

Abstract
Background
The Birt-Hogg-Dubé syndrome is a rare cancer susceptibility syndrome characterised by renal tumours, lung cysts and pneumothoraces, and fibrofolliculomas. It is caused by dominantly inherited mutations in FLCN. Our objective was to report renal tumour characteristics in a large series of patients with the Birt-Hogg-Dubé syndrome.
Methods
We studied French Birt-Hogg-Dubé patients with a history of renal tumour.
Results
We included 33 patients with 21 distinct germline FLCN mutations. Median age at diagnosis of first renal tumour was 46, and age varied from 20 to 83. Twenty cases had one renal tumour, the remainder had two or more tumours. Most cases (23/33, 70%) had oncocytoma or renal cell carcinoma of the chromophobe or hybrid chromophobe-oncocytoma type, three had clear cell carcinoma (9%), and the other seven had carcinoma of papillary, undifferentiated or undetermined histology. Four cases had metastatic disease, although none died of it.
Conclusions
Age at renal tumour diagnosis was highly variable, highlighting the need for regular surveillance from young adulthood to old age. Most cases had tumour types classically associated with Birt-Hogg-Dubé, i.e. oncocytoma or renal cell carcinoma of the chromophobe or hybrid type. Nevertheless, 9% had clear cell renal cell carcinoma. Geneticists, urologists and oncologists should therefore be alert to the possibility of Birt-Hogg-Dubé in patients with renal cell carcinoma of clear cell histology, especially if there are associated manifestations. Finally, the behaviour of metastatic carcinoma seemed more indolent than in sporadic renal cancers.
Keywords: Renal cell carcinoma, Neoplastic syndromes, Hereditary, Birt-Hogg-Dubé, Genetic predisposition to disease, Folliculin
Introduction
The Birt-Hogg-Dubé syndrome (BHD) is a rare cancer susceptibility syndrome characterised mainly by renal cell tumours, lung cysts and pneumothoraces, and skin papules named fibrofolliculomas [1]. It is caused by germline mutations in the FLCN gene, which are inherited in a dominant fashion [1]. FLCN codes for folliculin, a protein that operates in the mTOR pathway and leads, when inactivated, to increased mitochondrial oxidative metabolism [2,3]. Phenotype is variable among patients with BHD. Indeed, renal cell tumours affects up to 34% of mutation carriers, and consists predominantly of chromophobe renal cell carcinomas (RCC), benign oncocytomas, and RCC with hybrid chromophobe – oncocytoma histology [4-7]. Twenty-four to 37% of BHD cases present with at least one episode of pneumothorax in their lifetime, and 79% to 84% have skin fibrofolliculomas [4,5,8].
We describe herein renal cell tumour characteristics in 33 BHD patients, among them four cases with metastatic, albeit indolent disease.
Patients and methods
In France, patients are referred to accredited cancer genetics clinics throughout the country when genetic susceptibility to renal cell tumours is suspected. Not all patients present with a personal or family history of renal cell tumour. Indeed, some have non-renal manifestations suggestive of a syndrome in which renal cell tumour risk is increased, and others are healthy relatives of individuals with an established genetic predisposition. The genes most commonly analysed in hereditary RCC are VHL (associated with von Hippel-Lindau disease), FH (Hereditary Leiomyomatosis with RCC), FLCN (BHD), SDHB (PGL4 syndrome), and MET (Hereditary Papillary RCC) [9].
On November 15, 2013, we explored two overlapping sources, the PREDIR National Hereditary Kidney Cancer Centre database located at Hôpital Bicêtre near Paris, and the Molecular Genetics Laboratory at the Edouard Herriot University Hospital in Lyon. The PREDIR centre strives to collect clinical and molecular data on all French carriers of a mutation predisposing to renal cell tumours, whereas the Lyon laboratory is the main provider of FLCN germline analysis in France. We retrieved clinical data for all patients with a diagnosis of molecularly-proven BHD, and selected those with a history of renal cell tumour at the time of genetic testing. All patients had signed an informed consent form before genetic testing, as legally required. Cases reported in two previous papers are included in this study [10,11]. Follow-up data for included patients are up-to-date as of 29 September 2014, the day the final version of this manuscript was submitted.
