

Abstract
Purpose
CALGB80303 was a phase III trial of 602 patients with locally advanced or metastatic pancreatic cancer comparing gemcitabine/bevacizumab versus gemcitabine/placebo. The study found no benefit in any outcome from the addition of bevacizumab to gemcitabine. Blood samples were collected and multiple angiogenic factors were evaluated and then correlated with clinical outcome in general (prognostic markers) and with benefit specifically from bevacizumab treatment (predictive markers).
Experimental Design
Plasma samples were analyzed via a novel multiplex ELISA platform for 31 factors related to tumor growth, angiogenesis, and inflammation. Baseline values for these factors were correlated with overall survival (OS) using univariate Cox proportional hazard regression models and multivariable Cox regression models with leave-one-out cross validation. Predictive markers were identified using a treatment by marker interaction term in the Cox model.
Results
Baseline plasma was available from 328 patients. Univariate prognostic markers for OS were identified including: Ang2, CRP, ICAM-1, IGFBP-1, TSP-2 (all P < 0.001). These prognostic factors were found to be highly significant, even after adjustment for known clinical factors. Additional modeling approaches yielded prognostic signatures from multivariable Cox regression. The gemcitabine/bevacizumab signature consisted of IGFBP-1, interleukin-6, PDGF-AA, PDGF-BB, TSP-2; whereas the gemcitabine/ placebo signature consisted of CRP, IGFBP-1, PAI-1, PDGF-AA, P-selectin (both P < 0.0001). Finally, three potential predictive markers of bevacizumab efficacy were identified: VEGF-D (P <0.01), SDF1 (P <0.05), and Ang2 (P < 0.05).
Conclusion
This study identified strong prognostic markers for pancreatic cancer patients. Predictive marker analysis indicated that plasma levels of VEGF-D, Ang2, and SDF1 significantly predicted for benefit or lack of benefit from bevacizumab in this population.
Introduction
Pancreatic cancer is one of the leading causes of cancer-related death worldwide (1). Surgery is rarely curative and the benefit of gemcitabine and other treatments has been marginal. Only recently has the survival for patients with metastatic pancreatic adenocarcinoma moved beyond 1 year (2), and the combination chemotherapy regimen responsible for this improvement, FOLFIRINOX, may not be appropriate for many patients with pancreatic cancer (3). Recently, the addition of Nab-paclitaxel to gemcitabine improved overall survival (OS) for patients with metastatic pancreatic cancer, however, this improvement was relatively modest (4). The clinical hallmarks of pancreatic cancer include marked cachexia, hypercoaguability, and pain syndromes out of proportion to the tumor volume (5). The primary tumor is also notoriously desmoplastic, locally invasive, and metastatic early in its course. The pathophysiology underlying these conditions has been associated with multiple factors associated with tumor angiogenesis and inflammation (5).
CALGB80303 was a randomized, double-blind placebo controlled study of standard of care gemcitabine chemotherapy ± bevacizumab in patients with advanced or metastatic pancreatic cancer (6). This study included 602 patients and was conducted by CALGB, which has recently merged into the Alliance for Clinical Trials in Oncology (Alliance). Bevacizumab (Avastin; Genentech/ Roche Inc.) is a monoclonal antibody that binds all known isoforms of VEGFA (vascular endothelial growth factor-A, also commonly known as VEGF). Bevacizumab is associated with improved clinical outcomes in several cancers, including metastatic colorectal (7), non–small cell lung (8), renal cell (9), and glioblastoma (10, 11). However, despite promising phase II data (12), bevacizumab conferred no benefit in terms of OS or progression free survival in CALGB80303 (6).
Recognizing the potential value of biomarkers that might predict for sensitivity and resistance to bevacizumab, as well as prognostic markers that could also guide the management of patients with pancreatic cancer, plasma, serum, and urine were collected at baseline and at each restaging during treatment on CALGB80303. At the time the study was initiated, the analyses of the angiogenic and inflammatory factors felt to play a role in the prognosis of pancreatic cancer had typically been limited by the size and quality of the available datasets, and by the use of standard ELISA methodology, which significantly limits the number of factors that can be evaluated in a given sample.
Many of these limitations have since been overcome with the development of multiplex ELISA platforms (13). Multiplex approaches have the advantages of reduced sample volume requirements and a lower per analyte cost. This approach also facilitates analyses of multiple predictors and identifying patterns of expression among analytes. However, these approaches require rigorous technical optimization to account for variable concentrations across different analytes, the sensitivity and specificity of differing capture and detection antibodies, interference among analytes, and numerous other preanalytic considerations (14–16).
We report the results of our detailed angiome analysis that was conducted for greater than 30 markers of tumor angiogenesis and inflammation; the current analysis focuses on the pretreatment (baseline) plasma samples.
Materials and Methods
Patients
The design and results of CALGB80303 have been described previously (6). Briefly, eligible patients had histologically or cytologically confirmed pancreatic adenocarcinoma not amenable to curative surgery. Measurable disease was not required. Prior chemotherapy for metastatic disease was not permitted. Adjuvant chemotherapy was allowed if it did not contain gemcitabine or bevacizumab, if it was given more than 4 weeks before enrollment, and if the patient had subsequent disease progression. IRB-approved, written informed consent was obtained from patients who opted to participate in this correlative analysis of CALGB80303. This retrospective analysis conforms to the reporting guidelines established by the REMARK criteria.
Sample collection and analysis
Peripheral venous blood was collected into EDTA anticoagulant vacutainers. The tubes were centrifuged within 30 minutes of collection at 2,500 × g for 15 minutes. Plasma was aliquoted into cryovials and snap frozen, and samples were shipped on dry ice for centralized storage at −80° C at the CALGB Pathology Coordinating Office. Before analysis, all patients’ samples were shipped to our laboratory (Duke Molecular Reference Laboratory), thawed on ice, re-aliquoted based on specific assay requirements and stored at −80°C. All assays were performed in triplicate, limited to 2 freeze–thaw cycles only, and all analyses were conducted while blinded to clinical outcome.
Plasma samples were analyzed using the Searchlight platform (Aushon Biosystems, Inc.) or kit ELISAs following manufacturer’s protocol. Markers are listed in Table 1. Additional ELISA assays were conducted for IGF-1 (Immunodiagnostic Systems, Inc.) and TGF-βRIII (R&D Systems, Inc.). Plasma samples were thawed on ice, centrifuged at 20,000 × g for 5 minutes to remove precipitate, and subsequently loaded onto SearchLight plates with standard protein controls. Samples and standards were incubated at room temperature for 1 hour shaking at 950 rpm (Lab-Line Titer Plate Shaker, Model 4625). Plates were washed 3 times using a plate washer (Biotek Instruments, Inc., Model ELx405), biotinylated secondary antibody was added, and plates were incubated for 30 minutes. After washes, strep-tavidin-HRP was added, plates were incubated for 30 minutes, washed again, and SuperSignal substrate was added. Images were taken within 10 minutes, followed by image analysis using SearchLight array analyst software. All data represent the average of triplicate measures.
Table 1.
