

Abstract
Arsenic as a potential risk factor for type 2 diabetes has been received attention recently. However, the roles of arsenic on development of diabetes are unclear. In this study, we compared the influences of inorganic arsenic (iAs) on normal and diabetic mice by systems toxicology approaches. Although iAs exposure did not change glucose tolerance in normal mice, it caused the pancreatic β-cell dysfunction and increased gluconeogenesis and oxidative damages in liver. However, iAs exposure worsened the glucose tolerance in diabetic mice, which might be due to increased gluconeogenesis and impairment of pancreatic β-cell function. It is interesting that iAs exposure could improve the insulin sensitivity based on the insulin tolerance testing by the activation of glucose uptake-related genes and enzymes in normal and diabetic individuals. Our data suggested that iAs exposure could cause pre-diabetic effects by altering the lipid metabolism, gluconeogenesis and insulin secretion in normal individual, and worsen diabetic effects in diabetes individual by these processes. Insulin resistance might be not the reason of diabetic effects caused by iAs, indicating that mechanism of the diabetogenic effects of iAs exposure is different from the mechanism associated with traditional risk factors (such as obesity)-reduced type 2 diabetes.
Diabetes mellitus characterized by hyperglycemia is an increasing worldwide health problem1. It is largely classified into insulin-dependent diabetes mellitus (type 1 diabetes) and noninsulin-dependent diabetes mellitus (type 2 diabetes). The type 2 diabetes (T2D) makes up more than 90% of all diabetes cases2. Established risk factors for T2D, such as genetics, diet and lifestyle, do not fully explain the increase in this disease. There is considerable interest in understanding the contribution of nontraditional risk factors to the diabetes epidemic, including environmental pollutants3,4. Among these environmental pollutants, arsenic (As) exposure has been paid much attention5. As is a ubiquitous toxic metalloid in the environment. Epidemiological studies carried out in Bangladesh6, Taiwan7, Mexico8, and United States9 have shown a strong diabetogenic effect of As in human populations mainly through As-contaminated drinking water. Although some other researches concluded that the existing data were insufficient in support of an association between As and T2D10, data from human studies support an association between As and diabetes in population with arsenic drinking-water levels of >500 µg/L3.
The T2D is characterized by disruptions in whole-body glucose homeostasis due to insulin resistance and impaired β-cell dysfunction11. Individuals with β-cell unable to sustain increases insulin secretion to compensate for insulin resistance will develop T2D, and thus inadequate β-cell function is essential to the course of T2D12,13. In vitro studies using insulinoma cell lines implicate several pathways by which inorganic arsenic (iAs) can affect pancreatic β-cell function to inhibit insulin expression and/or secretion14,15. Fu et al.16 found that iAs exposure dampened reactive oxygen species (ROS) signaling involved in glucose-stimulated insulin secretion, and disturbed β-cell function. However, in vitro researches did not consider physiologically relevance. In animal studies, glucose levels, insulin regulation, insulin sensitivity and pancreatic effects were often the reported endpoint17,18. iAs and/or its methylated trivalent metabolites might cause insulin resistance by interacting with a number of elements involved in insulin signaling, including insulin receptor substrate (IRS), peroxisome proliferator-activated receptor-γ (PPAR-γ), protein kinase C (PKC) and phosphatidylinositol-3 kinase (PI3K)3,19. Although recent results supported a link between iAs exposure and diabetes, the findings from animal studies were inconsistent. The pre-diabetic effects caused by iAs and its mechanism of actions in normal individual are unclear. Moreover, the influence of iAs on the development of diabetes in diabetic individuals also need to be specifically evaluated. The comparison of diabetic effects between normal and diabetic individuals might provide new insights to the mechanism of actions of iAs.
In this study, we compared the influences of iAs on normal C57BLKS/J (db/m) mice and diabetic C57BKS/Leprdb (db/db) mice. The mice were exposed to iAs or deionized water for 16 weeks. The diabetes-related endpoints, including blood glucose levels, glucose tolerance, and insulin tolerance were analyzed. Oxidative stress and damage, enzyme activities, gene expression profiles, and metabolic profiles in mice were determined. The gluconeogenesis, lipid metabolism, insulin resistance and function of β-cell were also analyzed. Based on above information, the roles of As on diabetes development were characterized.
Results
Food Intake and body weight
iAs exposure decreased food intake of db/m mice, but increased the food intake of db/db mice (Figure 1A). After 16-week experiment period, all experimental animals gained weight, and the increases of diabetic mice were higher than normal mice. iAs exposure did not change the body weight of both types of mice (Figure 1B). However, it slightly decreased the relative liver weight, and slightly increased the relative pancreas weight in both type of mice (Figures 1C and 1D).
Figure 1. Influences of arsenic on food intake (A), body weight (B), relative liver weight (C) and relative pancreas weight (D).

CK means control mice. DB means diabetic mice. Values are mean values ± standard deviation (n = 7–8). Differences of results among groups were calculated using ANOVA followed by Tukey's post hoc test. * means significant difference compared to CK group or between special groups (p < 0.05).