Mutations were identified using both Sanger sequencing of exons and their flanking regions and multiplex ligation probe-dependent amplification (MLPA) on genomic DNA extracted from blood. Detailed protocols are available on request. Mutations were considered pathogenic if they had been reported and reliably identified as such in the FLCN mutation database (http://grenada.lumc.nl/LOVD2/shared1), or for never-reported mutations, if they led to a truncated protein.
Results
We ascertained a total of 124 BHD patients from 72 distinct families. Thirty-three had a history of renal cell tumour (Tables 1 and 2). Twenty-one were male, 12 female. Median age at diagnosis of first tumour was 46 (age range 20–83). Twenty cases had one tumour, four had two tumours, and nine had multifocal (three or more) tumours. Most cases (23/33, 70%) had oncocytoma or RCC of the chromophobe or hybrid chromophobe-oncocytoma type, three had clear cell RCC (9%), one had papillary RCC with interspersed eosinophilic cells, and one had undifferentiated RCC. Histopathology was unavailable for five patients, most often because their tumours were radiologically characteristic but too small to justify surgery. The most common histological types are illustrated in Figure 1.
Table 1.
Birt-Hogg-Dubé patients with renal tumours
| Patient | Gender | Mutation | 1st renal tumour, age | Number of renal tumours | Histology | Other BHD manifestations | Duration of F-U (months) | Latest F-U data |
|---|---|---|---|---|---|---|---|---|
| 11525 | M | c.1285dup, p.His429Profs*27 | 73 | Multifocal | Ch, H | FF | 30 | 05/2014 |
| 11528 | M | c.1285dup, p.His429Profs*27 | 83 | 2 | N/A | FF, PNO | 103 | 11/2013 |
| 11545 | M | c.1285dup, p.His429Profs*27 | 39 | 1 | N/A | Lung cysts | 65 | 05/2011 |
| 13851 | M | c.1285dup, p.His429Profs*27 | 35 | 1 (Mt) | Und | FF | 225 | 03/2003 (D) |
| 14381 | M | c.755dup, p.Cys253Valfs*39 | 62 | 1 | CC | FF | 150 | 05/2009 |
| 14840 | M | c.1285dup, p.His429Profs*27 | 20 | 1 | N/A | FF, PNO | 672 | 02/2011 |
| 15842 | F | c.828del, p.Ala277Leufs*16 | 38 | 1 | Ch | - | 176 | 02/2014 |
| 19276 | M | c.1062 + 2 T > G | 56 | 2 | Ch, H | FF | 96 | 05/2011 |
| 19398 | F | c.663dup, p.Met222Aspfs*26 | 42 | Multifocal (Mt) | H | PNO | 115 | 09/2014 |
| 20471 | F | c.1285dup, p.His429Profs*27 | 34 | Multifocal | Ch, O | FF, PNO | 248 | 01/2014 |
| 20472 | M | c.67G > T, p.Glu23* | 43 | 1 | Ch | PNO | 132 | 03/2014 |
| 21310 | F | c.1198G > A, p.Val400Ile | 46 | 1(Mt) | Ch | - | 99 | 07/2014 |
| 22973 | M | c.1523A > G, p.Lys508Arg | 56 | 1 (Mt) | Ch | FF, PNO | 96 | 03/2014 |
| 25891 | F | c.318C > G, p.Tyr106* | 25 | 1 | P | Lung cysts | 96 | 03/2014 |
| 26016 | M | c.1300G > A, p.Glu434Lys | 48 | Multifocal | Ch, H | FF | 16 | 05/2011 |