Analyte characteristics at baseline
| Analyte | Unit | %CV | N | Median | Min | Max |
|---|---|---|---|---|---|---|
| Ang-2 | (pg/mL) | 14.7 | 328 | 301.4 | 9.9 | 3,150 |
| CRP | (ng/mL) | 27.3 | 328 | 20,178.5 | 14.2 | 942,048 |
| bFGF | (pg/mL) | 20.1 | 328 | 25.6 | 0.9 | 1,120 |
| GRO-α | (pg/mL) | 24.3 | 328 | 70.8 | 7.3 | 769 |
| HGF | (pg/mL) | 8.1 | 328 | 817.4 | 251.6 | 100900 |
| ICAM-1 | (ng/mL) | 14.4 | 328 | 355.0 | 2.8 | 2,498 |
| IGF-1 | (pg/mL) | 3.7 | 328 | 697.0 | 56.6 | 3,398 |
| IGFBP1 | (pg/mL) | 14.5 | 328 | 13,253.3 | 86.7 | 1,028,290 |
| IGFBP3 | (ng/mL) | 7.9 | 328 | 709.6 | 4.2 | 2,184 |
| IL-6 | (pg/mL) | 10.7 | 328 | 17.5 | 2.8 | 1,105 |
| IL-8 | (pg/mL) | 24.7 | 328 | 49.2 | 0.6 | 2,061 |
| MCP-1 | (pg/mL) | 10.4 | 328 | 521.8 | 146.7 | 3,445 |
| OPN | (ng/mL) | 23.0 | 328 | 74.1 | 1.6 | 1,036 |
| PAI-1 Act | (ng/mL) | 11.3 | 328 | 2.3 | 3.8 | 31 |
| PAI-1 Tot | (ng/mL) | 10.1 | 328 | 27.9 | 0.2 | 259 |
| PDGF-AA | (pg/mL) | 17.7 | 328 | 239.2 | 0.5 | 5,746 |
| PDGF-BB | (pg/mL) | 12.2 | 328 | 185.4 | 0.3 | 2,226 |
| PEDF | (ng/mL) | 14.7 | 328 | 3,549.3 | 2.6 | 7,855 |
| P-Selectin | (ng/mL) | 7.5 | 328 | 53.6 | 3.6 | 625 |
| PlGF | (pg/mL) | 22.1 | 328 | 5.0 | 0.1 | 363 |
| SDF1 | (pg/mL) | 9.4 | 328 | 1,103.0 | 11.3 | 36,383 |
| TGF-β1 | (pg/mL) | 12.5 | 326 | 36,239.2 | 37.8 | 20,6971 |
| TGF-β2 | (pg/mL) | 15.2 | 328 | 56.7 | 5.6 | 1,287 |
| TGF-βRIII | (pg/mL) | 2.6 | 328 | 410.6 | 4.6 | 1011 |
| TSP-2 | (ng/mL) | 8.8 | 328 | 20.7 | 1.5 | 451 |
| VCAM-1 | (ug/mL) | 10.0 | 328 | 1,614.0 | 0.9 | 7,971 |
| VEGF | (pg/mL) | 17.9 | 328 | 89.6 | 1.9 | 12,678 |
| VEGF-C | (pg/mL) | 19.0 | 328 | 572.4 | 10.2 | 34,038 |
| VEGF-D | (pg/mL) | 10.7 | 328 | 1,558.1 | 70.3 | 25,382 |
| sVEGFR1 | (pg/mL) | 13.3 | 328 | 123.0 | 8.2 | 43,692 |
| sVEGFR2 | (ng/mL) | 14.9 | 328 | 3,790.5 | 34.2 | 39,033 |
Abbreviations: ANG-2, angiopoietin-2; bFGF, basic fibroblast growth factor; HGF, hepatocyte growth factor; IGF-1, insulin-like growth factor-1; IGFBP, insulin-like growth factor-binding protein; PDGF, platelet-derived growth factor; PEDF, pigment epithelium-derived factor; PlGF, placental growth factor; VEGF, vascular endothelial growth factor; sVEGFR, soluble vascular endothelial growth factor receptor; TGF-β, transforming growth factor β; sTGF-βRIII, soluble transforming growth factor β receptor type III; TSP, thrombospondin; CRP, c-reactive protein; PAI-1, plasminogen activator inhibitor-1; Gro-α; growth-regulated oncogene-α; ICAM-1, intercellular adhesion molecule 1; IL, interleukin; MCP-1, macrophage chemoattractant protein-1; SDF1, stromal cell–derived factor-1; VCAM-1, vascular cell adhesion molecule 1.
Statistical analysis
Spearman correlations among analytes were calculated and illustrated in a hierarchical clustering dendrogram. OS was the primary endpoint of the treatment trial and was measured from trial registration until death from any cause. The prognostic value of each analyte as a continuous measure was assessed by a univariate Cox proportional hazards model for OS. Results were presented for each treatment arm. Conservative Bonferroni and False Discovery Rate (FDR) multiple testing corrections were used for prognostic markers. Median survival time of two dichotomized subgroups of patients with median as the split point was presented. Their corresponding HRs and 95% confidence intervals (CI) were given in the results tables. Cox regression analyses with each analyte adjusting for clinical variables, including age, race, gender, and performance status were performed. Multivariable Cox prognostic regression models limited to the top 5 analytes were built using leave-one-out cross validation. The top 5 analytes were chosen within each training set using the score method (17). This method uses the branch-and-bound algorithm and the criterion used to select the top 5 analytes is based on the global score χ2 statistic. Using the training set models, the left out samples were classified into either the high- or the low-risk groups. Kaplan–Meier plots were used to illustrate survival curves. The predictive value of each analyte was assessed using Cox proportional hazards model with treatment by analyte (dichotomized at the median) interactions. To identify trends that may not have been apparent at median cutoffs, analytes were further dichotomized at the quartiles. Permutation analysis was performed in the identification of predictive markers. We generated 1,000 permuted datasets by shuffling the survival data and randomly matching them with the analyte data. The permutation P-value is the proportion of P-values from the 1,000 permuted data sets that are smaller than the observed P-value. The number of deaths was 326 of 328; for the 2 patients that were alive at the time of data lock, the median follow-up time was 2.4 months. All P-values are 2-sided. SAS Version 9.2 (SAS Institute) and R Version 2.8.0 were used to carry out the analyses. Statistical analyses were performed by the Alliance Statistics and Data Center.
Results
Patient characteristics
Of the 602 patients who accrued to the parent protocol, 535 were treated and baseline EDTA plasma samples were available for analysis on 328 patients, 159 patients in the gemcitabine/placebo group, and 169 in the gemcitabine/ bevacizumab group (Fig. 1). The clinical characteristics and outcomes of these patients were similar to those on the parent study. In addition, among the patients in this correlative study, characteristics across both treatment groups were similar, except for a minor imbalance in gender (Table 2).
Figure 1.