Arsenic intake, methylation and excretion
During the exposure period, db/db mice consumed more water than normal mice. iAs exposure did not significantly change the water intake in both types of mice (Figure 2A). Based on the average daily water intakes, mice in CK+As and DB+As groups ingested 13.35 and 30.76 μg iAs/day, respectively. However the concentrations of As in urine and feces in mice of DB+As group was lower than those in CK+As group, indicating that diabetic individual might increase the accumulation of arsenic (Figures 2B and 2C). For As species, in urine, the percentage of dimethyl-As (DMA) in DB+As group was higher than that in CK+As group, indicating the higher methylation in diabetic mice. However, in feces, the percentage of DMA in DB+As group was lower than CK+As group, which might indicate the potential different influences of gut microbiota.
Figure 2. Arsenic intake and excretion by mice exposed to deionized water with or without 3 mg/L sodium arsenite.
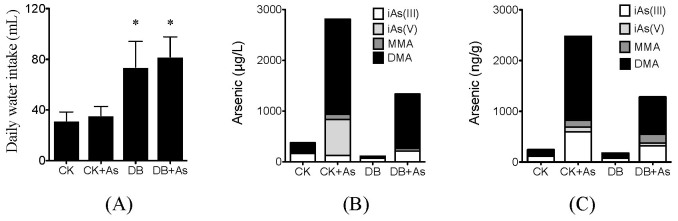
(A) Daily water intake; (B) Arsenic species in urine; (C) Arsenic species in feces. Four As species, including iAs (III), iAs (V), methyl-As (MMA) and dimethyl-As (DMA) were measured by HPLC-ICP-MS. CK means control mice. DB means diabetic mice. Values are mean values ± standard deviation (n = 7–8). Differences of results among groups were calculated using ANOVA followed by Tukey's post hoc test. * means significant difference (p < 0.05) compared to control mice.
Arsenic decreases glucose tolerance but increases insulin sensitivity
The fasting blood glucose (FBG) and fasting blood insulin (FBI) in db/db control mice were significantly higher than those in db/m control mice (Figures 3A and 3B). iAs exposure increased the FBG levels in db/db mice. However, it significantly increased the levels of FBI in db/m mice, and decreased its levels in db/db mice. Moreover, the FBG and FSI concentrations were further employed to calculate homeostasis model assessment-insulin resistance (HOMA-IR) and basic insulin secretion function index (HOMA-%β). The average HOMA-IR value in db/db control mice was higher than that in db/m control mice (Figure 3C). iAs exposure did not change HOMA-IR in both types of mice. Compared to db/m control mice, the value of HOMA-%β was significantly reduced in db/db control mice (Figure 3D). iAs exposure significantly increased the HOMA-%β values in db/m mice, but decreased its values in db/db mice.
Figure 3. Effects of arsenic exposure on blood glucose and insulin tolerance.

(A) Fasting blood glucose; (B) Fasting blood insulin; (C) HOMA-IR; (D) HOMA-%β; (E) and (F) OGTT; (G) OGTT AUC, calculated according to OGTT; (H) and (I) ITT; (J) ITT AUC, calculated according to ITT. CK means control mice. DB means diabetic mice. Values are mean values ± standard deviation (n = 7–8). Differences of results among groups were calculated using ANOVA followed by Tukey's post hoc test. In general, * means significant difference (p<0.05) compared to db/m control mice or between special groups. In (F) and (I), * means significant difference (p<0.05) compared to db/db control mice at the same time.
For db/m mice with or without iAs exposure, oral glucose tolerance test (OGTT) showed the characteristic rapid rise in blood glucose, peaking within 30 min of glucose challenge (Figure 3E). The peak was followed by a gradual decrease, indicating the uptake of glucose by liver and peripheral tissues, and blood glucose levels approached baseline levels by 120 min after glucose challenge. However, for db/db control mice, after 120 min, the blood glucose remained elevated compared to FBG (Figure 3F). iAs further reduced the glucose tolerance in db/db mice. To quantify glucose tolerance, we calculated the area under curve (AUC) for all treatment groups. The average AUC value was higher for db/db control mice compared to db/m control mice (Figure 3G). iAs exposure further increased the AUC in db/db mice, indicating the deteriorate of glucose tolerance. In the insulin tolerance test (ITT), insulin sensitivity was markedly worsened in db/db control mice than in db/m control mice (Figures 3H and 3I). iAs treatment significantly improved the insulin-induced reduction of blood glucose levels at 30 min in db/db mice. The ITT-AUC for db/db mice exposed to iAs was significantly lower than db/db control mice.
Arsenic increases inflammation and oxidative damage in diabetic mice
iAs exposure caused significant changes in expression of oxidative stress and inflammation-related genes catalase, IL-1β, Jnk2, and Mt1 in liver and pancreas of db/m mice, but decreased expression of genes IL-1β, Jnk2 in adipose (Figures 4A–4C). Most of these genes in db/db mice were also up-regulated in liver, adipose and pancreas. iAs exposure further increased their abnormal expression. We further chose liver as target organ to confirm the oxidative stress and damage based on pathological and biochemical analyses. Pathological analysis showed that iAs exposure caused inflammation of hepatocyte of db/m control mice (Figure 4D). In diabetic mice, besides inflammation, diffuse fatty degeneration and fat drop were observed in hepatocyte. iAs exposure worsened the impair of hepatocyte of diabetic mice, and cellular edema was also found. Based on biochemical analysis, the db/db control mice showed higher SOD activity and MDA level than db/m mice (Figures 4 E and 4F). But no significant changes in GSH and 8-OHdG levels were found between db/m control and db/db control mice (Figures 4 G and 4H). iAs increased the SOD activities, MDA levels and 8-OHdG levels in both types of mice, and higher levels were found in db/db mice exposed to iAs. For GSH, iAs decreased its activity in db/m mice, but increased its activity in db/db mice.
