

Abstract
Rationale
In cardiomyocytes from failing hearts, insufficient mitochondrial Ca2+ ([Ca2+]m) accumulation secondary to cytoplasmic Na+ overload decreases NAD(P)H/NAD(P)+ redox potential and increases oxidative stress when workload increases. These effects are abolished by enhancing [Ca2+]m with acute treatment with CGP-37157 (CGP), an inhibitor of the mitochondrial Na+/Ca2+ exchanger.
Objective
To determine if chronic CGP treatment mitigates contractile dysfunction and arrhythmias in an animal model of heart failure (HF) and sudden cardiac death (SCD).
Methods and Results
Here, we describe a novel guinea-pig HF/SCD model employing aortic constriction combined with daily β-adrenergic receptor stimulation (ACi) and show that chronic CGP treatment (ACi+CGP) attenuates cardiac hypertrophic remodeling, pulmonary edema, and interstitial fibrosis and prevents cardiac dysfunction and SCD. In the ACi group 4 weeks after pressure-overload, fractional shortening and the rate of left ventricular pressure development decreased by 36% and 32%, respectively, compared to sham-operated controls; in contrast, cardiac function was completely preserved in the ACi+CGP group. CGP treatment also significantly reduced the incidence of premature ventricular beats and prevented fatal episodes of ventricular fibrillation, but did not prevent QT prolongation. Without CGP treatment, mortality was 61% in the ACi group within 4 weeks of aortic constriction, while the death rate in the ACi+CGP group was not different from sham-operated animals.
Conclusions
The findings demonstrate the critical role played by altered mitochondrial Ca2+ dynamics in the development of HF and HF-associated SCD; moreover, they reveal a novel strategy for treating SCD and cardiac decompensation in HF.
Keywords: mitochondria, Na+/Ca2+ exchange, metabolism, NADH, reactive oxygen species, oxidative phosphorylation, Ca2+ regulation, CGP-37157, calcium regulation, oxidative stress
INTRODUCTION
The clinical syndrome of heart failure (HF), which can occur after acute myocardial ischemic injury or following longstanding chronic cardiovascular disease, is a progressive disease often involving a phase of compensatory hypertrophy followed by a transition to impaired contractility (decompensation) with systolic and/or diastolic dysfunction. While mortality in the later stages of the disease is most often attributed to impaired pump function, sudden cardiac death (SCD) is the leading cause of mortality in patients with mild to moderate HF, accounting for 35-50% of deaths1,2, prompting an increase in the use of implantable cardiac defibrillators (ICDs) as prophylactic devices3. The mechanisms underlying SCD, and the transition from compensated to decompensated HF, are incompletely understood, but neurohormonal abnormalities, ion channel/transporter changes, Ca2+ handling alterations, the activation of intra- and extra-cellular signaling pathways, and oxidative stress, have all been implicated in the pathophysiology4.
A limitation of energy production and/or substrate utilization is also thought to contribute to HF5-7 and several studies have reported HF-associated defects in mitochondrial function8-10. Mitochondria are not only the predominant source of ATP, upon which normal cardiac function depends, but also play a critical role in other biological processes, such as redox regulation, intermediary metabolism, cell signaling, and ion homeostasis. In the context of HF, the most detrimental consequences of mitochondrial dysfunction, either as the primary or as a secondary factor in the development of HF, are impaired bioenergetics and increased oxidative stress. Reactive oxygen species (ROS) have complex effects on the development of HF, playing a key role in cardiac remodeling by activating multiple signaling pathways involved in hypertrophic growth of cardiomyocytes, apoptosis, and proliferation of fibroblasts11. Increased oxidative stress also impairs cardiac function directly by modifying the proteins involved in excitation-contraction coupling12,13. Targeting mitochondria to improve bioenergetics and to maintain redox balance is emerging as a novel therapeutic strategy for HF14-16.
Upstream of oxidative stress, recent evidence has suggested that altered Na+ and Ca2+ gradients across the mitochondrial inner membrane may be a proximal factor in the imbalance of energy supply and demand17,18, as well as ROS balance19, in failing heart cells. The pathway of contractile and electrical dysfunction involves a vicious cycle whereby increased cytoplasmic Na+([Na+]c), along with impaired SR Ca2+ release, leads to blunted Ca2+ signaling to the mitochondria during increased work17-20. Consequently, the failure to stimulate Ca2+-dependent dehydrogenases of Krebs cycle21-25 results in the net oxidation of the matrix NADH pool. The decreased NADH/NAD+ redox potential, on the one hand, results in a deficiency of reducing equivalents that drive oxidative phosphorylation (OXPHOS) and ATP production, and, on the other hand, compromises the ability to scavenge ROS, owing to the interdependence of the NADH and NADPH redox pools20 (see schema in Online Figure I). Mitochondrial NADPH is crucial because it provides the reducing power to maintain the reduced glutathione (GSH), thioredoxin (Trx(SH)2) and glutaredoxin pools, all of which are vital for ROS scavenging and the prevention of protein thiol oxidative damage26.