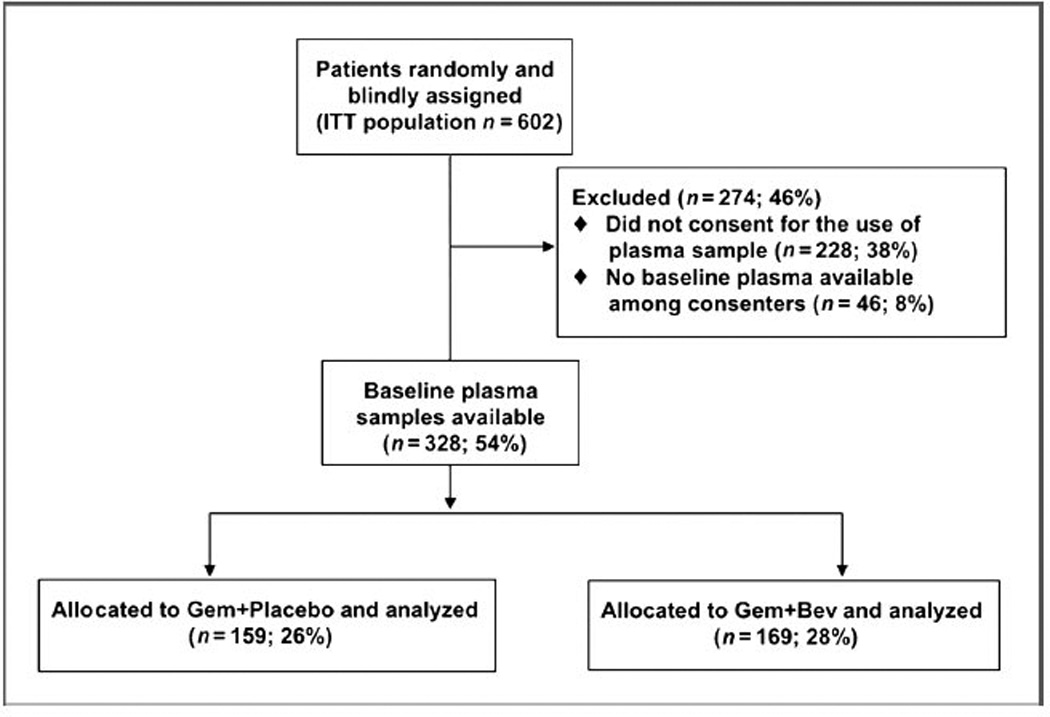
Consort diagram.
Table 2.
Patient characteristics
| Overall whole population |
% | Overall EDTA population No. |
% | Gem + Placebo No. |
% | Gem + Bev No. |
% | |
|---|---|---|---|---|---|---|---|---|
| No. patients | 602 | 328 | 159 | 169 | ||||
| Age (y) | ||||||||
| Median | 64.1 | 64.0 | 65.5 | 63.1 | ||||
| Range | 26.3–88.7 | 35.8–84.2 | 35.8–83.7 | 35.9–84.2 | ||||
| Gender male | 55 | 55 | 48 | 62 | ||||
| Race white | 88 | 89 | 91 | 88 | ||||
| ECOG PS | ||||||||
| 0 | 37 | 40 | 39 | 40 | ||||
| 1 | 52 | 50 | 53 | 48 | ||||
| 2 | 11 | 10 | 8 | 12 | ||||
| Extent of disease | ||||||||
| Locally adv. | 88 | 88 | 88 | 88 | ||||
| Metastatic | 12 | 12 | 12 | 12 | ||||
| Median OS (range) | 5.9 (5.3–6.5) | 6.1 (5.5–6.9) | 6.3 (5.1–8.0) | 5.9 (5.0–7.0) | ||||
| Median PFS (range) | 3.5 (3.0–3.8) | 3.8 (3.3–4.0) | 3.5 (2.4–4.2) | 3.8 (3.5–4.6) |
Baseline angiome factor measurement and correlation
Single and multiplex analyses demonstrated good sensitivity and coefficients of variation (CV) were generally in the range of 10% to 30%. Only 2 analytes (PlGF and bFGF) had levels below the limits of quantification for greater than 10% of patients evaluated. The medians and ranges for all analytes at baseline are provided in Table 1.
Spearman correlations were performed on all analytes and a dendrogram was generated to describe patterns of expression among analytes (Fig. 2). Strong correlation coefficients (>0.70) were noted for 2 pairs of analytes; TGF-β1 and PDGF-AA (0.76) and CRP and IL-6 (0.72). Moderate associations with correlation coefficients between 0.4 and 0.7 were noted among VEGF/PDGF family members, as well as among multiple inflammatory markers.
Figure 2.
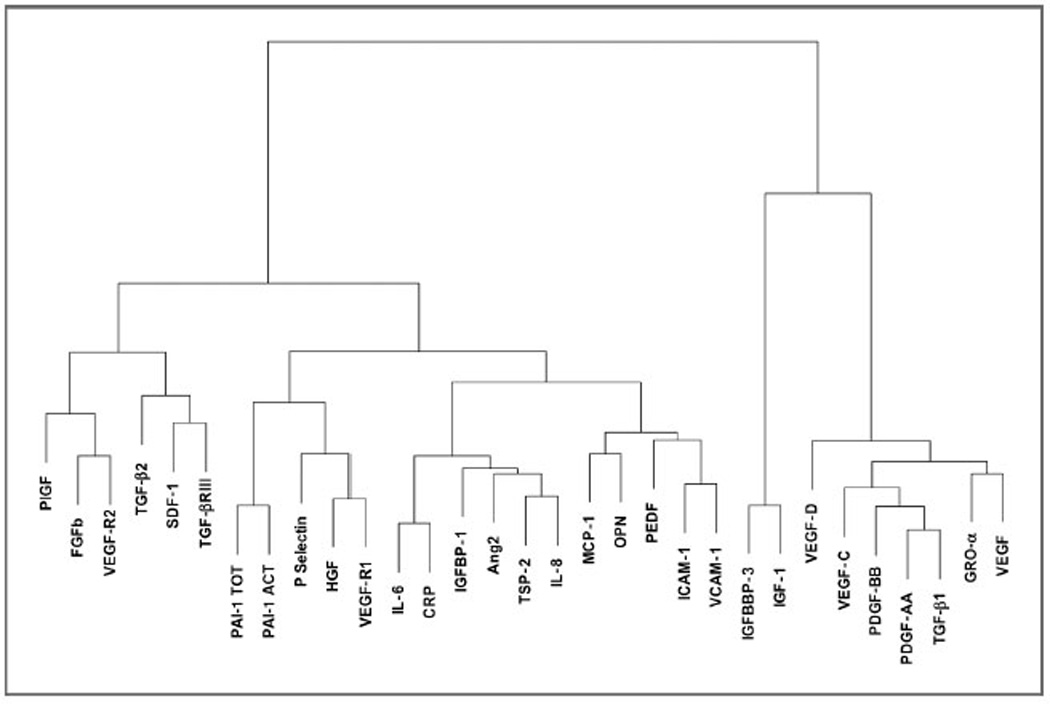
Spearman-based dendrogram.
