophysiology of major psychiatric disorders. Elevated levels of cytokines are a well-replicated finding in most major mental illnesses. Infusion of pro-inflammatory cytokines and interferon is perhaps the best experimental human model of depression (134), and elevated levels of cytokines are known to be associated with psychosis, depression and mania (135-137).
In parallel, there is a consistent body of evidence for an increase in oxidative stress in mood and psychotic disorders, including reduction in brain glutathione levels, changes in antioxidant enzymes, lipid peroxidation, protein carbonylation and DNA damage, as well as progressive structural changes consistent with oxidative damage (13,14,138).
Elevated levels of pro-inflammatory cytokines appear to precede the development of de novo disorder, suggesting that they play a role in its genesis (139). Inflammatory mediators have been thought to be related to processes underpinning stage-related structural and cognitive decline (140) and may be relevant to acute relapse and associated brain structural changes. The first data to support this hypothesis is the finding that the pro-inflammatory cytokines IL-6 and TNF-α were elevated in both early and late stage disorder, whereas the anti-inflammatory cytokine IL-10 was increased only in the early stage of the disorder (141). Additionally, TNF-α, while elevated throughout the course, was higher in later stages, suggesting that the inflammatory state is more perturbed later in the course of the disorder.
There are similar stage-dependent changes in oxidative parameters. In late stage patients, the activity of key enzymes in the glutathione pathway, glutathione peroxidase and glutathione-S-transferase, are increased compared to early stage patients and controls (142). Akin to the pattern seen with inflammatory markers, this stage-related change in oxidative biology may form part of the progressive failure of compensatory mechanisms over time, and may in part underlie the phenomenology of disease progression (143,144).
Consistent with a role for these pathways, there is evidence that most established psychotropic agents, including mood stabilizers and atypical antipsychotics, have substantive impacts on oxidative and inflammatory pathways (145,146). The selective COX-2 blocker celecoxib displays potential efficacy in the treatment of bipolar disorder (147) and schizophrenia (148). The use of statins, which have intrinsic anti-inflammatory and antioxidant properties, appears to be associated with lowered risks of mood disorders in community studies and in cohorts of individuals with cardiac disorders (139,149). N-acetylcysteine, which has core antioxidant and anti-inflammatory properties, shows preclinical and clinical efficacy in bipolar disorder and schizophrenia (13), and is a potential neuroprotective candidate (150). Aspirin appeared to reduce the symptoms of schizophrenia in a placebo-controlled trial (151), and was linked to less progression of disease in bipolar disorder in a pharmaco-epidemiological study (152). Minocycline, which has antioxidant and anti-inflammatory properties, has potential in diverse illness models (153).
FATTY ACID MARKERS
Phospholipids are the main structural elements of all cell membranes, and make up around 60% of the dry weight of the brain. The polyunsaturated fatty acids (PUFAs) play central roles in a broad range of physiological functions. For example, two particularly important PUFAs are eicosapentaenoic acid (EPA) and arachidonic acid (AA), which are key players in signal transduction, ion transport and receptor sensitivity (e.g., for serotonin, dopamine, endocannabinoids), as well as precursors in the biosynthesis of the eicosanoids (prostaglandins, leukotrienes, thromboxanes), which mediate the inflammatory response. Another key PUFA, docosahexaenoic acid (DHA), serves as a precursor for the docosanoids (resolvins, neuroprotectins), which have a neuroprotective effect.
PUFAs are essential fatty acids, and since humans are unable to synthesize them de novo, they must be sourced in the diet. The typical Western diet contains relatively low levels of anti-inflammatory omega-3 fatty acids (e.g., EPA) and high levels of pro-inflammatory omega-6 fatty acids (e.g., AA) and saturated fatty acids, leading to increased production of pro-inflammatory eicosanoids. This imbalance has numerous pathological consequences, and is a potent promoter of chronic disease (154).
In relation to mental health, the omega-3 PUFAs may play a role in the pathogenesis of major affective (155,156) and psychotic disorders (157). Alterations in fatty acids in major depression include a decrease in omega-3 PUFAs and increased omega-6/omega-3 PUFA ratios in plasma, erythrocytes, adipose tissue and post-mortem brain tissue (156,158,159). The patterns of these fatty acid alterations are not specific to depression, but are also found in other conditions accompanied by increased oxidative stress, such as Alzheimer's disease, bipolar disorder and schizophrenia, and during normal ageing (160). A recent meta-analysis of 18 studies in schizophrenia examining the four most frequently explored PUFAs (DHA, AA, docosapentaenoic acid, linoleic acid) concluded that decreased levels of DHA and AA were present in antipsychotic-naïve patients. Furthermore, antipsychotic medication may (partially) normalize PUFA measures (161). The reasons for these membrane PUFA deficits are not completely known, but the available data suggests that increased oxidative stress may be one of the mechanisms underlying the reduced levels of omega-3 PUFAs in people with major depressive or psychotic disorder (162,163).
Alterations in membrane PUFAs may be present before the manifestation of stage 2 disorders. A study in a cohort of 33,000 women from the general population found a relationship between the dietary intake of fish (the richest dietary source of PUFAs) and vitamin D and psychotic-like symptoms (164). There is also preliminary evidence that fatty acid deficits may be present during the early stages (stage 1b) of psychotic disorders. For instance, we have shown that supplementation with long-chain omega-3 fatty acids can reduce the risk of progression to psychotic disorder in individuals at ultra-high risk of psychosis (165). In addition, we have found that lower levels of omega-3 PUFAs correlated with more severe negative symptoms in ultra-high risk subjects (166). Together, these findings imply that omega-3 PUFA deficits may be present before the onset of schizophrenia. In fact, omega-3 fatty acids are the only treatment superior to placebo in preventing conversion from stage 1b to stage 2 psychosis (167). On the other hand, PUFA supplementation may have relatively less benefit in patients with established schizophrenia (168). As omega-3 PUFAs are potent anti-inflammatory agents, this suggests that neuroinflammation could be a stage-specific phenomenon that may precede the dopamine overactivity associated with a first psychotic episode (stage 2).
There is also evidence that nervonic acid (NA), a mono-unsaturated omega-9 fatty acid that has been reported to be decreased in people with schizophrenia (169), may serve as a biomarker to differentiate truly prodromal people who are at immediate risk of transition to psychosis from those who do not progress to stage 2 (166). As NA is the major constituent of the sphingolipids in myelin membranes, decreased levels of NA could reflect the suboptimal myelination status seen in ultra-high risk individuals who progress to a psychotic disorder. This view is supported by a recent diffusion tensor imaging study in people with recent-onset psychotic disorders, showing that a lower total PUFA concentration was associated with lower fractional anisotropy in the corpus callosum and bilateral parietal, occipital, temporal and frontal white matter (170).