Mitochondrial Ca2+ ([Ca2+]m) is determined by the balance of influx through the mitochondrial Ca2+ uniporter (MCU) and efflux through the mitochondrial Na+/Ca2+ exchanger (mNCE); hence, a change in either of these pathways can disrupt mitochondrial Ca2+ dynamics. The Km of mNCE for Na+ is ~5-10 mmol/L, falling within the range of [Na+]c levels in cardiomyocytes27. Therefore, the elevated [Na+]C consistently observed in HF can significantly accelerate the [Ca2+]m efflux rate. Studies of isolated heart mitochondria have shown that an increase of extra mitochondrial Na+ leads to decreased [Ca2+]m and compromises mitochondrial energetics, including oxidation of the NADH pool, decreased OXPHOS rate and ATP levels28,29. Similarly, elevated [Na+]c in isolated myocytes from failing guinea pig hearts19 (or ouabain-treated myocytes30) causes insufficient [Ca2+]m accumulation during increased work, net oxidation of NAD(P)H, and a massive increase of intracellular ROS31, ultimately resulting in abnormal Ca2+ regulation and arrhythmias30. The adverse effects of high [Na+]c are abolished by CGP-37157 (CGP), an inhibitor of mNCE19,30,31.
In the present study, we examine whether in vivo treatment with CGP prevents the development of HF and SCD using a novel guinea pig model of HF induced by left ventricular pressure overload combined with a daily β-adrenergic challenge. We show that partial inhibition of mNCE attenuates cardiac remodeling, preserves cardiac contractile function, and protects against HF- associated arrhythmias and SCD, significantly improving overall survival. The results highlight mNCE as a novel target for HF treatment.
METHODS
Methods are described in detail in the Online Supplement.
RESULTS
Daily β-adrenergic challenge accelerates the transition from LV hypertrophy to HF
A guinea pig HF model was developed by combining ascending aortic constriction (AC) with daily administration of the β-adrenergic agonist isoproterenol via i.p. injection (ACi). The rationale was to expose the pressure-overloaded heart to a period of increased work each day to simulate β-adrenergic stress. The isoproterenol dose was chosen so that it would have minimal effects on cardiac hypertrophic remodeling in the absence of AC. There was no evidence of cardiac hypertrophic remodeling in sham-operated animals exposed to the 4-week isoproterenol challenge (SHAMi) as compared with the sham-operated, untreated group (SHAM). However, in the presence of AC, the same isoproterenol treatment significantly exacerbated the decline in cardiac fractional shortening (FS) evoked by AC alone (Online Figure II). In ACi, FS decreased from 46.5±1.0% at week 0 (pre-banding) to 30.5±1.5% at week 4, whereas with AC alone, FS was not significantly decreased by week 4 (44.2±1.0% at week 0 versus 43.4±2.2% at week 4). Significant cardiac dysfunction in the AC group was observed only after 6 weeks of pressure overload (FS 38.4±1.2%), while at week 8, FS decreased to 29.3±3.3%, which was similar to the functional decline evident at week 4 in ACi (Online Figure II). Thus, the major effect of the daily isoproterenol challenge was to accelerate the transition from LV hypertrophy (LVH) to decompensated HF.
Inhibition of mNCErestores mitochondrial Ca2+ accumulation and prevents energetic mismatch and oxidative stress in myocytes from ACi hearts during a rapid increase in work
Impaired mitochondrial Ca2+ signaling secondary to cytoplasmic Na+ overload is a major cause of energy/redox imbalance in myocytes from pressure-overload induced failing hearts and interventions that raise [Ca2+]m above a required threshold prevent cellular ROS overload19,31. Therefore, we first examined whether the same defect was present in the ACi model and if acute CGP treatment could mitigate the problem in vitro. [Na+]c overload was also observed in the ACi model. [Na+]c in cardiomyocytes isolated from ACi heart was increased by 208% compared to that in myocytes from SHAMi hearts (4.8±1.2 mmol/L in Shami v.s. 14.8±1.1 mmol/L in ACi) (Online Figure III). [Ca2+]m dynamics, monitored with MityCam fluorescence, showed that mitochondria take up Ca2+ on a beat-to-beat basis (Fig. 1A). Upon stimulation (0.1 Hz), [Ca2+]m increased abruptly followed by decay during diastole. An increase of work from 0.1Hz to 1Hz stimulation resulted in a further increase in MityCam signal (1-F/F0) (Fig. 1A). Measurement of peak MityCam signal indicated that accumulation of [Ca2+]m during increased work was significantly less in myocytes from ACi hearts than SHAMi hearts, whereas 1μmol/L CGP treatment fully restored [Ca2+]m in the ACi myocytes (Fig. 1 A and B). Isolated cardiomyocytes were challenged with a rapid increase in work from the resting state to 4 Hz stimulation in the presence of 100 nmol/L isoproterenol. NAD(P)H autofluorescence was strongly oxidized during 4Hz-stimulation in ACi cardiomyocytes (Fig. 1A). NAD(P)H decreased from 67%±4.4 reduced at rest to 33%±5.7 at the end of the stimulation period, while in SHAMi myocytes subjected to the same protocol, NAD(P)H levels were well maintained (67%±5.1 reduced at pre-stimulation vs. 66%±4.8 at the end of stimulation). Notably, the oxidation of NAD(P)H in the ACi myocytes was prevented by treatment with 1μmol/L CGP-37157 (68%±5.5 reduced at pre-stimulation vs. 64%±9.1 at the end of stimulation) (Fig. 1A). CGP had no effect on the sarcolemmal Na+-Ca2+ exchange current at concentrations up to 10 μM (Online Figure IV A), confirming its specificity for the mitochondrial Na+-Ca2+ exchanger.