Prognostic marker identification
Univariate and multivariable Cox regression models were used to identify markers with significant prognostic impact. Because of observed treatment-by-analyte interactions for some analytes, both univariate and multivariable Cox prognostic regression analyses were conducted separately for the gemcitabine/placebo and the gemcitabine/bevacizumab groups. In the univariate analysis, IGFBP-1, ICAM-1, Ang-2, CRP, IL-8, TSP-2, VCAM-1, PAI-1 active, and IGF-1 were significantly correlated with OS in the gemcitabine/placebo group (Table 3). Similarly, TSP-2, CRP, IL-6, IGFBP-1, Ang-2, ICAM-1, and VCAM-1 were significantly correlated with OS in the gemcitabine/bevacizumab group (Table 3). The analytes presented in Table 3 have FDR q-values of less than 0.005 (18). The prediction results from multivariable leave-one-out cross-validation models with just the clinical factors age, gender, PS, and extent of disease are as follows. For gemcitabine/bevacizumab, the HR = 1.5 (95% CI, 1.1–2.0) with log-rank test P-value of 0.0104. For gemcitabine/placebo, the HR = 1.9 (95% CI, 1.4–2.6) with log-rank test P-value of 0.0001. In contrast to the numbers presented in Fig. 3, the analyte models were stronger than the models with just clinical covariates for both treatment arms. All of these markers remained significant (P < 0.05) after accounting for multiple testing using Bonferroni correction. Thirty-one tests within gemcitabine/placebo and gemcitabine/bevacizumab were performed for the prognostic variable analysis. In addition, all of these markers remained prognostic after correction for known clinical prognostic variables, including age, race, gender, and performance status (data not shown). With the exception of IGF-1, all of these factors were unfavorable prognostic markers. Favorable and unfavorable prognostic markers are markers where, as the level of the marker increased, the rate of death decreased or increased, respectively. To assess whether there were any potential major violations in the proportional hazards assumptions of the Cox model, Schoenfeld residuals were calculated. Only one marker for the gemcitabine/placebo group exhibited some departure from proportionality. However, the log-rank test comparing the survival curves of patients with low and high levels of the marker dichotomized at the median is consistent with the result from the Cox model. To assess violations of the linearity assumption, deviance and martingale residual plots were generated and no unusual patterns or extreme outliers were identified.
Table 3.
Baseline univariate EDTA prognostic markers (OS)
| < Median |
> Median |
|||||||
|---|---|---|---|---|---|---|---|---|
| Cohort | Analyte | P-valuea | q-valueb | Median survival (months) |
95% CI | Median survival (months) |
95% CI | < med vs. > med HR (95% CI) |
| Gemcitabine + | IGFBP-1 | <0.0001 | <0.0001 | 9.2 | (7.3–9.9) | 4.3 | (3.1–5.7) | 1.7 (1.2–2.4) |
| Placebo | ICAM-1 | <0.0001 | <0.0001 | 8.4 | (6.1–9.7) | 4.8 | (3.5–6.8) | 1.4 (1.01–1.90) |
| Ang2 | <0.0001 | <0.0001 | 9.6 | (7.7–10.4) | 4.6 | (3.3–5.8) | 2.4 (1.7–3.3) | |
| CRP | <0.0001 | <0.0001 | 9.7 | (8.1–10.6) | 3.8 | (2.9–4.8) | 2.3 (1.6–3.1) | |
| IL-8 | <0.0001 | <0.0001 | 8.7 | (6.1–9.7) | 5.0 | (3.5–6.9) | 1.3 (0.98–1.86) | |
| TSP-2 | <0.0001 | 0.0002 | 9.0 | (6.8–9.7) | 4.6 | (3.3–5.6) | 1.6 (1.1–2.1) | |
| VCAM-1 | 0.0002 | 0.0005 | 9.0 | (6.7–9.7) | 4.8 | (3.6–5.9) | 1.6 (1.2–2.3) | |
| PAI-1 Act | 0.0004 | 0.0010 | 8.1 | (6.7–9.2) | 4.8 | (3.4–5.9) | 1.3 (0.94–1.77) | |
| IGF-1 | 0.0012 | 0.0023 | 4.2 | (3.3–5.6) | 9.0 | (6.8–9.7) | 0.65 (0.47–0.89) | |
| Gemcitabine + | TSP-2 | <0.0001 | <0.0001 | 6.1 | (5.2–7.4) | 5.7 | (4.1–7.1) | 1.3 (0.9–1.7) |
| Bevacizumab | CRP | <0.0001 | <0.0001 | 7.4 | (5.9–9.5) | 4.6 | (3.5–5.8) | 1.9 (1.4–2.6) |
| IL-6 | <0.0001 | <0.0001 | 8.4 | (7.0–9.7) | 3.8 | (3.1–4.8) | 2.3 (1.7–3.2) | |
| IGFBP-1 | <0.0001 | <0.0001 | 8.0 | (6.5–9.7) | 4.1 | (3.2–5.3) | 2.2 (1.6–3.0) | |
| Ang2 | <0.0001 | <0.0001 | 7.0 | (5.5–15.0) | 4.8 | (3.8–6.5) | 1.4 (1.1–2.0) | |
| ICAM-1 | 0.0002 | 0.0003 | 6.8 | (5.5–8.3) | 5.0 | (4.0–6.8) | 1.5 (1.1–2.0) | |
| VCAM-1 | 0.0012 | 0.0014 | 7.1 | (5.5–8.1) | 5.3 | (4.1–6.5) | 1.5 (1.1–2.0) | |
| IGF-1 | 0.0019 | 0.0020 | 4.8 | (3.7–5.9) | 7.1 | (5.7–8.1) | 0.72 (0.53–0.97) | |
Above is a list of analytes that is prognostic univariately for OS using the Cox proportional hazard model. On the right-hand side, it indicates the median survival time and its 95% CI for less than median and greater than median level of the analytes. All analytes presented remain significant (P < 0.05) after accounting for multiple testing using Bonferroni correction methods.
From Cox proportional hazard model using continuous analyte values (primary analysis).
False Discovery Rate (FDR).
Figure 3.
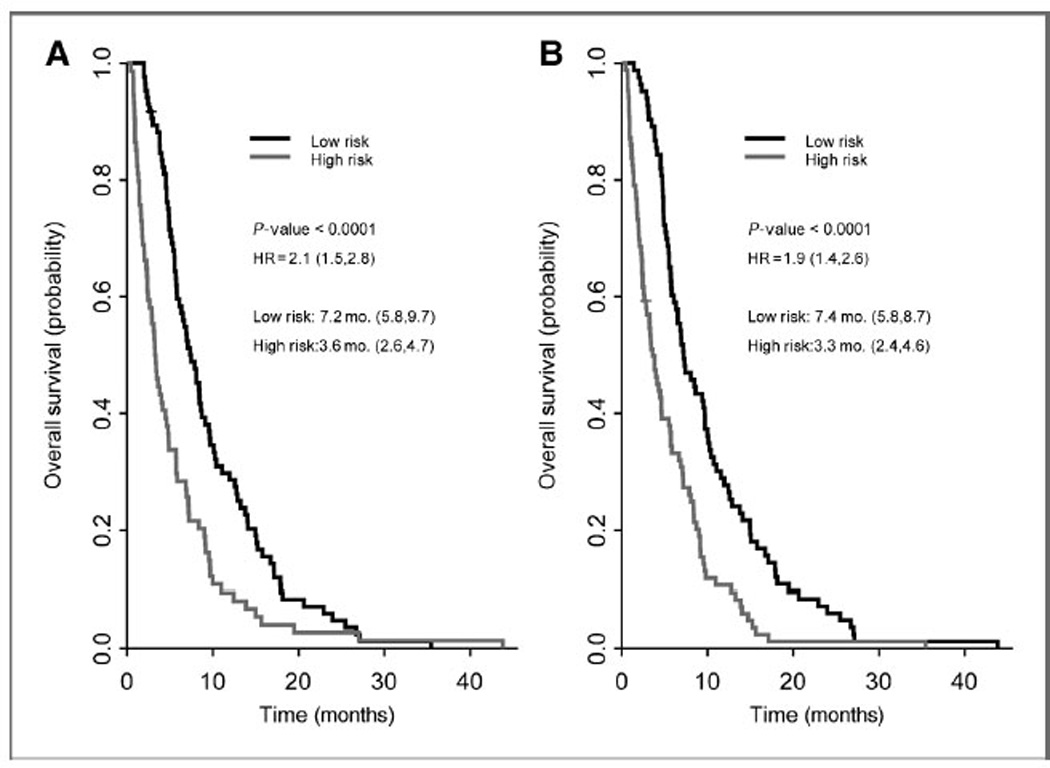
Kaplan–Meier estimates of OS for (A) gemcitabine/placebo and (B) gemcitabine/bevacizumab groups using leave-one-out cross-validated Cox proportional hazard models. Gemcitabine/placebo (A) model consists of: IGFBP-1 (100%), CRP (99.4%), PDGF-AA (99.4%), PAI-1 total (99.4%), and P-selectin (95.0%). Gemcitabine/ bevacizumab (B) model consists of: IGFBP-1 (100%), IL-6 (100%), PDGF-AA (99.4%), PDGF-BB (98.8%), and TSP-2 (97.6%). The percentages of the analytes picked in the training set of the leave-one-out cross validation are indicated in parentheses above.