Figure 4. Effects of arsenic on oxidative stress and inflammation.

(A) Gene expressions in liver; (B) Gene expressions in adipose; (C) Gene expressions in pancreas; (D) pathological maps of liver; (E) SOD activities; (F) MDA levels; (G) GSH levels and (H) 8-OHdG levels. SOD, MDA, GSH and 8-OHdG were measured by ELISA assays. CK means control mice. DB means diabetic mice. Values are mean values ± standard deviation (n = 7–8). Differences of results among groups were calculated using ANOVA followed by Tukey's post hoc test. * means significant difference (p<0.05) compared to db/m control mice.
Arsenic changes lipid metabolism in liver and adipose
iAs exposure decreased expression of lipid metabolism-related genes in liver of db/m mice, including CD36, Hmgcr, Fasn, Abca1 and Pgc, but increased expression of genes PPARr-2, Pck1, Ccnd1 and Gpihbp1 (Figure 5A). In db/db control mice, most of these genes in liver were up-regulated. iAs exposure further enhanced the up-regulation of these genes. For adipose, iAs exposure increased expression of genes CD36, Fasn, PPARr-2 and Pck1 in db/m mice, and decreased the expression of genes Hmgcr, Abca1, Pgc, and Gpihbp1 (Figure 5B). In db/db control mice, genes CD36, Fasn, PPARr-2 and Abca1 were up-regulated. But iAs exposure did not significantly change their expression in adipose of db/db mice. The activities of Na+K+-ATP, Ca2+Mg2+-ATP and levels of PPAR-r and free fatty acid (FFA) in liver were further analyzed by ELISA methods (Figures 5C–5F). For the four parameters, the db/db control mice showed higher levels than db/m control mice. iAs exposure increased hepatic PPAR-r level in db/m mice and FFA level in db/db mice, but did not increase PPAR-r levels in db/db mice and ATP activities in both type of mice.
Figure 5. Effects of arsenic on lipid metabolism.

(A) Gene expressions in liver; (B) Gene expressions in adipose; (C) PPAR-r levels; (D) Na+K+-ATP activities; (E) Ca2+Mg2+-ATP activities; and (F) free fatty acid (FFA) levels. PPAR-r, Na+K+-ATP, Ca2+Mg2+-ATP and FFA were measured by ELISA assays. CK means control mice. DB means diabetic mice. Values are mean values ± standard deviation (n = 7–8). Differences of results among groups were calculated using ANOVA followed by Tukey's post hoc test. * means significant difference (p<0.05) compared to db/m control mice.
Arsenic changes glucose metabolism and pancreatic function
Glucose metabolism-related genes, including Adipoq, Glut4 and Irs1 were analyzed. iAs exposure increased expression of the three genes in liver of db/m mice, and decreased expression of Adipoq and Glut4 in adipose (Figure 6A). In db/db mice, the three genes were down-regulated in liver and adipose. But, iAs exposure up-regulated their expressions in db/db mice. The levels of protein tyrosine phosphatase-1B (Ptp1b) and leptin in liver exert important effects on the insulin resistance. The Ptp1b level were increased in db/db mice (Figure 6B). iAs exposure decreased the Ptp1b levels in both types of mice. For leptin, because of the type of db/db mice, its levels in db/db control mice were not influenced (Figure 6C), but iAs exposure increased leptin levels, and significant increases were found in DB+As group (p<0.05).
Figure 6. Effects of arsenic on (A) glucose-related genes, (B) hepatic Ptp1b levels, (C) hepatic leptin levels and (D) impair of pancreas.

Ptp1b and leptin were measured by ELISA assays. CK means control mice. DB means diabetic mice. Values are mean values ± standard deviation (n = 7–8). Differences of results among groups were calculated using ANOVA followed by Tukey's post hoc test. * means significant difference (p < 0.05) compared to db/m control mice or between special groups.
In pancreas, pathological analysis showed that iAs exposure caused vacuolus and inflammation in db/m mice (Figure 6D). In diabetic mice, the structure of pancreas became irregularly arranged, and inflammation and hyperemia edema were also found. iAs exposure worsened the impair of pancreas in diabetic mice. For gene expression, iAs exposure decreased expression of genes Irs1 and Glut4, but increased the Adipoq expression in db/m mice. Similar changes were found in db/db control mice. iAs exposure enhanced the changes in expression of genes Glut4 and Irs1 in db/db mice.
Metabolic profiling of serum by 1H NMR spectrocopy
In order to better characterize variations of serum metabolic profiles, a partial least-squares discriminant analysis (PLS-DA) model was carried out on all individuals. The metabolic profiles in db/db mice exposed to iAs had higher similarity with those in db/db control mice, which were different with those in db/m mice with or without iAs exposure (Figure 7). Altered metabolites in treatment groups were identified following the criteria that p < 0.05 and fold change ≥ ± 1.2 (Table 1). A total of 6, 17, and 18 altered metabolites in CK+As, DB and DB+As groups were found, respectively. iAs exposure did not cause many changes in serum metabolic profiles in db/m mice, which are consistent with our previous results20. As expected, db/db mice with or without As exposure showed many altered diabetes-related metabolites such as branched-chain and aromatic amino acids (BCAA) including leucine, tyrosine and phenylalanine. Many glucose metabolism-related pathways, including TCA cycle, glycolysis, and propanoate metabolism were also significantly altered in db/db mice. But iAs exposure did not significantly alter the serum metabolites in db/db mice.