Finally, increased activity of phospholipase A2, a family of enzymes that catalyze the cleavage of PUFAs from the sn-2 position of phospholipids, is a robust biological finding in schizophrenia (171). Consistent with the staging model, phospholipase A2 activity appears to be higher in first-episode psychosis (stage 2), but not in multi-episode chronic schizophrenia (stages 3-5) (172). Our own research on the role of intracellular phospholipase A2 (inPLA2) in ultra-high risk subjects has shown that levels of membrane omega-3 and omega-6 PUFAs and inPLA2 activity were strongly correlated. However, some of the significant associations were in opposite directions in individuals who did (a positive correlation) and who did not (a negative correlation) transition to psychosis (173). These findings are suggestive of ongoing changes in the interactions of membrane metabolic markers during the course of the ultra-high risk phase of illness (stage 1b).
Thus, there is accumulating evidence that cell membrane PUFAs are involved in the pathophysiology of major mood and psychotic disorders. Lipid metabolism could provide a stage-specific phenomenon that is particularly relevant in the early stages of illness, and should be further investigated and considered for preventive interventions.
CONCLUSIONS
In summary, a large body of data already exists that may offer predictive and early diagnostic markers, as well as indicators of pathophysiological processes that may vary across the stages of the evolution of major psychiatric disorders. We have attempted to bring together evidence from several lines of neurobiological work to address problems of the complex overlaps and heterogeneity of course and outcome across psychiatric disorders. A neurobiologically-informed staging approach that crosses current diagnostic silos may bring clarity to such complexity. What we have proposed is a framework for the next steps in research, and we suggest that some caveats are worth considering as this field moves forward.
First, it is important for staging approaches to be agnostic to traditional symptom-based nosological boundaries. These may not respect the true biological boundaries of disease entities, and therefore may have low utility for predicting outcome or selecting specific interventions. It is perhaps more useful to examine specific neurobiological domains that cut across diagnoses, e.g., working memory, negative salience etc., an approach central to the recent concept of Research Domain Criteria (174). Such an approach is likely to help characterize differential predictors of outcome based on cross-cutting dimensions rather than categorical distinctions.
Second, research needs to address ways to anchor the staging scheme to optimum stage-specific interventions. Available data already suggest certain common pathophysiological mechanisms that could be targeted with benign interventions: e.g., oxidative/inflammatory changes may respond to N-acetylcysteine or fish oil; disturbances in the HPA axis to stress management or cognitive-behavioral therapy; and chronobiological changes to sleep hygiene, simple lifestyle interventions or melatonin. Evidently, for the very early stages, the “do no harm” imperative is critical.
Other biomarkers implicate a developmental component – particularly the structural/functional changes in the brain and neurocognitive impairments – and may respond to more specific treatments such as antidepressants, mood stabilizers, second generation antipsychotics or cognitive remediation.
Third, a staging approach would do well to distinguish pathophysiology from etiological factors. As in the rest of medicine, disease staging based on clinical/biomarker measures may not necessarily map onto etiologically-based classifications. Genetic risk should also be interpreted agnostically, as it is unlikely to provide diagnostic specificity, but may be useful as a guide to disturbances in neurodevelopmental trajectories or aspects of neurobiological functioning.
Fourth, a useful point of departure in staging across diagnoses might be a shift from the broad, non-specific presentation to the evolution of more specific syndromal pictures. Such an evolution may not only involve the emergence of new symptoms or the evolving syndromal coherence of symptom clusters, but also the development (or lack thereof) of disability and functional and social impacts.
Overall, there is a critical need for longitudinal studies of biology and clinical manifestations, agnostic to the traditional diagnoses, to chart the evolution of distinct trajectories. This will inform not only our treatment choices, but also our understanding of the pathophysiology of these complex illnesses. Elaborating the complex linkages between the disturbances in basic neurobiology and the symptomatology that we see in clinical psychiatry will be a major step forward in the development of a truly pre-emptive psychiatry.
References
- 1.Trusheim MR, Berndt ER, Douglas FL. Stratified medicine: strategic and economic implications of combining drugs and clinical biomarkers. Nat Rev Drug Discov. 2007;6:287–93. doi: 10.1038/nrd2251. [DOI] [PubMed] [Google Scholar]
- 2.McGorry PD. Issues for DSM-V: clinical staging: a heuristic pathway to valid nosology and safer, more effective treatment in psychiatry. Am J Psychiatry. 2007;164:859–60. doi: 10.1176/ajp.2007.164.6.859. [DOI] [PubMed] [Google Scholar]
- 3.McGorry PD, Nelson B, Goldstone S, et al. Clinical staging: a heuristic and practical strategy for new research and better health and social outcomes for psychotic and related mood disorders. Can J Psychiatry. 2010;55:486–97. doi: 10.1177/070674371005500803. [DOI] [PubMed] [Google Scholar]
- 4.McGorry PD. The next stage for diagnosis: validity through utility. World Psychiatry. 2013;12:213–5. doi: 10.1002/wps.20080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertelsen M, Jeppesen P, Petersen L, et al. Five-year follow-up of a randomized multicenter trial of intensive early intervention vs standard treatment for patients with a first episode of psychotic illness: the OPUS trial. Arch Gen Psychiatry. 2008;65:762–71. doi: 10.1001/archpsyc.65.7.762. [DOI] [PubMed] [Google Scholar]
- 6.Hegelstad WT, Larsen TK, Auestad B, et al. Long-term follow-up of the TIPS early detection in psychosis study: effects on 10-year outcome. Am J Psychiatry. 2012;169:374–80. doi: 10.1176/appi.ajp.2011.11030459. [DOI] [PubMed] [Google Scholar]
- 7.Norman RM, Manchanda R, Malla AK, et al. Symptom and functional outcomes for a 5 year early intervention program for psychoses. Schizophr Res. 2011;129:111–5. doi: 10.1016/j.schres.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Insel T. The arrival of preemptive psychiatry. Early Interv Psychiatry. 2007;1:5–6. doi: 10.1111/j.1751-7893.2007.00017.x. [DOI] [PubMed] [Google Scholar]