Using leave-one-out cross-validation analysis, 5-analyte prognostic models for OS were developed, one model for the gemcitabine/placebo group and the other for the gemcitabine/bevacizumab group (see Fig. 3). The gemcitabine/ placebo model for OS consisted of IGFBP-1, CRP, PDGF-AA, PAI-1 total, and P-selectin. The top 4 markers were selected in more than 99% of the training sets, with the exception of P-selectin, which was selected in 82% of the training sets. The median survivals of the high- and low-risk groups in this model were 3.3 and 7.4 months, respectively, and were associated with an HR of 1.9. The gemcitabine/ bevacizumab model for OS consisted of IGFBP-1, IL-6, PDGF-AA, PDGF-BB, and TSP-2. These markers were selected in more than 98% of the training sets. The median survivals of the high- and low-risk groups were 3.6 and 7.2 months, respectively, and were associated with an HR of 2.1. Kaplan–Meier plots of these models are shown in Fig. 3.
Predictive marker identification
Predictive markers were identified using the Cox proportional hazards model. Analyte values were evaluated using both median and quartile cutpoints. Three markers were identified as being predictive for benefit or lack of benefit from bevacizumab: VEGF-D, SDF1, and Ang-2. The treatment-by-analyte interaction P-values were 0.0040 for VEGF-D at Q1, 0.019 for SDF1 at median, and 0.036 for Ang-2 at median. Permutation P-values for all 3 markers were below 0.05 with 1,000 iterations. Lower levels of VEGF-D (below Q1, lowest 25%) predicted for benefit from bevacizumab whereas higher levels of VEGF-D (above Q1, top 75%) predicted for lack of benefit from bevacizumab. Conversely, below median levels of both Ang-2 and SDF1 predicted for greater benefit in the placebo group. These data are shown graphically in Fig. 4. The P-values for treatment-marker interaction terms remained significant after adjusting for clinical covariates.
Figure 4.
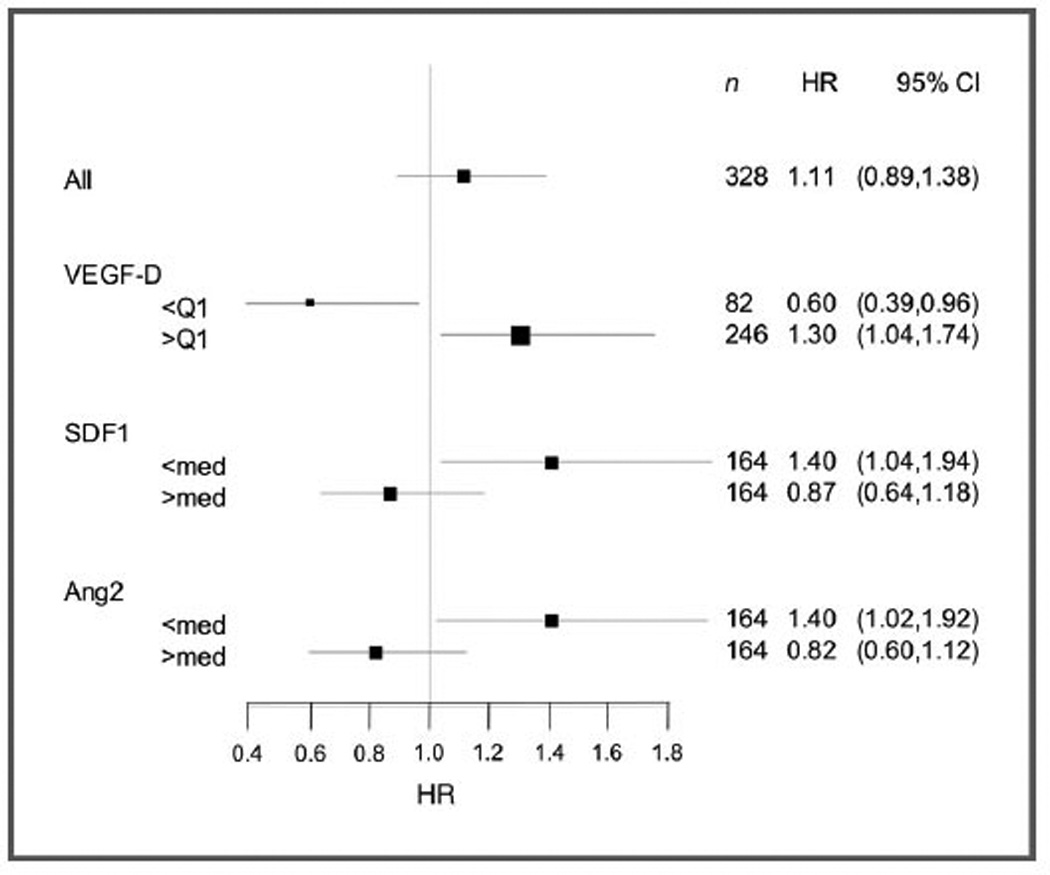
Forest plots of HRs (gemcitabine/placebo vs. gemcitabine/bevacizumab) for OS by biomarker. VEGF-D, by quartile; SDF1, by median; Ang-2, by median.
Discussion
Although analyses of tumor tissue are usually considered the gold standard for biomarker development, this approach is often limited by the availability and reliability of archived clinical samples, and research-only biopsies carry significant risks and costs. These issues are particularly important in studies of pancreatic adenocarcinoma, because the majority of patients are currently diagnosed by cytology or small gauge core biopsies.
For these reasons, the search for blood-based markers in this disease has begun in earnest and the multiplex angiome analysis of CALGB80303 is one of the largest such analyses reported to date. In this large multicenter study, technical analyses were robust with good sensitivity and low variability, which were generally comparable to single kit ELISAs. Unsupervised hierarchical clustering identified potential patterns of analyte expression; potentially biologically relevant groupings were observed across VEGF/ PDGF family members, TGF-β family members, and various inflammatory and coagulation factors. Such analyses may provide novel insights into the coregulation and counter regulation of these factors and their underlying biology.