| 27674 | M | c.616A > T, p.Lys206* | 56 | 1 | Ch | PNO | 93 | 05/2013 |
| 28190 | M | c.1367_1398del, p.Asp456Glyfs*19 | 53 | 1 | O | Lung cysts | 16 | 09/2012 |
| 28535 | F | c.1063-2A > G | 51 | 1 | Ch | FF, lung cysts | 72 | 04/2014 |
| 28537 | F | c.1300G > A, p.Glu434Lys | 47 | 2 | H | PNO | 20 | 02/2014 |
| 28590 | M | c.1285dup, p.His429Profs*27 | 47 | 1 | Ch | FF, lung cysts | 13 | 05/2013 |
| 31122 | F | c.1285dup, p.His429Profs*27 | 41 | Multifocal | CC | Lung cysts, PNO | 264 | 06/2005 |
| 36616 | M | c.1285del, p.His429Thrfs*39 | 40 | 1 | N/A | FF | 312 | 01/2008 |
| 37644 | M | c.958dup, p.Arg320Profs*70 | 41 | 2 | O | FF, PNO | 6 | 07/2008 |
| 37673 | M | c.610_611delinsTA, p.Ala204* | 44 | Multifocal | Ch | - | 9 | 07/2008 |
| 40154 | F | c.1528_1530del, p.Glu510del | 30 | Multifocal | O | - | N/A | 02/2009 |
| 40893 | M | c.323G > T, p.Ser108Ile | 51 | 1 | CC | FF, PNO | 45 | 03/2013 |
| 42207 | F | c.1300G > A, p.Glu434Lys | 54 | 1 | Ch | FF, PNO | 74 | 01/2014 |
| 42337 | M | c.1300G > A, p.Glu434Lys | 50 | 1 | H | FF, lung cysts, PNO | 60 | 02/2014 |
| 47950 | M | c.1458del, p.Ile486Metfs*5 | 44 | Multifocal | O | FF | 24 | 02/2014 |
| 47639 | F | c.57_58del, p.Phe20Leufs*16 | 44 | 1 | H | PNO | 43 | 05/2014 |
| 48746 | M | c.1318del, p.Glu440Argfs*28 | 54 | 1 | Ch | FF, lung cysts | 35 | 01/2014 |
| 48781 | F | c.1285dup, p.His429Profs*27 | 55 | 1 | N/A | FF, PNO | 80 | 02/2013 |
| 50335 | M | c.958dup, p.Arg320Profs*70 | 39 | Multifocal | O | PNO | 24 | 06/2014 |
All patients were alive at last follow-up, unless specified otherwise. CC, clear cell renal cell carcinoma. Ch, chromophobe renal cell carcinoma. D, deceased. FF, fibrofolliculomas, F-U, follow-up. H, hybrid chromophobe- oncocytoma renal cell carcinoma. Mt, metastatic disease. N/A, not available. O, oncocytoma. P, papillary renal cell carcinoma, type undetermined. PNO, pneumothorax. Und, undifferentiated.
Table 2.
Summary of clinical and histopathological characteristics of renal tumours among 33 patients with Birt-Hogg-Dubé
| Characteristics | Number of patients | |
|---|---|---|
| Gender | Male | 21 |
| Female | 12 | |
| Tumour number | 1 | 20 |
| 2 | 4 | |
| Multifocal | 9 | |
| Tumour histology | Oncocytoma/Chromophobe/Hybrid chromophobe- oncocytoma | 23 |
| Clear cell | 3 | |
| Papillary (type undetermined) | 1 | |
| Undifferentiated | 1 | |
| N/A | 5 | |
| Extra-renal manifestations § | Lung cysts/pneumothorax | 22 |
| Fibrofolliculomas | 19 | |
| None | 4 |
§twelve patients had both pulmonary and dermatological manifestations. RCC, renal call carcinoma.
Figure 1.
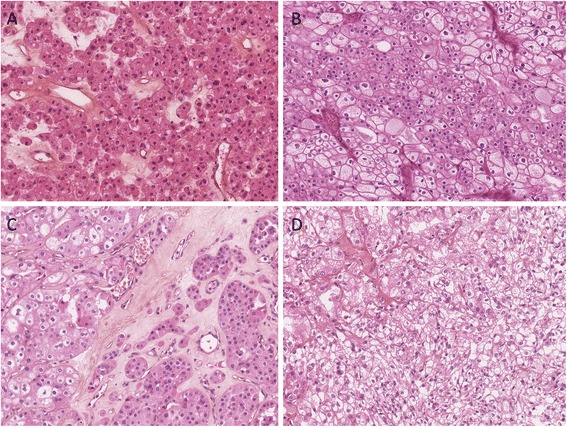
Main types of renal tumours seen in our patients with BHD. Oncocytoma (A), chromophobe carcinoma (B), hybrid chromophobe-oncytoma carcinoma (C), and clear cell carcinoma (D).