- 9.Wigman JT, van Os J, Thiery E, et al. Psychiatric diagnosis revisited: towards a system of staging and profiling combining nomothetic and idiographic parameters of momentary mental states. PloS One. 2013;8:e59559. doi: 10.1371/journal.pone.0059559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eaton WW, Badawi M, Melton B. Prodromes and precursors: epidemiologic data for primary prevention of disorders with slow onset. Am J Psychiatry. 1995;152:967–72. doi: 10.1176/ajp.152.7.967. [DOI] [PubMed] [Google Scholar]
- 11.Atkinson AJ, Colburne WA, DeGruttola VG, et al. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 12.Koutsouleris N, Davatzikos C, Bottlender R, et al. Early recognition and disease prediction in the at-risk mental states for psychosis using neurocognitive pattern classification. Schizophr Bull. 2012;38:1200–15. doi: 10.1093/schbul/sbr037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berk M, Ng F, Dean O, et al. Glutathione: a novel treatment target in psychiatry. Trends Pharmacol Sci. 2008;29:346–51. doi: 10.1016/j.tips.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Yao JK, Keshavan MS. Antioxidants, Redox signaling, and pathophysiology in schizophrenia: an integrative view. Antioxid Redox Signal. 2011;15:2011–35. doi: 10.1089/ars.2010.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Depp CA, Mausbach BT, Harmell AL, et al. Meta-analysis of the association between cognitive abilities and everyday functioning in bipolar disorder. Bipolar Disord. 2012;14:217–26. doi: 10.1111/j.1399-5618.2012.01011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fett AK, Viechtbauer W, Dominguez MD, et al. The relationship between neurocognition and social cognition with functional outcomes in schizophrenia: a meta-analysis. Neurosci Biobehav Rev. 2011;35:573–88. doi: 10.1016/j.neubiorev.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 17.Bartholomeusz CF, Allott K. Neurocognitive and social cognitive approaches for improving functional outcome in early psychosis: theoretical considerations and current state of evidence. Schizophr Res Treat. 2012;2012:815315. doi: 10.1155/2012/815315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brewer WJ, Wood SJ, Phillips LJ, et al. Generalized and specific cognitive performance in clinical high-risk cohorts: a review highlighting potential vulnerability markers for psychosis. Schizophr Bull. 2006;32:538–55. doi: 10.1093/schbul/sbj077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reichenberg A, Weiser M, Rabinowitz J, et al. A population-based cohort study of premorbid intellectual, language, and behavioral functioning in patients with schizophrenia, schizoaffective disorder, and nonpsychotic bipolar disorder. Am J Psychiatry. 2002;159:2027–35. doi: 10.1176/appi.ajp.159.12.2027. [DOI] [PubMed] [Google Scholar]
- 20.Woodberry KA, Giuliano AJ, Seidman LJ. Premorbid IQ in schizophrenia: a meta-analytic review. Am J Psychiatry. 2008;165:579–87. doi: 10.1176/appi.ajp.2008.07081242. [DOI] [PubMed] [Google Scholar]
- 21.Fusar-Poli P, Deste G, Smieskova R, et al. Cognitive functioning in prodromal psychosis: a meta-analysis. Arch Gen Psychiatry. 2012;69:562–71. doi: 10.1001/archgenpsychiatry.2011.1592. [DOI] [PubMed] [Google Scholar]
- 22.Seidman LJ, Giuliano AJ, Meyer EC, et al. Neuropsychology of the prodrome to psychosis in the NAPLS consortium: relationship to family history and conversion to psychosis. Arch Gen Psychiatry. 2010;67:578–88. doi: 10.1001/archgenpsychiatry.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mesholam-Gately RI, Giuliano AJ, Goff KP, et al. Neurocognition in first-episode schizophrenia: a meta-analytic review. Neuropsychology. 2009;23:315–36. doi: 10.1037/a0014708. [DOI] [PubMed] [Google Scholar]
- 24.Irani F, Kalkstein S, Moberg EA, et al. Neuropsychological performance in older patients with schizophrenia: a meta-analysis of cross-sectional and longitudinal studies. Schizophr Bull. 2011;37:1318–26. doi: 10.1093/schbul/sbq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bozikas VP, Andreou C. Longitudinal studies of cognition in first episode psychosis: a systematic review of the literature. Aust N Z J Psychiatry. 2011;45:93–108. doi: 10.3109/00048674.2010.541418. [DOI] [PubMed] [Google Scholar]
- 26.Szoke A, Trandafir A, Dupont ME, et al. Longitudinal studies of cognition in schizophrenia: meta-analysis. Br J Psychiatry. 2008;192:248–57. doi: 10.1192/bjp.bp.106.029009. [DOI] [PubMed] [Google Scholar]
- 27.Green MF, Bearden CE, Cannon TD, et al. Social cognition in schizophrenia, Part 1: performance across phase of illness. Schizophr Bull. 2012;38:854–64. doi: 10.1093/schbul/sbq171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arts B, Jabben N, Krabbendam L, et al. Meta-analyses of cognitive functioning in euthymic bipolar patients and their first-degree relatives. Psychol Med. 2008;38:771–85. doi: 10.1017/S0033291707001675. [DOI] [PubMed] [Google Scholar]
- 29.Bora E, Yucel M, Pantelis C. Cognitive endophenotypes of bipolar disorder: a meta-analysis of neuropsychological deficits in euthymic patients and their first-degree relatives. J Affect Disord. 2009;113:1–20. doi: 10.1016/j.jad.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 30.Bora E, Yucel M, Pantelis C, et al. Meta-analytic review of neurocognition in bipolar II disorder. Acta Psychiatr Scand. 2011;123:165–74. doi: 10.1111/j.1600-0447.2010.01638.x. [DOI] [PubMed] [Google Scholar]
- 31.Elshahawi HH, Essawi H, Rabie MA, et al. Cognitive functions among euthymic bipolar I patients after a single manic episode versus recurrent episodes. J Affect Disord. 2011;130:180–91. doi: 10.1016/j.jad.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 32.Lopez-Jaramillo C, Lopera-Vasquez J, Gallo A, et al. Effects of recurrence on the cognitive performance of patients with bipolar I disorder: implications for relapse prevention and treatment adherence. Bipolar Disord. 2010;12:557–67. doi: 10.1111/j.1399-5618.2010.00835.x. [DOI] [PubMed] [Google Scholar]
- 33.Torres IJ, DeFreitas VG, DeFreitas CM, et al. Neurocognitive functioning in patients with bipolar I disorder recently recovered from a first manic episode. J Clin Psychiatry. 2010;71:1234–42. doi: 10.4088/JCP.08m04997yel. [DOI] [PubMed] [Google Scholar]
- 34.Daban C, Martinez-Aran A, Torrent C, et al. Specificity of cognitive deficits in bipolar disorder versus schizophrenia. A systematic review. Psychother Psychosom. 2006;75:72–84. doi: 10.1159/000090891. [DOI] [PubMed] [Google Scholar]
- 35.Gale CR, Batty GD, McIntosh AM, et al. Is bipolar disorder more common in highly intelligent people? A cohort study of a million men. Mol Psychiatry. 2013;18:190–4. doi: 10.1038/mp.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer SE, Carlson GA, Wiggs EA, et al. A prospective study of the association among impaired executive functioning, childhood attentional problems, and the development of bipolar disorder. Dev Psychopathol. 2004;16:461–76. doi: 10.1017/s095457940404461x. [DOI] [PubMed] [Google Scholar]