Figure 1. The effects of CGP-37157 on [Ca2+]m dynamics, NAD(P)H oxidation, and ROS production in myocytes isolated from ACi hearts.
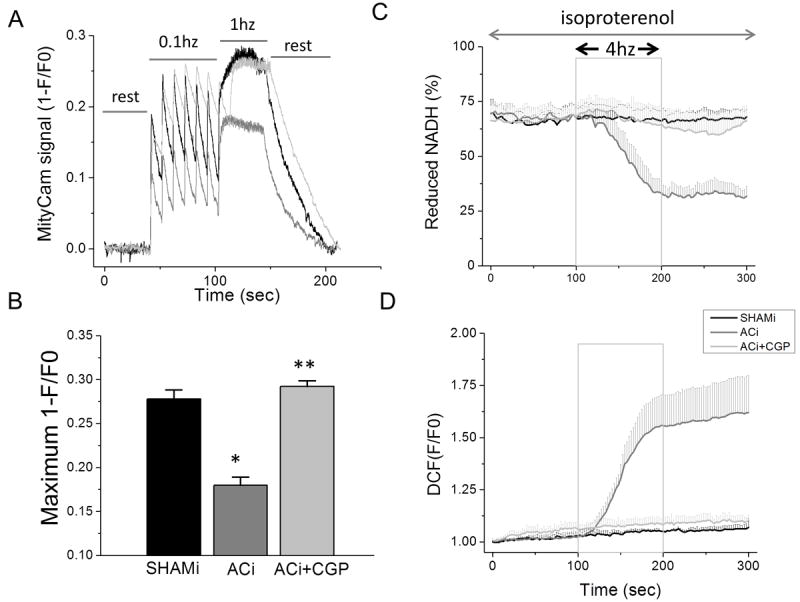
A) Representative recording of MityCam fluorescence (1-F/F0) in cardiomyocytes isolated from SHAMi (black) and ACi with (light gray) or without (gray) the presence of 1μmol/L CGP-37157 (CGP) in the perfusate. B) Mean peak MityCam fluorescence (1-F/F0) during 1Hz stimulation in SHAMi (n=30), ACi (n=29), and ACi + CGP (n=13). *, p<0.001 as compared to SHAMi; **, p<0.001 as compared to ACi. In the presence of 100nmol/L isoproterenol, NAD(P)H autofluorescence (A) and CM-DCF oxidation (B) were recorded in myocytes isolated from SHAMi hearts (n=3) or ACi hearts, with (n=3) or without (n=3) 1μmol/L CGP-37157 (CGP) in the perfusate. Box indicates duration of 4Hz field-stimulation.
The oxidation of the NAD(P)H pool during increased work in ACi cells was associated with increased oxidative stress. ROS accumulation was monitored with the H2O2-sensitive fluorescent probe 5-(and -6)-chloromethyl-2’,7’-dichlorodihydrofluorescein diacetate (CM-DCF). CM-DCF oxidation rate in SHAMi myocytes was low, with no significant change induced by the increased work (Fig. 1B). In contrast, ROS accumulation in ACi cells was markedly increased upon stimulation (Fig. 1B). During stimulation, the CM-DCF oxidation rate in ACi cells was ~10-fold higher than that in SHAMi cells. The enhanced intracellular ROS accumulation rate in ACi cells was abolished by CGP-37157 treatment (Fig. 1B).
Effects of chronic CGP-37157 treatment on ACi-induced cardiac remodeling and functional deterioration
Significant cardiac hypertrophy, pulmonary congestion, and interstitial fibrosis were present in the ACi group by the end of week 4. HW/TL was 53% higher in ACi (0.75±0.04 g/cm) versus SHAM (0.49±0.02 g/cm) and LW/TL was 66% higher in ACi (5.69±0.43 g/cm) versus SHAM (3.41±0.08 g/cm) (Fig. 2A). Histological analysis showed that interstitial fibrosis in ACi hearts was 12.5-fold higher than in SHAM (Fig. 2B). Isoproterenol challenge in the absence of pressure overload did not significantly increase fibrosis, although there was a trend towards more fibrosis in the SHAMi group relative to the SHAM controls (percent fibrotic tissue area was 0.53±0.2 in SHAM versus 1.1±0.5 in SHAMi, p=0.052) after 4-weeks of injections (Fig. 2B). CGP treatment protected against HF-associated cardiac remodeling: the ACi+CGP group developed less cardiac hypertrophy (HW/TL: 0.64±0.04 g/cm, p<0.05 versus ACi) and pulmonary congestion (LW/TL: 4.04±0.22 g/cm, p<0.001 versus ACi) at 4 weeks compared to the ACi group (Fig. 2A). CGP treatment also attenuated interstitial fibrosis, which was decreased by 50% in ACi+CGP group compared to that in ACi group (Fig. 2B).
Figure 2. The effects of CGP-37157 treatment on hypertrophic remodeling and cardiac function.