Figure 7. PLS-DA plot of 1H NMR spectra of serum metabolites from control (green), control+As (red), diabetes control (cyan) and diabetes+As (blue) groups.

PLS-DA was performed by MetaboAnalyst 2.0 (http://www.metaboanalyst.ca/MetaboAnalyst).
Table 1. Altered metabolites and biological pathways in different treatment groups. Altered metabolites were identified based on the criteria that p<0.05 and fold change ≥±1.2.
| Metabolites | CK+As | DB | DB+As | KEGG Pathway information |
|---|---|---|---|---|
| Amino acid metabolism | ||||
| Phenylalanine | 1.26 | 1.20 | 1.22 | Phenylalanine, tyrosine and tryptophan biosynthesis |
| Tyrosine | 1.49 | 1.32 | 1.36 | |
| Arginine | 0.74 | 0.70 | Arginine and proline metabolism | |
| Creatine | 0.76 | 0.77 | ||
| Leucine | 0.75 | 0.73 | Valine, leucine and isoleucine biosynthesis | |
| Methionine | 0.72 | 0.68 | Cysteine and methionine metabolism | |
| Energy metabolism | ||||
| Glutamine | 0.74 | 0.70 | Nitrogen metabolism | |
| Histidine | 1.30 | 1.31 | 1.47 | |
| Formate | 1.44 | 1.70 | Methane metabolism | |
| Lipid metabolism | ||||
| Taurine | 1.83 | 2.15 | Primary bile acid biosynthesis | |
| Carbohydrate metabolism | ||||
| Ascorbate | 1.85 | 1.83 | Ascorbate and aldarate metabolism | |
| Fumarate | 1.32 | 1.74 | 1.77 | Citrate cycle (TCA cycle) |
| Pyruvate | 0.71 | 0.64 | ||
| Glucose | 2.35 | 2.65 | Glycolysis | |
| Lactate | 0.68 | 0.65 | ||
| p-hydroxybutyrate | 1.34 | 1.20 | Propanoate metabolism | |
| Acetone | 0.73 | 0.65 | ||
| Nucleotide metabolism | ||||
| UTP | 1.41 | 1.54 | 1.83 | Pyrimidine metabolism |
Discussion
Arsenic as a nontraditional risk factor to the T2D has been given more and more attentions. Although there are many epidemiological researches between iAs and diabetes, the roles of iAs in development of diabetes are still unclear. More experimental data at environmentally relevant concentrations are needed. In this study, normal and diabetic mice were chosen as model animal to address the roles of iAs. Previous studies showed that mice might be less susceptible than human to arsenic toxicity, partly due to a faster metabolism and clearance of arsenic21. Therefore, it is necessary to use higher exposure concentration of iAs than the environmentally relevant concentrations in mouse experiment. In drinking source water, 1.5 mg/L iAs has been found22. So 3 mg/L was chosen as exposure levels of iAs in this study.
The db/db mice showed higher As intake and lower As excretion. Similar with previous literatures23, a high degree of methylation of As in urine was found in urine of both types of mice, but higher degree methylation of As in db/db mice was found. On the other side, iAs influenced the daily food intake of mice, and caused sight changes of relative liver and pancreas weight. These results showed that diabetic individuals have different excretion and metabolism of iAs, and iAs might actively contribute to diabetes development and progression. Therefore in-depth investigation of the effect of iAs on the process leading to development of diabetes is required.
The changes in glucose homeostasis, reduced insulin sensitivity and β-cell function are the core pathophysiological defects in T2D24. As expected, the db/db control mice had lower glucose tolerance than db/m control mice. iAs exposure slightly increased the glucose tolerance in db/m mice, but significantly worsened the glucose tolerance in db/db mice, indicating that iAs exposure could pose higher risk for diabetic individuals and need to be paid more attention. For insulin sensitivity, according to the calculation of HOMA-IR, iAs exposure did not change the insulin resistance. However, the ITT test showed that iAs exposure could improve the response of injected insulin in both types of mice, especially for db/db mice. The results suggested that iAs exposure could not worsen insulin resistance, even improve the insulin sensitivity to some extent. As for another symbol of T2D, the β-cell function, iAs stimulated the insulin secretion in db/m mice, but decreased the insulin secretion in db/db mice. The results showed that iAs could influence the β-cell function. The calculated HOMA-%β supported the results that db/m mice exposed to iAs had higher HOMA-%β values than db/m control mice. The value in db/db control mice was significantly lower than that of db/m control mice, indicating the impairment of β-cell function. iAs exposure further impaired β-cell function in db/db mice. Based on above results, it seems that iAs could cause or worsen diabetic effects by impairment of β-function instead of change of insulin resistance.