- 37.Ratheesh A, Lin A, Nelson B, et al. Neurocognitive functioning in the prodrome of mania – an exploratory study. J Affect Disord. 2013;147:441–5. doi: 10.1016/j.jad.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 38.Glahn DC, Almasy L, Barguil M, et al. Neurocognitive endophenotypes for bipolar disorder identified in multiplex multigenerational families. Arch Gen Psychiatry. 2010;67:168–77. doi: 10.1001/archgenpsychiatry.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Austin MP, Mitchell P, Goodwin GM. Cognitive deficits in depression: possible implications for functional neuropathology. Br J Psychiatry. 2001;178:200–6. doi: 10.1192/bjp.178.3.200. [DOI] [PubMed] [Google Scholar]
- 40.Baune BT, Czira ME, Smith AL, et al. Neuropsychological performance in a sample of 13-25 year olds with a history of non-psychotic major depressive disorder. J Affect Disord. 2012;141:441–8. doi: 10.1016/j.jad.2012.02.041. [DOI] [PubMed] [Google Scholar]
- 41.Douglas KM, Porter RJ. Longitudinal assessment of neuropsychological function in major depression. Aust N Z J Psychiatry. 2009;43:1105–17. doi: 10.3109/00048670903279887. [DOI] [PubMed] [Google Scholar]
- 42.Gualtieri CT, Johnson LG, Benedict KB. Neurocognition in depression: patients on and off medication versus healthy comparison subjects. J Neuropsychiatry Clin Neurosci. 2006;18:217–25. doi: 10.1176/jnp.2006.18.2.217. [DOI] [PubMed] [Google Scholar]
- 43.Naismith SL, Hickie IB, Turner K, et al. Neuropsychological performance in patients with depression is associated with clinical, etiological and genetic risk factors. J Clin Exp Neurosci. 2003;25:866–77. doi: 10.1076/jcen.25.6.866.16472. [DOI] [PubMed] [Google Scholar]
- 44.Purcell R, Maruff P, Kyrios M, et al. Neuropsychological function in young patients with unipolar major depression. Psychol Med. 1997;27:1277–85. doi: 10.1017/s0033291797005448. [DOI] [PubMed] [Google Scholar]
- 45.Snyder HR. Major depressive disorder is associated with broad impairments on neuropsychological measures of executive function: a meta-analysis and review. Psychol Bull. 2013;139:81–132. doi: 10.1037/a0028727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McDermott LM, Ebmeier KP. A meta-analysis of depression severity and cognitive function. J Affect Disord. 2009;119:1–8. doi: 10.1016/j.jad.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 47.Maalouf FT, Brent D, Clark L, et al. Neurocognitive impairment in adolescent major depressive disorder: state vs. trait illness markers. J Affect Disord. 2011;133:625–32. doi: 10.1016/j.jad.2011.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klimes-Dougan B, Ronsaville D, Wiggs EA, et al. Neuropsychological functioning in adolescent children of mothers with a history of bipolar or major depressive disorders. Biol Psychiatry. 2006;60:957–65. doi: 10.1016/j.biopsych.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 49.Micco JA, Henin A, Biederman J, et al. Executive functioning in offspring at risk for depression and anxiety. Depress Anxiety. 2009;26:780–90. doi: 10.1002/da.20573. [DOI] [PubMed] [Google Scholar]
- 50.Cannon M, Moffitt TE, Caspi A, et al. Neuropsychological performance at the age of 13 years and adult schizophreniform disorder: prospective birth cohort study. Br J Psychiatry. 2006;189:463–4. doi: 10.1192/bjp.bp.105.020552. [DOI] [PubMed] [Google Scholar]
- 51.McClintock SM, Husain MM, Greer TL, et al. Association between depression severity and neurocognitive function in major depressive disorder: a review and synthesis. Neuropsychology. 2010;24:9–34. doi: 10.1037/a0017336. [DOI] [PubMed] [Google Scholar]
- 52.Fossati P, Harvey PO, Le Bastard G, et al. Verbal memory performance of patients with a first depressive episode and patients with unipolar and bipolar recurrent depression. J Psychiatr Res. 2004;38:137–44. doi: 10.1016/j.jpsychires.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 53.Wood SJ, Yung AR, McGorry PD, et al. Neuroimaging and treatment evidence for clinical staging in psychotic disorders: from the at-risk mental state to chronic schizophrenia. Biol Psychiatry. 2011;70:619–25. doi: 10.1016/j.biopsych.2011.05.034. [DOI] [PubMed] [Google Scholar]
- 54.Boos HB, Aleman A, Cahn W, et al. Brain volumes in relatives of patients with schizophrenia: a meta-analysis. Arch Gen Psychiatry. 2007;64:297–304. doi: 10.1001/archpsyc.64.3.297. [DOI] [PubMed] [Google Scholar]
- 55.Gogtay N, Vyas NS, Testa R, et al. Age of onset of schizophrenia: perspectives from structural neuroimaging studies. Schizophr Bull. 2011;37:504–13. doi: 10.1093/schbul/sbr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diwadkar VA, Montrose DM, Dworakowski D, et al. Genetically predisposed offspring with schizotypal features: an ultra high-risk group for schizophrenia? Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:230–8. doi: 10.1016/j.pnpbp.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 57.Bhojraj TS, Francis AN, Montrose DM, et al. Grey matter and cognitive deficits in young relatives of schizophrenia patients. NeuroImage. 2011;54(Suppl. 1):S287–92. doi: 10.1016/j.neuroimage.2010.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhojraj TS, Sweeney JA, Prasad KM, et al. Gray matter loss in young relatives at risk for schizophrenia: relation with prodromal psychopathology. NeuroImage. 2011;54(Suppl. 1):S272–9. doi: 10.1016/j.neuroimage.2010.04.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Steen RG, Mull C, McClure R, et al. Brain volume in first-episode schizophrenia: systematic review and meta-analysis of magnetic resonance imaging studies. Br J Psychiatry. 2006;188:510–8. doi: 10.1192/bjp.188.6.510. [DOI] [PubMed] [Google Scholar]
- 60.Borgwardt SJ, McGuire PK, Aston J, et al. Structural brain abnormalities in individuals with an at-risk mental state who later develop psychosis. Br J Psychiatry. 2007;191(Suppl. 51):s69–75. doi: 10.1192/bjp.191.51.s69. [DOI] [PubMed] [Google Scholar]
- 61.Ellison-Wright I, Glahn DC, Laird AR, et al. The anatomy of first-episode and chronic schizophrenia: an anatomical likelihood estimation meta-analysis. Am J Psychiatry. 2008;165:1015–23. doi: 10.1176/appi.ajp.2008.07101562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun D, Phillips L, Velakoulis D, et al. Progressive brain structural changes mapped as psychosis develops in 'at risk' individuals. Schizophr Res. 2009;108:85–92. doi: 10.1016/j.schres.2008.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takahashi T, Wood SJ, Yung AR, et al. Progressive gray matter reduction of the superior temporal gyrus during transition to psychosis. Arch Gen Psychiatry. 2009;66:366–76. doi: 10.1001/archgenpsychiatry.2009.12. [DOI] [PubMed] [Google Scholar]