In addition to renal cell tumours, 29/33 cases also had documented extra-renal BHD manifestations. Twenty-two (67%) had lung cysts and/or pneumothoraces, and 19 (58%) had fibrofolliculomas (twelve cases had both pulmonary and dermatological manifestations).
There were 21 distinct mutations: frameshift (n = 10), missense (n = 4), nonsense (n = 4), splice site (n = 2) and in frame deletion (n = 1).
Among those 33 patients, four had metastatic RCC, either at the time of diagnosis or subsequently. All presented with symptoms that led to the diagnosis of RCC. Case 13851 had RCC age 35, he had undergone successful nephrectomy at the time. A solitary lung lesion was found incidentally age 58 on a CT scan that was prescribed after an episode of malignant hypertension. It was surgically removed and pathological examination showed it was a metastasis from a poorly differentiated RCC. The patient died of ischemic stroke four years after surgical resection of the metastasis, and there was no evidence of cancer relapse at the time. Case 19398 was diagnosed age 42 with bilateral multifocal hybrid chromophobe- oncocytoma RCC and liver metastases. Biopsy of a left renal tumour led to the pathological diagnosis. No further surgical procedure was carried out. For the first three years, she was under systemic therapy with sunitinib, everolimus and then temsirolimus, and disease stability was achieved. All treatments were then stopped. Eight years after the initial diagnosis, she is clinically well and the liver metastases are radiologically stable. Case 21310 had left nephrectomy age 46 for chromophobe RCC, and subsequently presented with liver and lung metastases. She is still alive five years after the metastases were first observed, even though slow radiological and clinical progression has justified treatment with sunitinib, axitinib, everolimus, bevacizumab and pazopanib. Finally, case 22973 had right nephrectomy age 56 for a chromophobe RCC. He received adjuvant radiotherapy for multiple, metastatic retroperitoneal lymph-nodes that could not be removed surgically. Seven years later, there are no radiological or clinical signs of disease progression.
Discussion
Our series of 33 cases is one of the largest ever published [4-8]. This study confirms that cases can have either unifocal disease, or multiple tumours, and that most renal cell tumours are oncocytomas or RCC of chromophobe or hybrid chromophobe-oncocytoma histology [4-6]. Other histologies are however associated with the syndrome, clear cell in particular. Indeed, among our cases, 3 (9%) had clear cell carcinomas. Given that they are not typical in BHD, all clear cell RCC were reviewed by one of our expert pathologists, and confirmation was obtained. This proportion is similar to what has been reported in the past, and clinicians should therefore remain alert to the possibility of BHD in patients with clear cell RCC, especially if there are associated manifestations [6].
The age at renal cell tumour diagnosis varied between 20 and 83, with a median age of 46. Of special interest was the fact that two patients presented with a renal cell tumour in their twenties, highlighting the need for regular surveillance from an early age [12]. This surveillance should be continued with no upper age limit and as long as the patients are in good condition, as one patient with two tumours in his eighties was successfully treated by percutaneous radiofrequency ablation.
These 33 cases with renal cell tumours represent 27% of all ascertained BHD patients (33/124), and 34% of those for which we have evidence of renal imaging (33/98). This observation is remarkably consistent with two large studies from the United States National Cancer Institute that reported overall tumour frequencies of 29% and 34% in BHD patients evaluated by CT scan [4,5]. We expect the proportion in our study to be representative of the true renal cell tumour risk associated with the syndrome, as French index cases are offered FLCN genetic analysis because they present with all types of clinical manifestations suggestive of BHD.
An exhaustive description of extrarenal manifestations is beyond the scope of this paper. Nevertheless, 67% and 58% of our cases also had pulmonary and dermatological manifestations respectively, in addition to RCC. These figures represent the minimal prevalence as pulmonary assessment by CT scan and examination by a dermatologist knowledgeable in the syndrome was not systematic.