Gene expression and metabolic profiles were further applied to characterize potential mechanism of actions of iAs on diabetic effects. We analyzed three types of gene expression in liver and adipocytes, involved in oxidative stress, lipid metabolism and glucose uptake. As known, iAs exposure up-regulated oxidative stress and inflammation-related genes, including Catalase, ikka, IL-1, Jnk2 and Mt1 in db/m mice. The enzyme test further identified the results. In addition, iAs also increased expressions of Ccnd1 in liver, indicating the cell proliferation (Figure 4). These results are consistent with previous researches25,26, indicating that iAs can increase the hepatic toxicity. In db/db control mice, these genes were also up-regulated in liver and adipocyte, indicating that diabetes can also cause oxidative damage and inflammation27,28. iAs exposure further increased oxidative damage and inflammation, and worsened hepatotoxicity in diabetic individual. In addition, it should be noted that iAs caused DNA damage based on 8-OHdG test in both types of mice, but the DNA damage was not found in db/db control mice, indicating the special toxicity caused by iAs29,30. But iAs caused higher 8-OHdG levels in db/m mice than db/db mice, which is different from the results of MDA levels. Since the MDA and 8-OHdG indicate the different endpoint of oxidative damage, we think the oxidative stress caused by iAs exposure in db/m and db/db mice could induce different oxidative damage, and the detail mechanism need to be further analyzed.
Some lipogenesis-related genes, including genes Fasn, PPARr-2, Hmgcr, CD36 and Abca1 were up-regulated in liver and adipocyte of db/db mice, indicating the increase of lipogenesis. The expression of PPAR-r was also verified by ELSIA test. iAs exposure further increased the expression of most genes in liver of db/db mice, but decreased them in adipocyte. It should be noted that iAs exposure decreased the expression of these genes in liver and adipocyte in db/m mice. These results suggested that iAs posed different influences on lipid metabolism of db/m and db/db mice. It is known that the lipogenesis is related to the gluconeogenesis31. We measured the expression of gene Pck1, which is related to gluconeogenesis32. Its overexpression could increase the gluconeogenesis in liver and blood glucose. We found that gene Pck1 was up-regulated in db/db mice. It is interesting that the gene had higher regulation in db/m and db/db mice exposed to iAs, indicating that iAs could increase the hepatic gluconeogenesis. The results could be identified by the ATP test and hepatic FFA level. The gluconeogenesis process needs energy. We found the increase of Na+K+-ATP, and Ca2+Mg2+-ATP in liver. It has also been proven that FFA delivery to liver could stimulate the expression of gene Pck1 and gluconeogenesis33,34. In db/m mice exposed to iAs, the gluconeogenesis did not increase blood glucose, which might be due to the increase of blood insulin. But, in db/db mice exposed to iAs, because of lack of blood insulin, the gluconeogenesis worsened the diabetic effects, which might be one of the reasons that caused higher FBG and lower glucose tolerance.
Besides gluconeogenesis, as is known, individuals with β-cell unable to secret enough insulin to compensate for insulin resistance will develop T2D. Thus inadequate β-cell function is essential to the course of the disease. Chronic oxidative stress leading to oxidative damage has long been implicated in β-cell dysfunction in diabetes35,36. Reactive oxygen species' signals produced during glucose metabolism are recognized as intracellular regulators of glucose-stimulated insulin secretion acting to increase insulin secretion. Our study found that the expression of genes related to oxidative stress and inflammation in pancreas of db/db mice were up-regulated. iAs exposure increased their expression in db/db mice, indicating higher oxidative damage and inflammation. Combined with the results of pathological analysis, FGB, FGI and HOMA-%β, the antioxidant and inflammatory responses appears to play paradoxical roles in β-cell function: in db/m mice exposed to iAs, influencing glucose-triggered ROS signaling and insulin secretion. In db/db mice exposed to iAs, the higher antioxidant and inflammatory responses than db/db control mice indicates the serious oxidative damage and subsequent potential apoptosis/necrosis, which might be verified by the significant up-regulation of gene Ccnd1. Therefore, the impairment of β-cell might be another reason of worse glucose tolerance in db/db mice exposed to iAs.
When insulin was injected, different results on blood glucose were found in different treatment groups. The results could be explained by the changes in insulin and glucose uptake-related gene expressions and enzyme activities. Db/db control mice had the lowest insulin resistance during insulin tolerance test. We found the genes Irs1, Glut4 and Adipoq were down-regulated in liver and adipocyte of db/db control mice, In addition, we found the significant increase of Ptp1b in liver. These results indicated the reduction of insulin signaling and increase of insulin resistance37,38,39,40. However, when iAs was exposed to db/m and db/db mice, above genes were up-regulated compared with db/m control or db/db control mice, and the levels of Ptp1b were inhibited, which increase the insulin sensitivity41.
Recent epidemiological studies have used metabolic profiles to predict incident diabetes and revealed some diabetes-related metabolites and metabolism pathways42,43. For metabolites, hydroxybutyrate and BCAA have shown a strong relationship with insulin resistance44,45. For metabolism pathway, several studies have demonstrated the final products of fatty acid degradation and glycolysis are included in TCA cycle, and the TCA cycle intermediates are involved in amino acid synthesis and degradation as well as gluconeogenesis46,47. In our study, we found that these metabolites and pathways have been altered in db/db control mice compared to db/m control mice, indicating the special metabolic profiles in diabetic individuals. However, iAs could not significantly change the metabolic profiles in db/m and db/db mice. The results indicates that effects of iAs on development of diabetes can not been effectively indicated by metabolic profiles.
Conclusions
The iAs exposure caused oxidative damage, DNA damage and inflammation in normal and diabetic individuals, indicating its toxicities. Based on the comparison of influences of iAs on db/m and db/db mice, iAs exposure did not change glucose tolerance in db/m mice, but caused the pancreatic β-cell dysfunction and increased gluconeogenesis. However, iAs exposure worsened the glucose tolerance in db/db mice, which might be due to increased gluconeogenesis and impairment of pancreatic β-cell function. Our data suggested that iAs exposure could cause pre-diabetic effects by altering the lipid metabolism, gluconeogenesis and insulin secretion in normal individual, and could worsen diabetic effects in diabetes individual by these processes. It is interesting that the iAs exposure improve the insulin sensitivity based on the ITT by the activation of glucose uptake-related genes and enzymes in normal and diabetic individuals, indicating mechanism of diabetogenic effects of iAs exposure is different from the mechanism associated with traditional risk factors (such as obesity)-reduced type 2 diabetes.