- 64.Takahashi T, Wood SJ, Soulsby B, et al. Follow-up MRI study of the insular cortex in first-episode psychosis and chronic schizophrenia. Schizophr Res. 2009;108:49–56. doi: 10.1016/j.schres.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 65.Takahashi T, Wood SJ, Yung AR, et al. Insular cortex gray matter changes in individuals at ultra-high-risk of developing psychosis. Schizophr Res. 2009;111:94–102. doi: 10.1016/j.schres.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 66.McIntosh AM, Owens DC, Moorhead WJ, et al. Longitudinal volume reductions in people at high genetic risk of schizophrenia as they develop psychosis. Biol Psychiatry. 2011;69:953–8. doi: 10.1016/j.biopsych.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 67.Vita A, De Peri L, Silenzi C, et al. Brain morphology in first-episode schizophrenia: a meta-analysis of quantitative magnetic resonance imaging studies. Schizophr Res. 2006;82:75–88. doi: 10.1016/j.schres.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 68.Velakoulis D, Wood SJ, Wong MT, et al. Hippocampal and amygdala volumes according to psychosis stage and diagnosis: a magnetic resonance imaging study of chronic schizophrenia, first-episode psychosis, and ultra-high-risk individuals. Arch Gen Psychiatry. 2006;63:139–49. doi: 10.1001/archpsyc.63.2.139. [DOI] [PubMed] [Google Scholar]
- 69.Wood SJ, Kennedy D, Phillips LJ, et al. Hippocampal pathology in individuals at ultra-high risk for psychosis: a multi-modal magnetic resonance study. NeuroImage. 2010;52:62–8. doi: 10.1016/j.neuroimage.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 70.Buehlmann E, Berger GE, Aston J, et al. Hippocampus abnormalities in at risk mental states for psychosis? A cross-sectional high resolution region of interest magnetic resonance imaging study. J Psychiatr Res. 2010;44:447–53. doi: 10.1016/j.jpsychires.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 71.Olabi B, Ellison-Wright I, McIntosh AM, et al. Are there progressive brain changes in schizophrenia? A meta-analysis of structural magnetic resonance imaging studies. Biol Psychiatry. 2011;70:88–96. doi: 10.1016/j.biopsych.2011.01.032. [DOI] [PubMed] [Google Scholar]
- 72.Hulshoff Pol HE, Kahn RS. What happens after the first episode? A review of progressive brain changes in chronically ill patients with schizophrenia. Schizophr Bull. 2008;34:354–66. doi: 10.1093/schbul/sbm168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ho BC, Andreasen NC, Ziebell S, et al. Long-term antipsychotic treatment and brain volumes: a longitudinal study of first-episode schizophrenia. Arch Gen Psychiatry. 2011;68:128–37. doi: 10.1001/archgenpsychiatry.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andreasen NC, Liu D, Ziebell S, et al. Relapse duration, treatment intensity, and brain tissue loss in schizophrenia: a prospective longitudinal MRI study. Am J Psychiatry. 2013;170:609–15. doi: 10.1176/appi.ajp.2013.12050674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arnone D, Cavanagh J, Gerber D, et al. Magnetic resonance imaging studies in bipolar disorder and schizophrenia: meta-analysis. Br J Psychiatry. 2009;195:194–201. doi: 10.1192/bjp.bp.108.059717. [DOI] [PubMed] [Google Scholar]
- 76.Usher J, Leucht S, Falkai P, et al. Correlation between amygdala volume and age in bipolar disorder – a systematic review and meta-analysis of structural MRI studies. Psychiatry Res. 2010;182:1–8. doi: 10.1016/j.pscychresns.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 77.Vita A, De Peri L, Sacchetti E. Gray matter, white matter, brain, and intracranial volumes in first-episode bipolar disorder: a meta-analysis of magnetic resonance imaging studies. Bipolar Disord. 2009;11:807–14. doi: 10.1111/j.1399-5618.2009.00759.x. [DOI] [PubMed] [Google Scholar]
- 78.Hickie IB, Naismith SL, Ward PB, et al. Psychomotor slowing in older patients with major depression: relationships with blood flow in the caudate nucleus and white matter lesions. Psychiatry Res. 2007;155:211–20. doi: 10.1016/j.pscychresns.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 79.Kalmar JH, Wang F, Spencer L, et al. Preliminary evidence for progressive prefrontal abnormalities in adolescents and young adults with bipolar disorder. J Int Neuropsychol Soc. 2009;15:476–81. doi: 10.1017/S1355617709090584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Salokangas RK, Cannon T, Van Erp T, et al. Structural magnetic resonance imaging in patients with first-episode schizophrenia, psychotic and severe non-psychotic depression and healthy controls. Results of the schizophrenia and affective psychoses (SAP) project. Br J Psychiatry. 2002;181(Suppl. 43):s58–65. doi: 10.1192/bjp.181.43.s58. [DOI] [PubMed] [Google Scholar]
- 81.Ketter TA, Wang PW, Becker OV, et al. Psychotic bipolar disorders: dimensionally similar to or categorically different from schizophrenia? J Psychiatr Res. 2004;38:47–61. doi: 10.1016/s0022-3956(03)00099-2. [DOI] [PubMed] [Google Scholar]
- 82.Strasser HC, Lilyestrom J, Ashby ER, et al. Hippocampal and ventricular volumes in psychotic and nonpsychotic bipolar patients compared with schizophrenia patients and community control subjects: a pilot study. Biol Psychiatry. 2005;57:633–9. doi: 10.1016/j.biopsych.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 83.De Peri L, Crescini A, Deste G, et al. Brain structural abnormalities at the onset of schizophrenia and bipolar disorder: a meta-analysis of controlled magnetic resonance imaging studies. Curr Pharmaceut Des. 2012;18:486–94. doi: 10.2174/138161212799316253. [DOI] [PubMed] [Google Scholar]
- 84.Ellison-Wright I, Bullmore E. Anatomy of bipolar disorder and schizophrenia: a meta-analysis. Schizophr Res. 2010;117:1–12. doi: 10.1016/j.schres.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 85.Umbricht D, Krljes S. Mismatch negativity in schizophrenia: a meta-analysis. Schizophr Res. 2005;76:1–23. doi: 10.1016/j.schres.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 86.Umbricht D, Koller R, Schmid L, et al. How specific are deficits in mismatch negativity generation to schizophrenia? Biol Psychiatry. 2003;53:1120–31. doi: 10.1016/s0006-3223(02)01642-6. [DOI] [PubMed] [Google Scholar]
- 87.He W, Chai H, Zheng L, et al. Mismatch negativity in treatment-resistant depression and borderline personality disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:366–71. doi: 10.1016/j.pnpbp.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 88.Kahkonen S, Yamashita H, Rytsala H, et al. Dysfunction in early auditory processing in major depressive disorder revealed by combined MEG and EEG. J Psychiatry Neurosci Jpn. 2007;32:316–22. [PMC free article] [PubMed] [Google Scholar]
- 89.Chang Y, Xu J, Shi N, et al. Dysfunction of processing task-irrelevant emotional faces in major depressive disorder patients revealed by expression-related visual MMN. Neurosci Lett. 2010;472:33–7. doi: 10.1016/j.neulet.2010.01.050. [DOI] [PubMed] [Google Scholar]