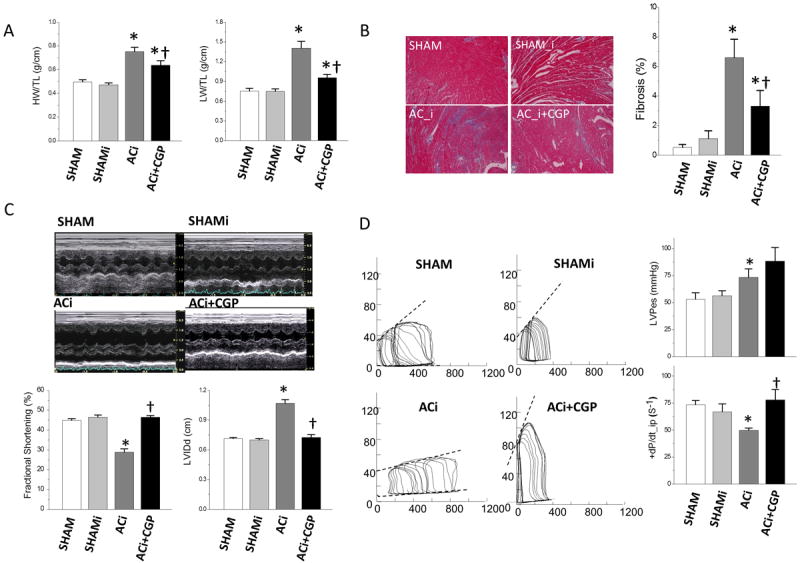
A) Heart weight, HW (left panel), and Lung weight, LW (right panel), were measured at 4 weeks after surgery and normalized to tibia length (TL). Isoproterenol challenge had no effects on either HW/TL or LW/TL in sham-operated animals (SHAMi vs. SHAM). Pressure overload with daily isoproterenol challenge (ACi) induced cardiac hypertrophy and lung congestion, and CGP-37157 treatment attenuated both cardiac hypertrophy and lung congestion (ACi+CGP). B) Interstitial fibrosis was analyzed with Masson’s Trichrome staining. Left panel: representative images from each experimental group; right panel: measurements of interstitial fibrous tissue showing that pressure overload, together with isoproterenol challenge, induced significant fibrosis in ACi group, which was attenuated by CGP-37157 treatment (ACi+CGP). C) Cardiac function is preserved by CGP-37157. Upper panel: representative pictures of echocardiographic study; lower panel: measurements of fractional shortening (FS) (left) and diastolic LV internal dimension (LVIDd) (right) showing reduced FS and LV dilation in ACi group but not in CGP-37157 treated group. D) Left panel: representative LV pressure-volume loops. Heart from ACi group shows increased end-diastolic volume and depressed end-systolic pressure-volume relation. Right panel: summary data of end-systolic LV pressure (upper panel) and dP/dt_ip (dP/dt normalized to pressure) (lower panel). Isoproterenol treatment in the absence of aortic constriction (SHAMi) had no effect on cardiac function.
Pressure overload with daily β-adrenergic challenge induced LVH and progressive cardiac dilation and impaired contractility within 4 weeks of surgery (Fig. 2C). Compared to SHAM, FS in the ACi group was decreased by 36% (28.9±1.6% in ACi vs. 44.9±0.9% in SHAM, p<0.001) and diastolic LV internal dimension (LVIDd) was increased by 51% (1.07±0.04 cm in ACi vs. 0.71±0.01 cm in SHAM, p<0.001) (Fig. 2C). Contractile impairment was also confirmed by in vivo hemodynamic studies: +dP/dt normalized to pressure (+dP/dt_ip) in the ACi group was decreased by 32% compared to that of SHAM controls (73.5±4.0 in SHAM vs. 49.9±2.1 in ACi, p<0.001) (Fig. 2D). In contrast, animals treated with CGP displayed well-maintained cardiac function without LV dilation. No significant differences in FS, LVIDd, or +dP/dt_ip between the SHAM and ACi+CGP groups were observed. β-adrenergic treatment alone (SHAMi vs. SHAM) had no effect on cardiac hemodynamic parameters.
Effects of β-adrenergic challenge, pressure overload and CGP-37157 on heart rate
Heart rate (HR) responses to the daily isoproterenol injection were recorded in a subset (3-6 guinea pigs) of animals from each experimental group by means of implanted biopotential telemetric transmitters. Baseline and maximal HR, determined by analysis of ECG acquired during and after isoproterenol injections, were compared at 1 and 4 weeks post-surgery (Online Figure V). Baseline HR was unaffected by chronic β-adrenergic treatment in the absence of pressure overload (SHAMi versus SHAM); however, the ACi group had higher baseline HR at weeks 1 and 4 after pressure overload compared to SHAMi controls (Online Figure V). Chronic treatment with CGP-37157 (ACi +CGP), administered continuously at dose of 0.015 mg/kg/hour by means of an implanted osmotic pump, attenuated, but did not completely eliminate, the increase of baseline HR at week 1 but abolished it at week 4 (baseline HR at week 4: 218.8±9.0 bpm in SHAMi vs. 206.2±7.5 bpm in ACi+CGP). Following injection of isoproterenol, HR rapidly increased to a maximum of ~390-400 bpm in all groups and then slowly declined back to the baseline rate in 4-5 hours (Online Figure V). The peaks and kinetics of the normalized chronotropic response to isoproterenol were not significantly different between the experimental groups.