Methods
Mice
Seven-week-old male C57BKS/Leprdb (db/db) mice were purchased from Model Animal Research Center of Nanjing University. C57BLKS/J db/m mice were chosen as normal control mice. All mice were housed in stainless-steel cages under controlled conditions with 25 ± 3°C, 50 ± 5% humidity and 12/12 h light/dark cycle. Following acclimation for one week, 16 db/db mice were randomly assigned to two groups. One group was fed with deionized water, and another group with 3 mg/L sodium arsenite solution. In addition, 16 db/m mice were also randomly divided into two groups given deionized water with or without 3 mg/L sodium arsenite. The exposure duration was 16 weeks. Sodium arsenite was obtained from National Standard Material Center (China). The sodium arsenite solution was prepared every week to minimize oxidation of As (III) to As (V). Daily water consumption was measured in all exposure groups. Body weights were determined every two weeks. All experimental processes were in accordance with NIH Guide for the Care and Use of Laboratory Animals. And the protocol was approved by the Committee on the Ethics of Animal Experiments of the Nanjing Military General Hospital.
Glucose hemostasis analysis
After 16-week exposure of iAs, FBG and FBI were measured. The foods for mice were deprived for 12 h. Then blood samples (2–3 μL each) were collected from tails to measure FBG using a Glucose Monitoring System (Simcare, China). Additional samples of blood (100 μL) from tails were applied to measure FBI by the Mouse Insulin ELISA kit (Jiancheng, China). HOMA-IR and HOMA-%β were calculated by the following equations48:
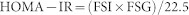 |
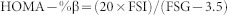 |
Where units of FSI and FSG were mU/L and mmol/L, respectively.
The OGTT and ITT were carried out according to previously described methods49. OGTT were performed after 12 h of food deprivation. Glucose was dissolved in diH2O and orally administered to the fasted mice (2 g/kg of body weight). Blood samples (2–3 μL each) were collected from tail to measure the glucose levels before and 15, 30, 60, 90, and 120 min after glucose administration. The ITT was conducted by intraperitoneal injection of 1.5 U/kg body weight insulin. Blood glucose was measured as described for the OGTT.
Speciation analysis of arsenic
Urine and feces samples were collected every two weeks and kept at −80°C. High performance liquid chromatography coupled with inductively coupled plasma-mass spectrometry (HPLC-ICP-MS) methods described by Van de Wiele et al50 were applied to detect and quantify iAs (III), iAs (V), MMA and DMA in urine and feces.
Pathological analysis
Parts of liver and pancreas were taken and fixed in 10% formalin solution. After 24–28 h they were dehydrated in a grade alcohol series and embedded in paraffin wax. Sections of 4–5 μm thickness were stained with hematoxylin-eosin and taken photos by microscope for pathological analysis.
Hepatic toxicity test
Hepatic oxidative stress and damage was determined by measurement of SOD, GSH, MDA, and 8-OHdG. For SOD, GSH, and MDA, part of liver was homogenized, and supernatants were used for various estimations. The protein contents, SOD, GSH, and MDA were measured using ELISA kits (Jiancheng, China). For 8-OHdG, genomic DNA of mouse livers were isolated by Genomic DNA Mini Preparation Kit (Beyotime, China), and the 8-OHdG levels were determined using a mouse 8-OHdG ELISA kit (Jiancheng, China).
The levels of hepatic PPAR-γ, FFA, Ppt1b, leptin, Na+K+-ATP, and Ca2+Mg2+-ATP were also determined by ELISA kits (Jiancheng, China). Each experiment was performed in triplicate.
Quantitative real time PCR (qPCR)
Total RNA in liver, adipocytes and pancreas were prepared using the Takara RNA Kit (Takara Bio. Japan). A total of 16 genes related to oxidative stress, inflammation, lipid metabolism, and glucose hemostasis were chosen. We performed qPCR analysis on an ABI7500 Real-Time PCR system using SYBR Select Master Mix qPCR reagent kit. The cycling parameters were 50°C for 2 min, 95°C for 2 min, followed by 45 cycles of 95°C for 15 sec, 58–60°C (slight difference for different genes) for 60 sec. Threshold cycles (Ct) and dissociation curves were determined with 7500 Software v2.0.6, and gene expression levels were normalized to gene β-actin. Standard curves and primer efficiencies were determined for all genes analyzed by qPCR.
Metabolomics analysis
Serum samples were applied to identify changes in metabolic profiles in mice. Serum samples were centrifuged at 3000 rpm for 10 min. Then, 75 μL phosphate buffer (pH 7.4) and 75 μL D2O containing 0.1% NaN3 and 0.05% TSP were added to 350 μL serum. The prepared serum samples were manually transferred into Bruker 1.7 mm NMR tubes. The 1H NMR spectra of metabolites were collected on a Bruker 600 MHz spectrometer (Bruker, Germany) using a cold probe. Fourier transformed 1H NMR spectra were manually phased and baseline was corrected using MestReC software (MestreC Research, Spain). Each spectrum was segmented into 0.005-ppm bins. A total of 44 metabolites were extracted and analyzed. Water resonance (5.0–4.5 ppm) was removed from data sets prior to normalization. Data analyses of spectra were performed on MetaboAnalyst 2.0 (http://www.metaboanalyst.ca/MetaboAnalyst), which provides PLS-DA and KEGG pathway analysis used in this study51.