Multiple markers were identified as highly prognostic. These laboratory-based prognostic markers were much more powerful than traditional clinical factors and remained statistically significant after adjustment for known clinical factors. Most of these markers are involved in tumor-related angiogenesis, inflammation, and coagulation, supporting the clinical importance of these factors in the underlying pathophysiology of pancreatic cancer. Indeed, ras mutations, which are present in approximately 90% of pancreatic adenocarcinomas, have been associated with the upregulation of multiple factors related to inflammation and inflammatory angiogenesis (19, 20). To the degree these prognostic factors are important drivers of tumor biology rather than nonspecific host responses, these factors may be considered potential therapeutic targets. Agents targeting some of these factors have already entered clinical trials, including trials focused on pancreatic cancer (21, 22). Building models with a combination of analytes and clinical factors can be investigated further in future research.
Candidate markers of sensitivity and resistance to bevacizumab were also identified. The ability to assess for treatment interactions that differentiate prognostic and predictive markers is a key advantage of randomized studies. However, these results need to be validated in other datasets given the number of patients analyzed the number of analytes tested, the borderline statistical significance of some results, and the overall lack of benefit from bevacizumab in unselected patients. These caveats notwithstanding, VEGF-D, Ang2, and SDF1 were found to be predictive of benefit or lack of benefit from bevacizumab.
VEGF-D is known to be upregulated by multiple factors associated with pancreatic cancer, including those related to angiogenesis, inflammation, and hypoxia (23, 24). Although the biology of VEGF-D is commonly linked to its role in lymphangiogenesis, processed forms of VEGF-D can bind with high affinity to VEGFR2 leading to activation of the receptor, endothelial cell proliferation, and tumor angiogenesis (25). Proteolytic processing is known to modulate the affinity of many angiogenic factors for their cognate receptor and other binding partners. Importantly, this phenomenon may partially underlie the recently reported importance of small isoforms and processed forms of VEGF-A as predictive markers for bevacizumab (26). Moreover, VEGF-D, when measured by immunohistochemistry on formalin-fixed paraffin embedded tumor samples, was also recently reported to predict for benefit and lack of benefit from bevacizumab in the phase III MAX study in metastatic colorectal cancer (27). Plasma VEGF-C and -D have also been reported to increase in the setting of tumor progression, albeit to date these reports have been limited to relatively modest sized, nonrandomized studies (28, 29).
In addition to VEGF-D, low levels of both SDF1 and Ang2 were identified by univariate analyses as potential predictors of primary resistance to bevacizumab. These findings are consistent with the context dependence of Ang2 and SDF1 signaling. Ang2 is known to play a critical role in vascular maturation, where its effects are known to be VEGF dependent (30). SDF1, is also known to act in a context and VEGF-dependent fashion, both in mobilization of endothelial cell precursors from the bone marrow and in the homing of these and other cells to the tumor metastatic niche (31). Both Ang2 and SDF1 are also known to be regulated by multiple inflammatory mediators and to play key roles in the trafficking of myeloid cells implicated in VEGF resistance (32). SDF1 exists in 2 processed forms, SDF1a and SDF1β, which are tightly regulated and which have differing affinities for their cognate receptor CXCR4, other cell surface glycoproteins, and heparins (33). Similar to many reports in the literature, the reagents used here were not able to distinguish SDF1α from SDF1β.
Other groups have also reported multiplex analysis of angiogenesis factors. Scherer and colleagues have evaluated multiple angiogenesis markers across a series of studies with bevacizumab using a novel VEGF assay that preferentially detects shorter isoforms of VEGF-A (e.g., VEGF110 and VEGF121). This VEGF-A assay was found to be predictive of benefit for bevacizumab in the AVITA trial of gemcitabine and erlotinib ± bevacizumab in metastatic pancreatic cancer. Similar results were seen in trials in metastatic breast (34) and gastric cancers (35), but not in non–small cell lung, colorectal, or renal cell cancers (36). In the phase III trial of pazopanib versus best supportive care in metastatic renal cell cancer, Tran and colleagues found IL-6 to be a significant predictor of benefit from pazopanib (37). These findings have also been recently confirmed by our group for patients with renal cell carcinoma treated with bevacizumab (38). The reasons for differences in prognostic and predictive markers across studies may reflect differences in tumor biology, the specific analytes measured, type of plasma analyzed, analytic methods, and other preanalytic considerations, such as the number of freeze–thaw cycles. A common theme emerging from these studies is the growing importance of different isoforms and/or alternatively processed forms within VEGF family of ligands and the importance of mediators of inflammation and inflammatory angiogenesis (39).
There are several limitations to the analysis presented. First, many known preanalytic considerations will apply to our analyses. Although we had defined procedures and SOPs in the parent protocol for sample processing at each individual site, processing methods were not directly monitored in this study. Variability in processing can influence the measurement of several analytes, particularly those that are affected by platelet degranulation and clotting. Laboratory methods also vary across different reports. The laboratory that performed this work is an Alliance Molecular Reference Laboratory, specifically approved to conduct multiplex analyses for circulating markers, with a particular emphasis on angiogenesis factors. Assay methods were highly standardized, including the number of freeze–thaw cycles, sample type tested (EDTA plasma), consistent reagents from single batch plate printing, and reference standards applied to every microtiter plate evaluated. Second, even though this was a phase III randomized study, and the largest such analysis to date in pancreatic cancer, this study nevertheless had only modest power to detect predictive marker effect sizes, and little power to detect multivariable predictive signatures. Of the 602 patients who accrued to the parent study, only 328 of patients were available for the biomarker analysis, leading to an additional potential source of bias. To test for a treatment interaction, only 169 and 159 of these patients were in the experimental and control groups, respectively. Approximately half of all patients would be expected to be above or below the median for any one factor, with very few patients having any permutation of "high" or "low" levels for multiple factors. With multiple analytes, statistical significance requires adjustment for multiple testing. To account for these issues, we used permutation analyses, limited our prognostic signature to only 5 features, and distinguished between adjusted and unadjusted P-values. Our prognostic results were found to be highly significant, even after correction for multiple testing. However, our predictive analyses were not corrected for multiple testing and therefore should be considered exploratory. Finally, many factors seem to be coregulated, as shown by our dendrogram analysis (Fig. 2). These considerations may lead to over-fitting of some models. For these reasons, our results require external validation in other datasets.
In conclusion, multiple factors with strong prognostic impact for patients with pancreatic cancer were identified in the current analysis. In addition, VEGF-D, Ang2, and SDF1 were identified as candidates for predicting sensitivity and resistance to bevacizumab in this population. If validated in additional studies, these markers could potentially be used to select those patients who might benefit from anti-VEGF therapy. These markers also suggest potential combination regimens that may overcome resistance to anti-VEGF therapy. These results may be applicable to other tumor types and warrant further testing.
Translational Relevance.
In this analysis, we provide data defining several statistically significant prognostic markers. Many of these prognostic markers were much more powerful than traditional clinical factors, even after adjustment for these known factors. Three markers (Ang-2, SDF1, VEGF-D) were also identified as potential predictors of benefit or lack of benefit for bevacizumab. Low levels of Ang-2 and SDF1 both predicted for a lack of benefit to bevacizumab. Interestingly, low levels of VEGF-D were observed to predict for benefit to bevacizumab whereas high levels predicted for a lack of benefit. In the context of bevacizumab-treatment and VEGF-A depletion, VEGF-D can bind and activate VEGFR2, eliciting a potential resistance mechanism for bevacizumab treatment in this disease. Although similar observations for low levels of VEGF-D predicting for bevacizumab benefit have been made in colon cancer using immunohistochemical approaches, confirmation and validation of these results will be needed before any of these markers can be used to direct clinical care.