- 90.Takei Y, Kumano S, Hattori S, et al. Preattentive dysfunction in major depression: a magnetoencephalography study using auditory mismatch negativity. Psychophysiology. 2009;46:52–61. doi: 10.1111/j.1469-8986.2008.00748.x. [DOI] [PubMed] [Google Scholar]
- 91.Jahshan C, Cadenhead KS, Rissling AJ, et al. Automatic sensory information processing abnormalities across the illness course of schizophrenia. Psychol Med. 2012;42:85–97. doi: 10.1017/S0033291711001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ahveninen J, Jaaskelainen IP, Osipova D, et al. Inherited auditory-cortical dysfunction in twin pairs discordant for schizophrenia. Biol Psychiatry. 2006;60:612–20. doi: 10.1016/j.biopsych.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 93.Magno E, Yeap S, Thakore JH, et al. Are auditory-evoked frequency and duration mismatch negativity deficits endophenotypic for schizophrenia? High-density electrical mapping in clinically unaffected first-degree relatives and first-episode and chronic schizophrenia. Biol Psychiatry. 2008;64:385–91. doi: 10.1016/j.biopsych.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bodatsch M, Ruhrmann S, Wagner M, et al. Prediction of psychosis by mismatch negativity. Biol Psychiatry. 2011;69:959–66. doi: 10.1016/j.biopsych.2010.09.057. [DOI] [PubMed] [Google Scholar]
- 95.Devrim-Ucok M, Keskin-Ergen HY, Ucok A. Mismatch negativity at acute and post-acute phases of first-episode schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2008;258:179–85. doi: 10.1007/s00406-007-0772-9. [DOI] [PubMed] [Google Scholar]
- 96.Salisbury DF, Shenton ME, Griggs CB, et al. Mismatch negativity in chronic schizophrenia and first-episode schizophrenia. Arch Gen Psychiatry. 2002;59:686–94. doi: 10.1001/archpsyc.59.8.686. [DOI] [PubMed] [Google Scholar]
- 97.Salisbury DF, Kuroki N, Kasai K, et al. Progressive and interrelated functional and structural evidence of post-onset brain reduction in schizophrenia. Arch Gen Psychiatry. 2007;64:521–9. doi: 10.1001/archpsyc.64.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Todd J, Michie PT, Schall U, et al. Deviant matters: duration, frequency, and intensity deviants reveal different patterns of mismatch negativity reduction in early and late schizophrenia. Biol Psychiatry. 2008;63:58–64. doi: 10.1016/j.biopsych.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 99.Umbricht DS, Bates JA, Lieberman JA, et al. Electrophysiological indices of automatic and controlled auditory information processing in first-episode, recent-onset and chronic schizophrenia. Biol Psychiatry. 2006;59:762–72. doi: 10.1016/j.biopsych.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 100.Valkonen-Korhonen M, Purhonen M, Tarkka IM, et al. Altered auditory processing in acutely psychotic never-medicated first-episode patients. Brain Res Cogn Brain Res. 2003;17:747–58. doi: 10.1016/s0926-6410(03)00199-x. [DOI] [PubMed] [Google Scholar]
- 101.Rasser PE, Schall U, Todd J, et al. Gray matter deficits, mismatch negativity, and outcomes in schizophrenia. Schizophr Bull. 2011;37:131–40. doi: 10.1093/schbul/sbp060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Oades RD, Wild-Wall N, Juran SA, et al. Auditory change detection in schizophrenia: sources of activity, related neuropsychological function and symptoms in patients with a first episode in adolescence, and patients 14 years after an adolescent illness-onset. BMC Psychiatry. 2006;6:7. doi: 10.1186/1471-244X-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shaikh M, Valmaggia L, Broome MR, et al. Reduced mismatch negativity predates the onset of psychosis. Schizophr Res. 2012;134:42–8. doi: 10.1016/j.schres.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 104.Shin KS, Kim JS, Kang DH, et al. Pre-attentive auditory processing in ultra-high-risk for schizophrenia with magnetoencephalography. Biol Psychiatry. 2009;65:1071–8. doi: 10.1016/j.biopsych.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 105.Hermens DF, Ward PB, Hodge MA, et al. Impaired MMN/P3a complex in first-episode psychosis: cognitive and psychosocial associations. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:822–9. doi: 10.1016/j.pnpbp.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 106.Glozier N, Martiniuk A, Patton G, et al. Short sleep duration in prevalent and persistent psychological distress in young adults: the DRIVE study. Sleep. 2010;33:1139–45. doi: 10.1093/sleep/33.9.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hickie IB, Rogers NL. Novel melatonin-based therapies: potential advances in the treatment of major depression. Lancet. 2011;378:621–31. doi: 10.1016/S0140-6736(11)60095-0. [DOI] [PubMed] [Google Scholar]
- 108.Angst J, Merikangas KR. Multi-dimensional criteria for the diagnosis of depression. J Affect Disord. 2001;62:7–15. doi: 10.1016/s0165-0327(00)00346-3. [DOI] [PubMed] [Google Scholar]
- 109.Keshavan MS, Reynolds CF, Kupfer DJ. Electroencephalographic sleep in schizophrenia: a critical review. Compr Psychiatry. 1990;31:34–47. doi: 10.1016/0010-440x(90)90052-t. [DOI] [PubMed] [Google Scholar]
- 110.Rao U, Hammen CL, Poland RE. Risk markers for depression in adolescents: sleep and HPA measures. Neuropsychopharmacology. 2009;34:1936–45. doi: 10.1038/npp.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Keshavan MS, Diwadkar VA, Montrose DM, et al. Premorbid characterization in schizophrenia: the Pittsburgh High Risk Study. World Psychiatry. 2004;3:163–8. [PMC free article] [PubMed] [Google Scholar]
- 112.Sarkar S, Katshu MZ, Nizamie SH, et al. Slow wave sleep deficits as a trait marker in patients with schizophrenia. Schizophr Res. 2010;124:127–33. doi: 10.1016/j.schres.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 113.Ruhrmann S, Schultze-Lutter F, Salokangas RK, et al. Prediction of psychosis in adolescents and young adults at high risk: results from the prospective European prediction of psychosis study. Arch Gen Psychiatry. 2010;67:241–51. doi: 10.1001/archgenpsychiatry.2009.206. [DOI] [PubMed] [Google Scholar]
- 114.Poulin J, Daoust AM, Forest G, et al. Sleep architecture and its clinical correlates in first episode and neuroleptic-naive patients with schizophrenia. Schizophr Res. 2003;62:147–53. doi: 10.1016/s0920-9964(02)00346-8. [DOI] [PubMed] [Google Scholar]
- 115.Keshavan MS, Miewald J, Haas G, et al. Slow-wave sleep and symptomatology in schizophrenia and related psychotic disorders. J Psychiatr Res. 1995;29:303–14. doi: 10.1016/0022-3956(95)00023-x. [DOI] [PubMed] [Google Scholar]
- 116.Keshavan MS, Reynolds CF, 3rd, Miewald JM, et al. A longitudinal study of EEG sleep in schizophrenia. Psychiatry Res. 1996;59:203–11. doi: 10.1016/0165-1781(95)02810-2. [DOI] [PubMed] [Google Scholar]