Antiarrhythmic effects of CGP-37157
Telemetry studies revealed that animals in the ACi HF group had a high incidence of premature ventricular beats (PVB) (Fig. 3). PVB frequency was significantly decreased by CGP treatment. To evaluate the antiarrhythmic effects of CGP, PVB were counted in a 2 hour-period at baseline and a 4 hour-period following isoproterenol injection at weeks 1 (Fig. 3A) and 4 (Fig. 3B) after surgery. At baseline, occasional PVB were detected in all groups, but the incidence of PVB was very low in SHAM and SHAMi groups. Moreover, the rates did not change from week 1 to week 4 (week1: 3.3±2.0; week 4: 4.0±2.5 counts/hour). In contrast, at baseline, the ACi group had a much higher incidence of PVB (23.5±5.3 counts/hour in week 1) and the frequency increased as HF developed (51.3±10.1 count/hour in week 4). CGP treatment significantly reduced the baseline frequency of PVB as compared to ACi (ACi+CGP PVB frequency: 9.2±6.3 counts/hr in week 1 and 13.0±3.7 counts/hr in week 4). Isoproterenol increased PVB frequency in all groups. At week 1, the PVB frequency in SHAMi after isoproterenol (1 mg/kg) was 104.0±36.2 counts/hour, while in ACi, the PVB frequency increased to 388.4±35.1 counts/hour after isoproterenol injection (Figs. 3A and 3B, right panels). CGP treatment inhibited the arrhythmogenic effect of the β-adrenergic challenge: PVB frequency following isoproterenol injection in the ACi+CGP group was 219.8±31.2 count/hour, a decrease of 43% compared to the ACi group. Although, as mentioned above, the HR response to isoproterenol was similar in all of the groups, the increase in PVB frequency following the injection was much less in week 4 than in week 1 (30.6±8.1 counts/hr in SHAMi, 105.9±12.5 in ACi, and 32.5±8.0 in ACi+CGP) (Fig. 3A and 3B, right panels), even though the isoproterenol dose was higher (2mg/kg). Increased intrinsic sympathetic drive related to post-operative stress in week 1 or β-adrenergic receptor desensitization in week 4 might explain this difference.
Figure 3. The antiarrhythmic effect of CGP-37157.
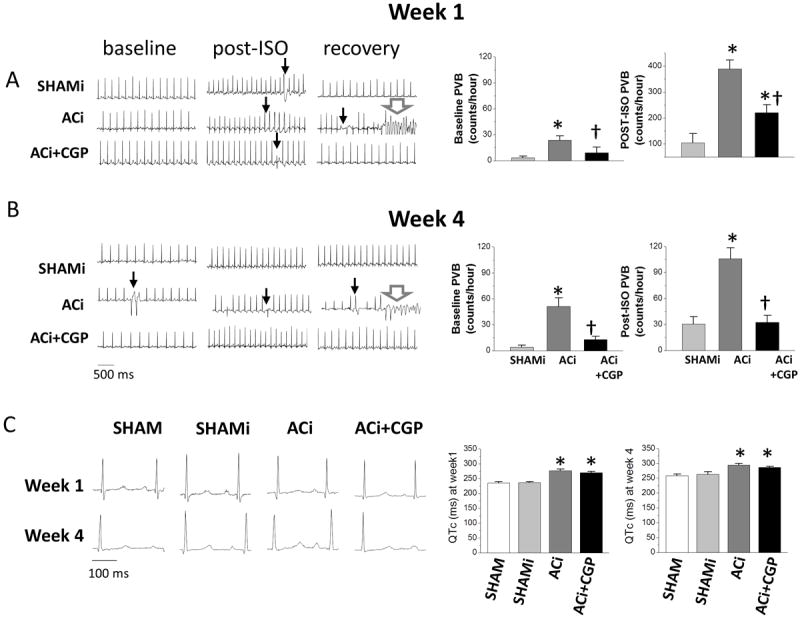
A and B) Left panels: Representative telemetric ECG recordings at week 1 (A) and week 4 (B) after aortic constriction showing baseline records before isoproterenol injection, records after isoproterenol injection (post-ISO), and during recovery 4 to 5 hours after isoproterenol injection. Black arrows indicate PVBs; white arrows indicate VF. Right panels: PVB incidence at baseline and after isoproterenol injection in week 1 and week 4. *, p<0.05 compared to SHAMi group, †, p<0.05 ACi+CGP compared to ACi group. C) Left panels: representative ECG recordings at baseline for each group at weeks 1 and 4; right panels: summary measurements of QTc at week 1 and week 4 showing that long QT was induced by pressure overload. Neither CGP-37157 treatment nor isoproterenol challenge had any effect on QTc.
The antiarrhythmic effect of CGP was greater in week 4, both at baseline and after isoproterenol injection (Fig. 3A and 3B). CGP treatment (comparing ACi+CGP with ACi) suppressed baseline PVB frequency by 60% in week 1 and 75% in week 4. In the contex of acute isoproterenol injection, CGP decreased PVB frequency by 43% in week 1 and 67% in week 4. Analysis of ECG data revealed prolonged QT intervals (QTc is corrected for HR) in both ACi and ACi+CGP groups (Fig. 3C). There was no difference in QTc between ACi and ACi+CGP groups at either week 1 or week 4, indicating that CGP treatment did not reverse QT prolongation in the HF model (Fig. 3C; right panels). QTc was also unaffected by the daily β-adrenergic challenge; there was no difference in QTc interval between SHAM and SHAMi groups.