Acknowledgments
The research for CALGB80303 (Alliance) was supported, in part, by grants from the National Cancer Institute (CA31946) to the Alliance for Clinical Trials in Oncology (Monica M. Bertagnolli, M.D., Chair) and to the Alliance Statistics and Data Center (Daniel J. Sargent, Ph.D., CA33601). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.
We gratefully acknowledge the invaluable contributions of the patients, their families and all the investigators who participated in this trial. We give special recognition to Donna Hollis for clinical data management as well as to A. Bulusu and J. Marcello for additional statistical support.
The following institutions participated in this study:
Christiana Care Health Services, Inc. CCOP, Wilmington, DE, Stephen Grubbs, M.D., supported by CA45418
Dana-Farber Cancer Institute, Boston, MA, Harold J. Burstein, M.D., Ph. D., supported by CA32291
Dartmouth Medical School-Norris Cotton Cancer Center, Lebanon, NH, Konstantin Dragnev, M.D., supported by CA04326
Duke University Medical Center, Durham, NC, Jeffrey Crawford, M.D., supported by CA47577
Georgetown University Medical Center, Washington, DC, Bruce Cheson, M.D., supported by CA77597
Greenville CCOP, see above Cancer Center of Carolinas
Hematology-Oncology Associates of CNY CCOP, Syracuse, NY, Jeffrey Kirshner, M.D., supported by CA45389
Massachusetts General Hospital, Boston, MA, Jeffrey W. Clark, M.D., supported by CA32291
Memorial Sloan-Kettering Cancer Center, New York, NY, Clifford A. Hudis, M.D., supported by CA77651
Missouri Baptist Medical Center, St. Louis, MO, Alan P. Lyss, M.D., supported by CA114558-02
Mount Sinai Medical Center, Miami, FL, Michael A. Schwartz, M.D., supported by CA45564
Mount Sinai School of Medicine, New York, NY, Lewis R. Silverman, M. D., supported by CA04457
Nevada Cancer Research Foundation CCOP, Las Vegas, NV, John A. Ellerton, M.D., supported by CA35421
New Hampshire Oncology-Hematology PA, Concord, NH, Douglas J. Weckstein, M.D.
Northern Indiana Cancer Research Consortium CCOP, South Bend, IN, Rafat Ansari, M.D., supported by CA86726
Rhode Island Hospital, Providence, RI, William Sikov, M.D., supported by CA08025
Roswell Park Cancer Institute, Buffalo, NY, Ellis Levine, M.D., supported by CA59518
Southeast Cancer Control Consortium Inc. CCOP, Goldsboro, NC, James N. Atkins, M.D., supported by CA45808
State University of New York Upstate Medical University, Syracuse, NY, Stephen L. Graziano, M.D., supported by CA21060
The Ohio State University Medical Center, Columbus, OH, Clara D. Bloomfield, M.D., supported by CA77658
University of California at San Diego, San Diego, CA, Barbara A. Parker, M.D., supported by CA11789
University of Chicago, Chicago, IL, Hedy L. Kindler, M.D., supported by CA41287
University of Illinois MBCCOP, Chicago, IL, David J. Peace, M.D., supported by CA74811
University of Iowa, Iowa City, IA, Daniel A. Vaena, M.D., supported by CA47642
University of Massachusetts Medical School, Worcester, MA, William V. Walsh, M.D., supported by CA37135
University of Minnesota, Minneapolis, MN, Bruce A. Peterson, M.D., supported by CA16450
University of Missouri/Ellis Fischel Cancer Center, Columbia, MO, Karl E. Freter, M.D., supported by CA12046
University of Nebraska Medical Center, Omaha, NE, Apar Ganti, M.D., supported by CA77298
University of North Carolina at Chapel Hill, Chapel Hill, NC, Thomas C. Shea, M.D., supported by CA47559
University of Vermont, Burlington, VT, Steven M. Grunberg, M.D., supported by CA77406
Wake Forest University School of Medicine, Winston-Salem, NC, David D. Hurd, M.D., supported by CA03927
Walter Reed Army Medical Center, Washington, DC, David C. Van Echo, M.D., supported by CA26806
Washington University School of Medicine, St. Louis, MO, Nancy Bartlett, M.D., supported by CA77440
Grant Support
This work was supported by the Alliance Foundation.
A.B. Nixon has commercial research grants from Tracon Pharmaceuticals, F Hoffman-La Roche, Amgen, and Pfizer and is a consultant/advisory board member of GlaxoSmithKline and Novartis. H.L. Kindler has commercial research grants from Genentech and is a consultant/advisory board member of Genentech. R.M. Goldberg has other commercial research support from Sanofi, Bayer, Yakult, and Fresenius Kobi and is a consultant/advisory board member of Sanofi, Bayer, and Jennerex. A.P. Venook has commercial research grants from Genentech/Roche and is a consultant/advisory board member of Genentech/Roche. H.I. Hurwitz has commercial research grants from F Hoffman-La Roche, Amgen, Pfizer, Tracon Pharmaceuticals, Genentech, Sanofi, Morphotek, GSK, and Tracon. Also he is a consultant/advisory board member of GlaxoSmithKline, Novartis, Genetech, Roche, Sanofi, Regeneron, BMS, Bayer, and Tracon.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: A.B. Nixon, H. Pang, P.N. Friedman, H.L. Kindler, R.M. Goldberg, H.I. Hurwitz
Development of methodology: A.B. Nixon, H. Pang, M.D. Starr, R.M. Goldberg, H.I. Hurwitz
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): A.B. Nixon, M.D. Starr, P.N. Friedman, M.M. Bertagnolli, H.L. Kindler, R.M. Goldberg, A.P Venook, H.I. Hurwitz
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): A.B. Nixon, H. Pang, M.M. Bertagnolli, H. L. Kindler, R.M. Goldberg, H.I. Hurwitz
Writing, review, and/or revision of the manuscript:A.B. Nixon, H. Pang, M.D. Starr, P.N. Friedman, M.M. Bertagnolli, H.L. Kindler, R.M. Goldberg, H. I. Hurwitz
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): M.D. Starr, R.M. Goldberg, A.P. Venook
Study supervision: H. Pang, M.M. Bertagnolli, H.I. Hurwitz
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Philip PA, Mooney M, Jaffe D, Eckhardt G, Moore M, Meropol N, et al. Consensus report of the national cancer institute clinical trials planning meeting on pancreas cancer treatment. J Clin Oncol. 2009;27:5660–5669. doi: 10.1200/JCO.2009.21.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Von Hoff DD, Ervin TJ, Arena FP, Chiorean EG, Infante JR, Moore MJ, et al. Randomized phase III study of weekly nab-paclitaxel plus gemcitabine versus gemcitabine alone in patients with metastatic adenocarcinoma of the pancreas (MPACT) J Clin Oncol. 2013;30(suppl 34) abstr LBA148. [Google Scholar]
- 5.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 6.Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the cancer and leukemia group B (CALGB 80303) J Clin Oncol. 2010;28:3617–3622. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 8.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 9.Escudier B, Bellmunt J, Negrier S, Bajetta E, Melichar B, Bracarda S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol. 2010;28:2144–2150. doi: 10.1200/JCO.2009.26.7849. [DOI] [PubMed] [Google Scholar]