- 117.Phillips LJ, McGorry PD, Garner B, et al. Stress, the hippocampus and the hypothalamic-pituitary-adrenal axis: implications for the development of psychotic disorders. Aust N Z J Psychiatry. 2006;40:725–41. doi: 10.1080/j.1440-1614.2006.01877.x. [DOI] [PubMed] [Google Scholar]
- 118.Zhang TY, Labonte B, Wen XL, et al. Epigenetic mechanisms for the early environmental regulation of hippocampal glucocorticoid receptor gene expression in rodents and humans. Neuropsychopharmacology. 2013;38:111–23. doi: 10.1038/npp.2012.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Watson S, Gallagher P, Ritchie JC, et al. Hypothalamic-pituitary-adrenal axis function in patients with bipolar disorder. Br J Psychiatry. 2004;184:496–502. doi: 10.1192/bjp.184.6.496. [DOI] [PubMed] [Google Scholar]
- 120.Pariante CM, Dazzan P, Danese A, et al. Increased pituitary volume in antipsychotic-free and antipsychotic-treated patients of the AEsop first-onset psychosis study. Neuropsychopharmacology. 2005;30:1923–31. doi: 10.1038/sj.npp.1300766. [DOI] [PubMed] [Google Scholar]
- 121.Ryan MC, Sharifi N, Condren R, et al. Evidence of basal pituitary-adrenal overactivity in first episode, drug naive patients with schizophrenia. Psychoneuroendocrinology. 2004;29:1065–70. doi: 10.1016/j.psyneuen.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 122.Myin-Germeys I, Delespaul P, van Os J. Behavioural sensitization to daily life stress in psychosis. Psychol Med. 2005;35:733–41. doi: 10.1017/s0033291704004179. [DOI] [PubMed] [Google Scholar]
- 123.Walder DJ, Walker EF, Lewine RJ. Cognitive functioning, cortisol release, and symptom severity in patients with schizophrenia. Biol Psychiatry. 2000;48:1121–32. doi: 10.1016/s0006-3223(00)01052-0. [DOI] [PubMed] [Google Scholar]
- 124.Garner B, Phassouliotis C, Phillips LJ, et al. Cortisol and dehydroepiandrosterone-sulphate levels correlate with symptom severity in first-episode psychosis. J Psychiatr Res. 2011;45:249–55. doi: 10.1016/j.jpsychires.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 125.Mondelli V, Pariante CM, Navari S, et al. Higher cortisol levels are associated with smaller left hippocampal volume in first-episode psychosis. Schizophr Res. 2010;119:75–8. doi: 10.1016/j.schres.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Aas M, Dazzan P, Mondelli V, et al. Abnormal cortisol awakening response predicts worse cognitive function in patients with first-episode psychosis. Psychol Med. 2011;41:463–76. doi: 10.1017/S0033291710001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cohrs S, Roher C, Jordan W, et al. The atypical antipsychotics olanzapine and quetiapine, but not haloperidol, reduce ACTH and cortisol secretion in healthy subjects. Psychopharmacology. 2006;185:11–8. doi: 10.1007/s00213-005-0279-x. [DOI] [PubMed] [Google Scholar]
- 128.Nicolo JP, Berger GE, Garner BA, et al. The effect of atypical antipsychotics on pituitary gland volume in patients with first-episode psychosis: a longitudinal MRI study. Schizophr Res. 2010;116:49–54. doi: 10.1016/j.schres.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 129.Garner B, Pariante CM, Wood SJ, et al. Pituitary volume predicts future transition to psychosis in individuals at ultra-high risk of developing psychosis. Biol Psychiatry. 2005;58:417–23. doi: 10.1016/j.biopsych.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 130.Walker EF, Brennan PA, Esterberg M, et al. Longitudinal changes in cortisol secretion and conversion to psychosis in at-risk youth. J Abnorm Psychol. 2010;119:401–8. doi: 10.1037/a0018399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Thompson KN, Phillips LJ, Komesaroff P, et al. Stress and HPA-axis functioning in young people at ultra high risk for psychosis. J Psychiatr Res. 2007;41:561–9. doi: 10.1016/j.jpsychires.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 132.Collip D, Nicolson NA, Lardinois M, et al. Daily cortisol, stress reactivity and psychotic experiences in individuals at above average genetic risk for psychosis. Psychol Med. 2011;41:2305–15. doi: 10.1017/S0033291711000602. [DOI] [PubMed] [Google Scholar]
- 133.Mondelli V, Dazzan P, Gabilondo A, et al. Pituitary volume in unaffected relatives of patients with schizophrenia and bipolar disorder. Psychoneuroendocrinology. 2008;33:1004–12. doi: 10.1016/j.psyneuen.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 134.McNutt MD, Liu S, Manatunga A, et al. Neurobehavioral effects of interferon-alpha in patients with hepatitis-C: symptom dimensions and responsiveness to paroxetine. Neuropsychopharmacology. 2012;37:1444–54. doi: 10.1038/npp.2011.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Miller BJ, Buckley P, Seabolt W, et al. Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry. 2011;70:663–71. doi: 10.1016/j.biopsych.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Raedler TJ. Inflammatory mechanisms in major depressive disorder. Curr Opin Psychiatry. 2011;24:519–25. doi: 10.1097/YCO.0b013e32834b9db6. [DOI] [PubMed] [Google Scholar]
- 137.Wadee AA, Kuschke RH, Wood LA, et al. Serological observations in patients suffering from acute manic episodes. Hum Psychopharmacol. 2002;17:175–9. doi: 10.1002/hup.390. [DOI] [PubMed] [Google Scholar]
- 138.Do KQ, Cabungcal JH, Frank A, et al. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol. 2009;19:220–30. doi: 10.1016/j.conb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 139.Pasco JA, Jacka FN, Williams LJ, et al. Clinical implications of the cytokine hypothesis of depression: the association between use of statins and aspirin and the risk of major depression. Psychother Psychosom. 2010;79:323–5. doi: 10.1159/000319530. [DOI] [PubMed] [Google Scholar]
- 140.Brietzke E, Kapczinski F. TNF-alpha as a molecular target in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1355–61. doi: 10.1016/j.pnpbp.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 141.Kauer-Sant'Anna M, Kapczinski F, Andreazza AC, et al. Brain-derived neurotrophic factor and inflammatory markers in patients with early- vs. late-stage bipolar disorder. Int J Neuropsychopharmacol. 2009;12:447–58. doi: 10.1017/S1461145708009310. [DOI] [PubMed] [Google Scholar]
- 142.Andreazza AC. Mitchondrial dysfunction and oxidative stress in bipolar disorder. Eur Psychiatry. 2009;24:S15. [Google Scholar]
- 143.Berk M, Kapczinski F, Andreazza AC, et al. Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev. 2011;35:804–17. doi: 10.1016/j.neubiorev.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 144.Moylan S, Maes M, Wray NR, et al. The neuroprogressive nature of major depressive disorder: pathways to disease evolution and resistance, and therapeutic implications. Mol Psychiatry. 2013;18:595–606. doi: 10.1038/mp.2012.33. [DOI] [PubMed] [Google Scholar]
- 145.Goldstein BI, Kemp DE, Soczynska JK, et al. Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. J Clin Psychiatry. 2009;70:1078–90. doi: 10.4088/JCP.08r04505. [DOI] [PubMed] [Google Scholar]