Prevention of HF-associated SCD by CGP-37157 and overall mortality benefit
The ACi model was associated with a remarkably high incidence of SCD. 61.3% of ACi animals died by the end of 4 weeks (Fig. 4) without overt HF symptoms (e.g, cyanosis, labored breathing, inactivity, piloerection or cachexia). ECG analysis showed that death was preceded by sustained polymorphic ventricular tachycardia and/or fibrillation (VT/VF) (Fig. 3A and 3B; left panels). Interestingly, SCD did not occur during the peak of the HR response after isoproterenol injection, but usually happened several hours after the injection (5/6 SCD events occurred between 2-5 hrs post injection, 1 occurred 9 hrs post-injection), when the HR had recovered almost to baseline. CGP treatment (ACi+CGP) significantly reduced the 4-week death rate to just 14.3%, which was not significantly different from that of the SHAMi control (Fig. 4). Moreover, all of the deaths in CGP-treated group and in the SHAMi group occurred in the first week after surgery, perhaps attributable to the post-operative stress making the animals more susceptible to arrhythmias. Survival analysis of the ACi group indicated that the death rate was higher in week 1 (22.4%) and week 4 (26.9%) compared to that in week 2 (15.8%) and week 3 (18.8%). Similar to the other groups, the higher death rate in week 1 in ACi may reflect the overall effects of post-operative stress, along with a higher susceptibility to isoproterenol-induced arrhythmias. Considering that the PVB frequency following isoproterenol injection in week 4 is only 27% of that in week 1, the higher death rate (26.9%) in week 4 suggests that PVBs may be more life-threatening due to HF-associated changes in the arrhythmogenic substrate.
Figure 4. Survival curves over 4 weeks after the time of surgery for animals in SHAM, SHAMi, ACi, and ACi+CGP groups.
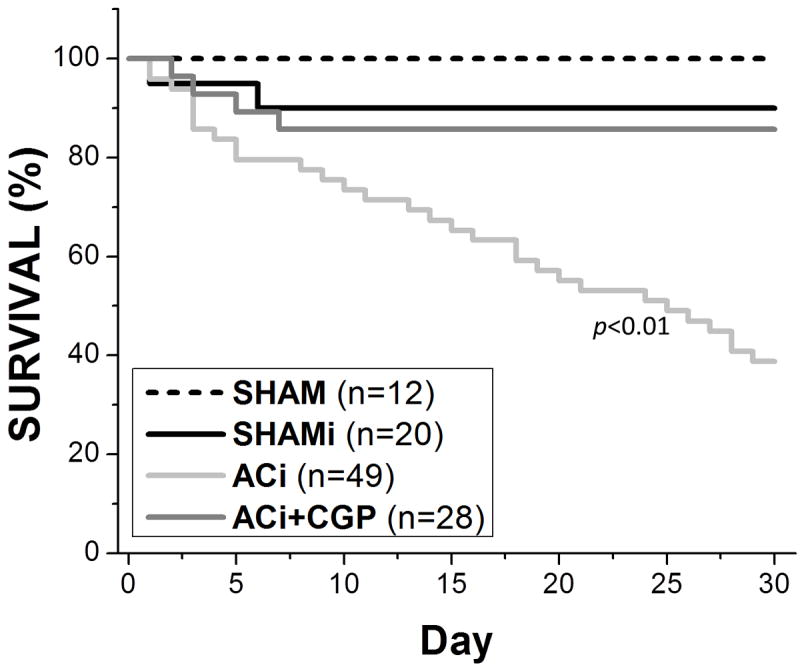
Survival of animals with pressure overload plus isoproterenol challenge was significantly increased by treatment with CGP-37157.
Antiarrhythmic effects of CGP-37157 on the isolated failing heart
We next determined if arrhythmia susceptibility and cardiac function in ACi failing hearts could be rescued by acute treatment with 1 μM CGP-37157 in isolated perfused hearts. Langendorff-perfused SHAMi and ACi hearts, isolated at week 4, were challenged with 10μmol/L isoproterenol for 15 min. Cardiac contractility and ECG were analyzed before (pre-ISO), during (ISO), and after (post-ISO) isoproterenol challenge. Baseline contractility before isoproterenol was significantly reduced in ACi hearts (Fig. 5A); LVDP, +dP/dt, and −dP/dt of ACi hearts were only 37%, 38%, and 36%, respectively, of values in SHAMi hearts (Table 1). Administration of isoproterenol increased cardiac contractility by a similar percentage in both ACi and SHAMi groups. However, ACi hearts were more sensitive to the stress of isoproterenol challenge; contractility of the ACi hearts was depressed more following isoproterenol washout. In the post-ISO state, LVDP, +dP/dt, and –dP/dt of ACi hearts were 12%, 16%, and 16% of values in SHAMi hearts (Table 1). Treatment with CGP-37157 had no statistically significant effects on cardiac contractility during pre-ISO or ISO states, but attenuated contractile dysfunction following isoproterenol treatment (Table 1). Treatment with CGP-37157 improved LVDP, +dP/dt, and −dP/dt recovery in the post-ISO state by 205%, 135%, and 120%, as compared to ACi hearts without CGP-37157 treatment (Table 1).
Figure 5. Effects of acute CGP-37157 treatment on isolated perfused failing hearts.