- 10.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 11.Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kindler HL, Friberg G, Singh DA, Locker G, Nattam S, Kozloff M, et al. Phase II trial of bevacizumab plus gemcitabine in patients with advanced pancreatic cancer. J Clin Oncol. 2005;23:8033–8040. doi: 10.1200/JCO.2005.01.9661. [DOI] [PubMed] [Google Scholar]
- 13.Leng SX, McElhaney JE, Walston JD, Xie D, Fedarko NS, Kuchel GA. ELISA and multiplex technologies for cytokine measurement in inflammation and aging research. J Gerontol A Biol Sci Med Sci. 2008;63:879–884. doi: 10.1093/gerona/63.8.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toedter G, Hayden K, Wagner C, Brodmerkel C. Simultaneous detection of eight analytes in human serum by two commercially available platforms for multiplex cytokine analysis. Clin Vaccine Immunol. 2008;15:42–48. doi: 10.1128/CVI.00211-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lash GE, Scaife PJ, Innes BA, Otun HA, Robson SC, Searle RF, et al. Comparison of three multiplex cytokine analysis systems: Luminex, SearchLight and FAST Quant. J Immunol Methods. 2006;309:205–208. doi: 10.1016/j.jim.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 16.de Jager W, Rijkers GT. Solid-phase and bead-based cytokine immunoassay: a comparison. Methods. 2006;38:294–303. doi: 10.1016/j.ymeth.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Furnival GM, WIlson RW. Regression by leaps and bounds. Technometrics. 1974;16:499–511. [Google Scholar]
- 18.Storey JD. A direct approach to false discovery rates. J R Statist Soc B. 2002;64:479–498. [Google Scholar]
- 19.Ancrile BB, O’Hayer KM, Counter CM. Oncogenic ras-induced expression of cytokines: a new target of anti-cancer therapeutics. Mol Interv. 2008;8:22–27. doi: 10.1124/mi.8.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714–1719. doi: 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bayliss TJ, Smith JT, Schuster M, Dragnev KH, Rigas JR. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin Biol Ther. 2011;11:1663–1668. doi: 10.1517/14712598.2011.627850. [DOI] [PubMed] [Google Scholar]
- 22.Schuster M, Rigas JR, Orlov SV, Milovanovic B, Prabhash K, Smith JT, et al. ALD518, a humanized anti-IL-6 antibody, treats anemia in patients with advanced non-small cell lung cancer (NSCLC): results of a phase II, randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2010;28(suppl) abstr 7631. [Google Scholar]
- 23.Von Marschall Z, Scholz A, Stacker SA, Achen MG, Jackson DG, Alves F, et al. Vascular endothelial growth factor-D induces lymphangiogenesis and lymphatic metastasis in models of ductal pancreatic cancer. Int J Oncol. 2005;27:669–679. [PubMed] [Google Scholar]
- 24.Kurahara H, Takao S, Maemura K, Shinchi H, Natsugoe S, Aikou T. Impact of vascular endothelial growth factor-C and -D expression in human pancreatic cancer: its relationship to lymph node metastasis. Clin Cancer Res. 2004;10:8413–8420. doi: 10.1158/1078-0432.CCR-04-0379. [DOI] [PubMed] [Google Scholar]
- 25.Achen MG, Stacker SA. Molecular control of lymphatic metastasis. Ann N Y Acad Sci. 2008;1131:225–234. doi: 10.1196/annals.1413.020. [DOI] [PubMed] [Google Scholar]
- 26.Jayson GC, de Haas S, Delmar P, Miles DW, Shah MA, Van Cutsem E, et al. Evaluation of plasma VEGFA as a potential predictive pan-tumour biomarker for bevacizumab. 2011 European Multidisciplinary Cancer Congress. 2011 Presented September 24, 2011:Abstract 804. [Google Scholar]
- 27.Weickhardt AJ, Williams D, Lee C, Simes J, Murone C, Wilson K, et al. Vascular endothelial growth factors (VEGF) and VEGF receptor expression as predictive biomarkers for benefit with bevacizumab in metastatic colorectal cancer (mCRC): Analysis of the phase III MAX study. J Clin Oncol. 2011;29(3531) suppl abstr. [Google Scholar]
- 28.Kopetz S, Hoff PM, Morris JS, Wolff RA, Eng C, Glover KY, et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J Clin Oncol. 2010;28:453–459. doi: 10.1200/JCO.2009.24.8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lieu CH, Tran HT, Fiorentino S, Jiang Z, Mao M, Overman MJ, et al. Relative impact of chemotherapy with or without bevacizumab on cytokines and angiogenic factors (CAFs) in metastatic colorectal cancer. J Clin Oncol. 2011;29(suppl 4) abstr 401. [Google Scholar]
- 30.Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10:165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- 31.Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferrara N. Role of myeloid cells in vascular endothelial growth factor-independent tumor angiogenesis. Curr Opin Hematol. 2010;17:219–224. doi: 10.1097/MOH.0b013e3283386660. [DOI] [PubMed] [Google Scholar]
- 33.De La Luz Sierra M, Yang F, Narazaki M, Salvucci O, Davis D, Yarchoan R, et al. Differential processing of stromal-derived factor-1alpha and stromal-derived factor-1β explains functional diversity. Blood. 2004;103:2452–2459. doi: 10.1182/blood-2003-08-2857. [DOI] [PubMed] [Google Scholar]
- 34.Miles DW, de Haas SL, Dirix L, Chan A, Pivot X, Tomczak P, et al. Plasma biomarker analyses in the AVADO phase III randomized study of First-Line Bevacizumab + Docetaxel in patients with human epidermal growth factor receptor (HER) 2-negative metastatic breast cancer. San Antonio Breast Cancer Symposium. 2010 December 8–12, 2010. [Google Scholar]
- 35.Van Cutsem E, de Haas S, Kang YK, Ohtsu A, Tebbutt NC, Ming Xu J, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a biomarker evaluation from the AVAGAST randomized phase III trial. J Clin Oncol. 2012;30:2119–2127. doi: 10.1200/JCO.2011.39.9824. [DOI] [PubMed] [Google Scholar]
- 36.Jayson GC, Hicklin DJ, Ellis LM. Antiangiogenic therapy—evolving view based on clinical trial results. Nat Rev Clin Oncol. 2012;9:297–303. doi: 10.1038/nrclinonc.2012.8. [DOI] [PubMed] [Google Scholar]
- 37.Tran HT, Liu Y, Zurita AJ, Lin Y, Baker-Neblett KL, Martin AM, et al. Prognostic or predictive plasma cytokines and angiogenic factors for patients treated with pazopanib for metastatic renal-cell cancer: a retrospective analysis of phase 2 and phase 3 trials. Lancet Oncol. 2012;13:827–837. doi: 10.1016/S1470-2045(12)70241-3. [DOI] [PubMed] [Google Scholar]
- 38.Nixon AB, Halabi S, Shterev I, Starr MD, Brady JC, Dutcher J, et al. Identification of predictive biomarkers of overall survival (OS) in patients (pts) with advanced renal cell carcinoma (RCC) treated with interferon alpha (I) +/− bevacizumab (B): results from CALGB 90206 (Alliance) J Clin Oncol. 2013;31(suppl) abstr 4520. [Google Scholar]
- 39.Ferrara N. Binding to the extracellular matrix and proteolytic processing: two key mechanisms regulating vascular endothelial growth factor action. Mol Biol Cell. 2010;21:687–690. doi: 10.1091/mbc.E09-07-0590. [DOI] [PMC free article] [PubMed] [Google Scholar]