Statistical analysis
Differences of results among groups were calculated using ANOVA test followed by Tukey's post hoc test. All analyses were performed by Graphpad Prism 5.
Author Contributions
S.L., X.C.G. and H.Y.Y. researched data. X.X.Z. contributed to discussion and reviewed/edited manuscript. M.L. and B.W. wrote the manuscript and researched data. All authors reviewed the paper.
Acknowledgments
The authors thank Dr. Shifeng Yun (Nanjing General Hospital of Nanjing Military Command) for technical assistance with animal care, monitoring, and data collection, Dr. Gary Cherr (University of California, Davis) for helpful advice regarding the data analyses. We also thank Dr. Jiamei Li (Nanjing University) for her assistance in the preparation of the manuscript. This work was supported by Natural Science Foundation of Jiangsu Province (BK20131270), Foundation of State Key Laboratory of Pollution Control and Resource Reuse, and Science Foundation of Nanjing University.
References
- Inceoglu B. et al. Acute augmentation of epoxygenated fatty acid levels rapidly reduces pain-related behavior in a rat model of type I diabetes. Proc Natl Acad Sci U S A 109, 11390–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmet P., Alberti K. G. & Shaw J. Global and societal implications of the diabetes epidemic. Nature 414, 782–7 (2001). [DOI] [PubMed] [Google Scholar]
- Maull E. A. et al. Evaluation of the association between arsenic and diabetes: A national toxicology program workshop review. Environ Health Perspect 120, 1658–1670 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hectors T. L. M. et al. Environmental pollutants and type 2 diabetes: a review of mechanisms that can disrupt beta cell function. Diabetologia 54, 1273–1290 (2011). [DOI] [PubMed] [Google Scholar]
- Navas-Acien A. et al. Arsenic exposure and type 2 diabetes: A systematic review of the experimental and epidemiologic evidence. Environ Health Perspect 114, 641–648 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M. R. et al. Association between type 2 diabetes and chronic arsenic exposure in drinking water: A cross sectional study in Bangladesh. Environ Health 11, 38 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C. H. et al. Long-term arsenic exposure and incidence of non-insulin-dependent diabetes mellitus: A cohort study in arseniasis-hyperendemic villages in Taiwan. Environ Health Perspect 108, 847–851 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Razo L. M. et al. Exposure to arsenic in drinking water is associated with increased prevalence of diabetes: a cross-sectional study in the Zimapan and Lagunera regions in Mexico. Environ Health 10, 73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meliker J. R., Wahl R. L., Cameron L. L. & Nriagu J. O. Arsenic in drinking water and cerebrovascular disease, diabetes mellitus, and kidney disease in Michigan: a standardized mortality ratio analysis. Environ Health 6, 4 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. F. et al. Arsenic and diabetes: current perspectives. Kaohsiung J Med Sci 27, 402–10 (2011). [DOI] [PubMed] [Google Scholar]
- Kahn S. E., Hull R. L. & Utzschneider K. M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–6 (2006). [DOI] [PubMed] [Google Scholar]
- Mathis D., Vence L. & Benoist C. beta-Cell death during progression to diabetes. Nature 414, 792–8 (2001). [DOI] [PubMed] [Google Scholar]
- Rhodes C. J. Type 2 diabetes-a matter of beta-cell life and death? Science 307, 380–4 (2005). [DOI] [PubMed] [Google Scholar]
- Lu T. H. et al. Arsenic induces pancreatic beta-cell apoptosis via the oxidative stress-regulated mitochondria-dependent and endoplasmic reticulum stress-triggered signaling pathways. Toxicol Lett 201, 15–26 (2011). [DOI] [PubMed] [Google Scholar]
- Diaz-Villasenor A. et al. Arsenic exposure and Calpain-10 polymorphisms impair the function of pancreatic beta-cells in humans: A pilot study of risk factors for T2DM. PLoS One 8, e51642 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J. Q. et al. Low-level arsenic impairs glucose-stimulated insulin secretion in pancreatic beta cells: Involvement of cellular adaptive response to oxidative stress. Environ Health Perspect 118, 864–870 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett J. R. A different diabetes arsenic plus high-fat diet yields an unusual diabetes phenotype in mice. Environ Health Perspect 119, A354–A354 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D. S., Walton F. S., Saunders R. J. & Styblo M. Characterization of the impaired glucose homeostasis produced in C57BL/6 mice by chronic exposure to arsenic and high-fat diet. Environ Health Perspect 119, 1104–1109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D. S., Harmon A. W., Devesa V., Thomas D. J. & Styblo M. Molecular mechanisms of the diabetogenic effects of arsenic: inhibition of insulin signaling by arsenite and methylarsonous acid. Environ Health Perspect 115, 734–42 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. et al. Impact of iron precipitant on toxicity of arsenic in water: A combined in vivo and in vitro study. Environ Sci Technol 47, 3432–3438 (2013). [DOI] [PubMed] [Google Scholar]
- Mazumder D. N. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol Appl Pharmacol 206, 169–75 (2005). [DOI] [PubMed] [Google Scholar]