In our series, four patients had metastatic, albeit indolent RCC. Comprehensive family data was collected for all patients in this series, and we have no knowledge of other relatives (deceased or alive) with metastatic RCC who were not tested for FLCN mutations, and who were therefore not included in this study. Very few cases of metastatic RCC associated with BHD have been described. Pavlovich et al. and Houweling et al. reported respectively two and five such cases; all died rapidly despite multidisciplinary management that included surgery, radiotherapy, immunotherapy and chemotherapy, and the good prognosis associated with metastatic disease in our study contrasts with these earlier reports [8,13]. Three of the four patients reported here are still alive more than five years after the metastases were diagnosed, while the fourth died of unrelated causes four years after a lung metastasis had been removed. Whether these observations reflect a specific behaviour only seen in BHD requires further investigation. Another hypothesis is that metastatic RCC in BHD patients is more sensitive to tyrosine kinase and mTOR inhibitors such as sunitinib and everolimus (two recently-developed drugs that are now routinely used in the treatment of metastatic RCC), as suggested for example by the mTOR signalling pathway activation seen in kidney tumours associated with the syndrome, but that would not account for the good prognosis of cases 13851 and 22973 who received no systemic treatment [14]. As shown in Table 1, most patients in our study are followed up medically, with relevant information being collected by the team in charge on a regular basis. There is however a minority from which we have not heard in the last few years, and we cannot rule out with certainty the possibility of aggressive metastatic disease in these patients.
Conclusions
The eponym syndrome was first described over 35 years ago by Birt, Hogg and Dubé [15]. However, the FLCN gene was only identified in 2002 [16]. The recent molecular characterisation of the syndrome, added to the fact that like most genetic diseases BHD is rare in the general population, means there are only limited data on the topic in the literature. As a result, this large and multicentric study will improve knowledge of the syndrome among geneticists, urologists and oncologists involved in the assessment and management of patients with a suspected or established genetic predisposition to renal cell tumours. It is of utmost importance to identify individuals with a high probability of developing renal cell tumours, so that they, and their at-risk relatives can benefit from personalised surveillance and management.
Acknowledgments
This project was funded by the Ligue nationale contre le Cancer (Comités de l’Indre et du Cher), the Myrovlytis Trust (BHD Foundation) and the Fondation Gustave Roussy.
Kamal Achkar, Pascaline Berthet, Olivier Binet, Virginie Bubien, Nacera Brahimi, Olivier Caron, Bertrand Collin, Marie-Agnès Collonge-Rame, Isabelle Coupier, Albert David, Benedicte Demuynck, Yves Denoux, Catherine Dugast, Bernard Escudier, Thierry Flam, Jean-Marie Ferrière, Jean Friedel, Brigitte Gilbert-Dussardier, Sophie Lejeune-Dumoulin, Michel Longy, Gwenael Nadeau, Nicolas Poulhalon, Olivier Rixe, Stanislas Ropert, Olivier Rouvière, Jean-Michel Salé, Ayhan Ulusakarya, Laurent Yonneau, Hélène Zattara are contributing collaborators. They are part of the French National Cancer Institute «Inherited predisposition to Kidney Cancer» network and should be acknowledged as such in the author list.
We are also grateful to Hélène Chomarat (Nantes), Jean-François Emile (Boulogne-Billancourt), Françoise Galateau-Salle (Caen), Catherine Mazerolles (Toulouse), and Pierre Validire (Paris) who sent us paraffin embedded tissue upon request for pathological review.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PRB, SD, AM, JMC, DJ, MOT, CA, DC, DL, MFA, JFC and SR were involved in the multidisciplinary assessement and clinical management of patients. SG performed FLCN genetic analyses. SF and VV were the pathologists of the study. PRB collected and analysed the data, and wrote the manuscript with support from JFC, SR and JMC. All authors read and approved the final manuscript.
Contributor Information
Patrick R Benusiglio, Email: pbenusiglio.wk@gmail.com.
Sophie Giraud, Email: sophie.giraud@chu-lyon.fr.
Sophie Deveaux, Email: sophie.deveaux@bct.aphp.fr.
Arnaud Méjean, Email: arnaud.mejean@egp.aphp.fr.
Jean-Michel Correas, Email: jean-michel.correas@nck.aphp.fr.