- 146.Meyer JM, McEvoy JP, Davis VG, et al. Inflammatory markers in schizophrenia: comparing antipsychotic effects in phase 1 of the clinical antipsychotic trials of intervention effectiveness study. Biol Psychiatry. 2009;66:1013–22. doi: 10.1016/j.biopsych.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nery FG, Monkul ES, Hatch JP, et al. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum Psychopharmacol. 2008;23:87–94. doi: 10.1002/hup.912. [DOI] [PubMed] [Google Scholar]
- 148.Muller N, Krause D, Dehning S, et al. Celecoxib treatment in an early stage of schizophrenia: results of a randomized, double-blind, placebo-controlled trial of celecoxib augmentation of amisulpride treatment. Schizophr Res. 2010;121:118–24. doi: 10.1016/j.schres.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 149.Stafford L, Berk M. The use of statins after a cardiac intervention is associated with reduced risk of subsequent depression: proof of concept for the inflammatory and oxidative hypotheses of depression? J Clin Psychiatry. 2011;72:1229–35. doi: 10.4088/JCP.09m05825blu. [DOI] [PubMed] [Google Scholar]
- 150.Dean O, Giorlando F, Berk M. N-acetylcysteine in psychiatry: current therapeutic evidence and potential mechanisms of action. J Psychiatry Neurosci Jpn. 2011;36:78–86. doi: 10.1503/jpn.100057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Laan W, Grobbee DE, Selten JP, et al. Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2010;71:520–7. doi: 10.4088/JCP.09m05117yel. [DOI] [PubMed] [Google Scholar]
- 152.Stolk P, Souverein PC, Wilting I, et al. Is aspirin useful in patients on lithium? A pharmacoepidemiological study related to bipolar disorder. Prostagland Leukotr Essent Fatty Acids. 2010;82:9–14. doi: 10.1016/j.plefa.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dean OM, Data-Franco J, Giorlando F, et al. Minocycline: therapeutic potential in psychiatry. CNS Drugs. 2012;26:391–401. doi: 10.2165/11632000-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 154.Simopoulos AP. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med. 2008;233:674–88. doi: 10.3181/0711-MR-311. [DOI] [PubMed] [Google Scholar]
- 155.Horrobin DF. Phospholipid metabolism and depression: the possible roles of phospholipase A2 and coenzyme A-independent transacylase. Hum Psychopharmacol. 2001;16:45–52. doi: 10.1002/hup.182. [DOI] [PubMed] [Google Scholar]
- 156.Parker G, Gibson NA, Brotchie H, et al. Omega-3 fatty acids and mood disorders. Am J Psychiatry. 2006;163:969–78. doi: 10.1176/ajp.2006.163.6.969. [DOI] [PubMed] [Google Scholar]
- 157.Horrobin DF. The membrane phospholipid hypothesis as a biochemical basis for the neurodevelopmental concept of schizophrenia. Schizophr Res. 1998;30:193–208. doi: 10.1016/s0920-9964(97)00151-5. [DOI] [PubMed] [Google Scholar]
- 158.Assies J, Pouwer F, Lok A, et al. Plasma and erythrocyte fatty acid patterns in patients with recurrent depression: a matched case-control study. PloS One. 2010;5:e10635. doi: 10.1371/journal.pone.0010635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.McNamara RK, Jandacek R, Rider T, et al. Fatty acid composition of the postmortem prefrontal cortex of adolescent male and female suicide victims. Prostagland Leukotr Essent Fatty Acids. 2009;80:19–26. doi: 10.1016/j.plefa.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 160.Ng F, Berk M, Dean O, et al. Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. Int J Neuropsychopharmacol. 2008;11:851–76. doi: 10.1017/S1461145707008401. [DOI] [PubMed] [Google Scholar]
- 161.Hoen WP, Lijmer JG, Duran M, et al. Red blood cell polyunsaturated fatty acids measured in red blood cells and schizophrenia: a meta-analysis. Psychiatry Res. 2013;207:1–12. doi: 10.1016/j.psychres.2012.09.041. [DOI] [PubMed] [Google Scholar]
- 162.Mahadik SP, Evans D, Lal H. Oxidative stress and role of antioxidant and omega-3 essential fatty acid supplementation in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:463–93. doi: 10.1016/s0278-5846(00)00181-0. [DOI] [PubMed] [Google Scholar]
- 163.Zamaria N. Alteration of polyunsaturated fatty acid status and metabolism in health and disease. Reprod Nutr Develop. 2004;44:273–82. doi: 10.1051/rnd:2004034. [DOI] [PubMed] [Google Scholar]
- 164.Hedelin M, Lof M, Olsson M, et al. Dietary intake of fish, omega-3, omega-6 polyunsaturated fatty acids and vitamin D and the prevalence of psychotic-like symptoms in a cohort of 33,000 women from the general population. BMC Psychiatry. 2010;10:38. doi: 10.1186/1471-244X-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Amminger GP, Schafer MR, Papageorgiou K, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2010;67:146–54. doi: 10.1001/archgenpsychiatry.2009.192. [DOI] [PubMed] [Google Scholar]
- 166.Amminger GP, Schafer MR, Klier CM, et al. Decreased nervonic acid levels in erythrocyte membranes predict psychosis in help-seeking ultra-high-risk individuals. Mol Psychiatry. 2012;17:1150–2. doi: 10.1038/mp.2011.167. [DOI] [PubMed] [Google Scholar]
- 167.Nasrallah H. Beyond dopamine: the "other" effects of antipsychotics. Curr Psychiatry. 2013;12:8–9. [Google Scholar]
- 168.Fusar-Poli P, Berger G. Eicosapentaenoic acid interventions in schizophrenia: meta-analysis of randomized, placebo-controlled studies. J Clin Psychopharmacol. 2012;32:179–85. doi: 10.1097/JCP.0b013e318248b7bb. [DOI] [PubMed] [Google Scholar]
- 169.Assies J, Lieverse R, Vreken P, et al. Significantly reduced docosahexaenoic and docosapentaenoic acid concentrations in erythrocyte membranes from schizophrenic patients compared with a carefully matched control group. Biol Psychiatry. 2001;49:510–22. doi: 10.1016/s0006-3223(00)00986-0. [DOI] [PubMed] [Google Scholar]
- 170.Peters BD, Machielsen MW, Hoen WP, et al. Polyunsaturated fatty acid concentration predicts myelin integrity in early-phase psychosis. Schizophr Bull. 2013;39:830–8. doi: 10.1093/schbul/sbs089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Berger GE, Smesny S, Amminger GP. Bioactive lipids in schizophrenia. Int Rev Psychiatry. 2006;18:85–98. doi: 10.1080/09540260600583072. [DOI] [PubMed] [Google Scholar]
- 172.Smesny S, Kinder D, Willhardt I, et al. Increased calcium-independent phospholipase A2 activity in first but not in multiepisode chronic schizophrenia. Biol Psychiatry. 2005;57:399–405. doi: 10.1016/j.biopsych.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 173.Smesny S, Milleit B, Hipler UC, et al. Omega-3 fatty acid supplementation changes intracellular phospholipase A activity and membrane fatty acid profiles in individuals at ultra-high risk for psychosis. Mol Psychiatry. 2014;19:317–24. doi: 10.1038/mp.2013.7. [DOI] [PubMed] [Google Scholar]
- 174.Insel T, Cuthbert B, Garvey M, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167:748–51. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]