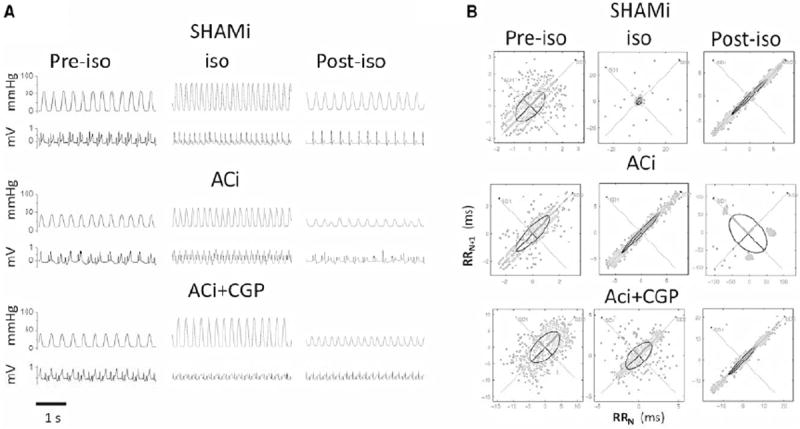
A) Representative traces of LV pressure and ECG simultaneously recorded before (pre-ISO), during (ISO), and after (post-ISO) isoproterenol (10μM) exposure in hearts of shami (n=3), ACi (n=3), and ACi with CGP-37157 (1μM) treatment (ACi+CGP) (n=3). B) Representative Poincaré plots of SHAMi, ACi, and ACi+CGP hearts during pre-ISO, ISO, and post-ISO states, respectively. Ectopic activity is evident as clustered of points only in the ACi heart post-ISO (middle right panel).
Table 1.
Measurements of cardiac contractility in isolated perfused heart.
| Shami | ACi | ACi+CGP | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Pre-iso | iso | Post-iso | Pre-iso | iso | Post-iso | Pre-iso | iso | Post-iso | |
| LVDP | 66.7±11.1 | 89.1±14.0 | 46.2±12.4 | 24.5±11.6 | 33.1±13.6* | 5.6±1.1* | 41.5±9.1 | 51.2±3.8* | 17.1±2.8*† |
| +dP/dt | 1255±137 | 3372±218 | 1114±160 | 483±198* | 988±373* | 181±36* | 759±122* | 1514±86* | 425±65*† |
| -dP/dt | -983±155 | -2247±352 | -927±354 | -355±139* | -730±323* | -151±33 | -563±102 | -938±101* | -330±48† |
p<0.05 when compared to contrl;
p<0.05 when compared to ACi.
Heart rate variability (HRV) analysis was applied to quantify changes in the ECG in the SHAMi and ACi hearts under the different experimental conditions. Treatment with 10μmol/L isoproterenol did not induce overt arrhythmias during the period of maximum LVPD in either group. There were no differences in RR interval, standard deviation of RR interval (SDRR), root mean square of successive difference (rMSSD), short term dispersion (SD1), or long term dispersion (SD2) between SHAMi, ACi, and ACi+CGP treatment groups during pre-ISO and ISO states (Table 2). However, following washout of isoproterenol, HRV of ACi hearts was dramatically increased compared to that of SHAMi hearts (clusters of points are indicative of ectopic activity in the Poincaré plots in Fig. 5B), whereas CGP treatment completely inhibited the arrhythmias in ACi hearts in the post-ISO state (Fig. 5B and Table 2). There were no differences in RR interval, SDRR, rMSSD, SD1, or SD2 between SHAMi and ACi+CGP treated hearts (Table 2).
Table 2.
HRV analysis. (SDNN, standard deviation of normal RR interval; RMSSD, root mean square of the successive differences)
| Shami | ACi | ACi+CGP | |
|---|---|---|---|
| Time domain Results | |||
| Mean RR (ms): | 260±18 | 336±25 | 307±36 |
| SDNN (ms): | 3.85±1.39 | 42.08±8.51* | 2.76±1.69† |
| RMSSD (ms): | 0.88±0.29 | 65.60±15.60* | 1.14±0.59† |
| Nonlinear Results | |||
| SD1 (ms): | 0.626±0.207 | 46.418±11.048* | 0.804±0.415† |
| SD2 (ms): | 5.394±1.968 | 36.542±6.167* | 3.812±2.347† |
p<0.05 when compared to contrl;
p<0.05 when compared to ACi.
DISCUSSION
The major findings of the present study were that: 1) the transition from compensated pressure-overload hypertrophy to HF is exacerbated by a daily β-adrenergic challenge; 2) SCD is a major contributor to mortality in the ACi HF model; 3) the mitochondrial Na+/Ca2+ exchange inhibitor, CGP-37157, prevents cardiac contractile decompensation, blunts hypertrophic remodeling, decreases arrhythmia incidence, and prevents SCD associated with HF.
The present study was motivated by our earlier findings that cytoplasmic [Na+]c overload, by increasing the driving force for mNCE-mediated Ca2+ extrusion from the mitochondrial matrix, results in insufficient [Ca2+]m accumulation during increased work17,19,30. If the Ca2+ signal to the mitochondria falls below a critical threshold level19, Ca2+-dependent activation of Krebs cycle enzymes is disrupted29, causing the mitochondrial pyridine nucleotide redox potential (NAD(P)H/NAD(P)+) to become more oxidized. The consequent decrease in reducing equivalents driving oxidative phosphorylation (NADH) and the antioxidant pathways (NADPH) causes both an impairment of energy supply and ROS overload (Fig. 1 C and D). This pathological mechanism was demonstrated in normal myocytes overloaded with [Na+]c17, myocytes treated with the Na+/K+ inhibitor ouabain30, and in myocytes isolated from failing (AC model) guinea pig hearts19. In the present study, we demonstrated that the net oxidation of NAD(P)H pool and increased oxidative stress during rapid pacing (4 Hz) in the presence of isoproterenol is also present in the ACi model. The increase in NAD(P)H oxidation, ROS accumulation, and arrhythmias associated with [Na+]c overload, can be prevented by either lowering [Na+]c19 or by inhibition of mNCE with CGP-3715719,30,31 (and the present work; Fig. 1C and D).