- Smith A. H. & Steinmaus C. M. Health effects of arsenic and chromium in drinking water: recent human findings. Annu Rev Public Health 30, 107–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbrey J. M. et al. Reduced arsenic clearance and increased toxicity in aquaglyceroporin-9-null mice. Proc Natl Acad Sci U S A 106, 15956–60 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio D. M. & Newgard C. B. Molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 9, 193–205 (2008). [DOI] [PubMed] [Google Scholar]
- Ghatak S. et al. Oxidative stress and hepatic stellate cell activation are key events in arsenic induced liver fibrosis in mice. Toxicol Appl Pharmacol 251, 59–69 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchin K. T. & Conolly R. Arsenic-induced carcinogenesis-oxidative stress as a possible mode of action and future research needs for more biologically based risk assessment. Chem Res Toxicol 23, 327–335 (2010). [DOI] [PubMed] [Google Scholar]
- Smith B. W. & Adams L. A. Nonalcoholic fatty liver disease and diabetes mellitus: pathogenesis and treatment. Nat Rev Endocrinol 7, 456–65 (2011). [DOI] [PubMed] [Google Scholar]
- Dey A. & Lakshmanan J. The role of antioxidants and other agents in alleviating hyperglycemia mediated oxidative stress and injury in liver. Food Funct. 4, 1148–84 (2013). [DOI] [PubMed] [Google Scholar]
- Takumi S. et al. In vivo mutagenicity of arsenite in the livers of gpt delta transgenic mice. Mutat Res 760, 42–7 (2014). [DOI] [PubMed] [Google Scholar]
- De Vizcaya-Ruiz A., Barbier O., Ruiz-Ramos R. & Cebrian M. E. Biomarkers of oxidative stress and damage in human populations exposed to arsenic. Mutat Res 674, 85–92 (2009). [DOI] [PubMed] [Google Scholar]
- Laplante M. & Sabatini D. M. mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc Natl Acad Sci U S A 107, 3281–3282 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beale E. G., Hammer R. E., Antoine B. & Forest C. Disregulated glyceroneogenesis: PCK1 as a candidate diabetes and obesity gene. Trends Endocrinol Metab 15, 129–135 (2004). [DOI] [PubMed] [Google Scholar]
- Boden G. Effects of free fatty acids on gluconeogenesis and glycogenolysis. Life Sci 72, 977–88 (2003). [DOI] [PubMed] [Google Scholar]
- Staehr P. et al. Effects of free fatty acids per se on glucose production, gluconeogenesis, and glycogenolysis. Diabetes 52, 260–7 (2003). [DOI] [PubMed] [Google Scholar]
- Rains J. L. & Jain S. K. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med 50, 567–575 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavodnik I. B., Dremza I. K., Lapshina E. A. & Cheshchevik V. T. Diabetes mellitus: metabolic effects and oxidative stress. Biol Membr 28, 83–94 (2011). [Google Scholar]
- Cho H. Protein tyrosine phosphatase 1B (PTP1B) and obesity. Vitam Horm 91, 405–24 (2013). [DOI] [PubMed] [Google Scholar]
- Leto D. & Saltiel A. R. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol 13, 383–96 (2012). [DOI] [PubMed] [Google Scholar]
- Guo S. D. Insulin signaling, resistance, and metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol 220, T1–T23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez J. J. & Iglesias P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur J Endocrinol 148, 293–300 (2003). [DOI] [PubMed] [Google Scholar]
- Carbone C. J. et al. Protein tyrosine phosphatase 1B is a key regulator of IFNAR1 endocytosis and a target for antiviral therapies. Proc Natl Acad Sci U S A 109, 19226–19231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. et al. Correlative and quantitative 1H NMR-based metabolomics reveals specific metabolic pathway disturbances in diabetic rats. Anal Biochem 383, 76–84 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian N. et al. NMR-based metabonomic study of Chinese medicine Gegen Qinlian Decoction as an effective treatment for type 2 diabetes in rats. Metabolomics 9, 1228–1242 (2013). [Google Scholar]
- Friedrich N. Metabolomics in diabetes research. J Endocrinol 215, 29–42 (2012). [DOI] [PubMed] [Google Scholar]
- Gall W. E. et al. alpha-hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population. PLoS One 5, e10883 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S. C. et al. Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab 5, 313–320 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champagne C. D., Houser D. S., Fowler M. A., Costa D. P. & Crocker D. E. Gluconeogenesis is associated with high rates of tricarboxylic acid and pyruvate cycling in fasting northern elephant seals. Am J Physiol Regul Integr Comp Physiol 303, R340–R352 (2012). [DOI] [PubMed] [Google Scholar]
- Wallage T. M., Levy J. C. & Matthews D. R. Use and Abuse of HOMA Modeling. Diabetes Care 27, 1487–1495 (2004). [DOI] [PubMed] [Google Scholar]
- Zhang X. et al. Structural changes of gut microbiota during berberine-mediated prevention of obesity and insulin resistance in high-fat diet-fed rats. PLoS One 7, e42529 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiele T. V. d. et al. Arsenic metabolism by human gut microbiota upon in vitro digestion of contaminated soils. Environ Health Perspect 118, 1004–1009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J., Mandal R., Sinelnikov I. V., Broadhurst D. & Wishart D. S. MetaboAnalyst 2.0--a comprehensive server for metabolomic data analysis. Nucleic Acids Res 40, W127–33 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]