Dominique Joly, Email: doume.joly@gmail.com.
Marc-Olivier Timsit, Email: marc-olivier.timsit@egp.aphp.fr.
Sophie Ferlicot, Email: sophie.ferlicot@bct.aphp.fr.
Virginie Verkarre, Email: virginie.verkarre@nck.aphp.fr.
Caroline Abadie, Email: caroline.abadie@chu-rennes.fr.
Dominique Chauveau, Email: chauveau.d@chu-toulouse.fr.
Dominique Leroux, Email: dleroux@chu-grenoble.fr.
Marie-Françoise Avril, Email: marie-francoise.avril@cch.aphp.fr.
Jean-François Cordier, Email: jean-francois.cordier@chu-lyon.fr.
Stéphane Richard, Email: stephane.richard@u-psud.fr.
References
- 1.Menko FH, van Steensel MA, Giraud S, et al. Birt-Hogg-Dubé syndrome: diagnosis and management. Lancet Oncol. 2009;10(12):1199–1206. doi: 10.1016/S1470-2045(09)70188-3. [DOI] [PubMed] [Google Scholar]
- 2.Hartman TR, Nicolas E, Klein-Szanto A, et al. The role of the Birt-Hogg-Dubé protein in mTOR activation and renal tumorigenesis. Oncogene. 2009;28(13):1594–1604. doi: 10.1038/onc.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hasumi H, Baba M, Hasumi Y, et al. Regulation of mitochondrial oxidative metabolism by tumor suppressor FLCN. J Natl Cancer Inst. 2012;104(22):1750–1764. doi: 10.1093/jnci/djs418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidt LS, Nickerson ML, Warren MB, et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet. 2005;76(6):1023–1033. doi: 10.1086/430842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toro JR, Wei MH, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45(6):321–331. doi: 10.1136/jmg.2007.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pavlovich CP, Walther MM, Eyler RA, et al. Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol. 2002;26(12):1542–1552. doi: 10.1097/00000478-200212000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Kunogi M, Kurihara M, Ikegami TS, et al. Clinical and genetic spectrum of Birt-Hogg-Dube syndrome patients in whom pneumothorax and/or multiple lung cysts are the presenting feature. J Med Genet. 2010;47(4):281–287. doi: 10.1136/jmg.2009.070565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houweling AC, Gijezen LM, Jonker MA, et al. Renal cancer and pneumothorax risk in Birt-Hogg-Dubé syndrome; an analysis of 115 FLCN mutation carriers from 35 BHD families. Br J Cancer. 2011;105(12):1912–1919. doi: 10.1038/bjc.2011.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maher ER. Genetics of familial renal cancers. Nephron Exp Nephrol. 2011;118(1):e21–e26. doi: 10.1159/000320892. [DOI] [PubMed] [Google Scholar]
- 10.Khoo SK, Giraud S, Kahnoski K, et al. Clinical and genetic studies of Birt-Hogg-Dubé syndrome. J Med Genet. 2002;39(12):906–912. doi: 10.1136/jmg.39.12.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kluger N, Giraud S, Coupier I, et al. Birt-Hogg-Dubé syndrome: clinical and genetic studies of 10 French families. Br J Dermatol. 2010;162(3):527–537. doi: 10.1111/j.1365-2133.2009.09517.x. [DOI] [PubMed] [Google Scholar]
- 12.Stamatakis L, Metwalli AR, Middelton LA, et al. Diagnosis and management of BHD-associated kidney cancer. Fam Cancer. 2013;12(3):397–402. doi: 10.1007/s10689-013-9657-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pavlovich CP, Grubb RL, III, Hurley K, et al. Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol. 2005;173(5):1482–1486. doi: 10.1097/01.ju.0000154629.45832.30. [DOI] [PubMed] [Google Scholar]
- 14.Hasumi Y, Baba M, Ajima R, et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci U S A. 2009;106(44):18722–18727. doi: 10.1073/pnas.0908853106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1987;113(12):1674–1677. doi: 10.1001/archderm.1977.01640120042005. [DOI] [PubMed] [Google Scholar]
- 16.Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2(2):157–164. doi: 10.1016/S1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]