The findings are the first to demonstrate that chronic treatment with CGP-37157 prevents the progressive decline in contractile function and SCD in vivo in an animal model of HF. The ACi model was developed to incorporate both hemodynamic stress (left ventricular pressure overload) and chronic sympathetic hyperstimulation, two factors which are known to contribute to HF and SCD in humans32,33. Compared to pressure overload alone, the ACi model accelerated the decline in function and pathology of HF, while isoproterenol treatment in the absence of pressure overload (SHAMi) had no significant effects on cardiac function and remodeling in control animals.
A unique feature of the ACi model is the high incidence of SCD (~60% at 4 weeks), which is manifested as an increased death rate even in the first week post-aortic constriction, when function is largely preserved, and continues over the course of heart failure progression. Notably, the ACi model shows QTc prolongation even after only 1 week of pressure overload, prior to overt contractile dysfunction, as well as a large increase in spontaneous premature ventricular beats. The PVB rate and the incidence of VT/VF were both suppressed by CGP-37157 treatment; however, the QTc prolongation was not reversed, suggesting that the alterations in sarcolemmal ionic currents during hypertrophy, by themselves, are not sufficient to account for the increased automaticity and tendency towards reentry underlying SCD. Presumably, the combined effects of ion channel remodeling, oxidative stress, and the change in the myocardial substrate were all involved in setting the stage for VT/VF. In this light, it is still not clear if long QTc is a useful predictor of SCD in human HF. Breidthardt et al34 reported prolonged QTc interval (≥440ms) in 72% of patients with acute destabilized heart failure, but 720-day all-cause mortality was no different in patients with prolonged versus normal QTc intervals. Patients with prolonged QRS interval, however, had a significant 2-fold increase in mortality. Similarly, QTc was determined to have no independent prognostic value on 1 year mortality in a subset of patients with congestive heart failure in the DIAMOND trial35. In general, though, the overall risk of malignant arrhythmias increases with the magnitude of QTc prolongation36 and more refined analyses, such as QTc variability, may have predictive value for SCD37, perhaps as a better indicator of QT dispersion38.
The present results suggest that prolonged QTc may provide the substrate for potentially fatal arrhythmias, but that a secondary event - we propose an oxidative stress event secondary to an energy/redox supply and demand mismatch - would be required to initiate SCD. The non-linear nature of the secondary event could therefore account for the unpredictability of SCD.
Apart from genetically engineered animals39, the ACi model is, to our knowledge, one of the few animal models of HF-associated acquired long QT that displays a high incidence of spontaneous SCD. The accelerated transition between the compensated and decompensated states should be useful for investigating the mechanisms behind progressive systolic and diastolic dysfunction, as well as vulnerability to arrhythmias, in future studies.
Altered β-adrenergic receptor (β-AR) signaling is known to be a key player in the development of HF40,41. In addition to impairment of the inotropic, chronotropic and lusitropic effects of β-AR stimulation in HF, chronic hyperactivation of the receptor results in receptor desensitization, detrimental effects on cardiac function, and structural remodeling, which can be mitigated by β-blockade. The impact of selective β-AR signal activation on the heart depends on the dose and duration of β-AR agonist administration42. In our study, the chronic effects of isoproterenol on cardiac remodeling were minimized by selecting an isoproterenol dose that had no independent hypertrophic effects in sham operated animals and by administrating isoproterenol with repeated injections that induced only transient activation of β-AR signaling. The β-AR challenge had detrimental functional effects (arrhythmias and impaired post-ISO contractility) on LV pressure-overloaded animals, but not on controls, suggesting that the pressure-overloaded heart is more vulnerable to β-AR activation. This is consistent other animal studies. For example, in spontaneously hypertensive rats with LVH, infusion of isoproterenol initiated a transition from compensated hypertrophy to heart failure, whereas the same infusion had no significant effects on cardiac function and remodeling in control rats43. Genetically-modified mouse lines that have constitutively increased β-AR drive with normal cardiac function and structure are also more susceptible to pressure overload44,45. Our finding that CGP-37157 treatment protects pressure overloaded hearts (both in vivo and ex vivo) from the detrimental effects of β-AR challenge supports the hypothesis that blunted [Ca2+]m accumulation leading to oxidative stress is a key event underlying increased vulnerability of the failing heart to β-AR-mediated cardiac dysfunction.
In addition to exacerbating contractile dysfunction, β-AR challenge also predisposed the pressure overloaded heart to VT/VF, and CGP-37157 protected ACi animals from both high PVB rate and SCD. Increased sympathetic activity during physical or emotional stress is known to provoke VT/VF in catecholaminergic polymorphic ventricular tachycardia (CPVT), and elevated diastolic Ca2+ and hyperactive ryanodine receptors (RyR) are thought to be critical factors leading to SCD in CPVT animal models46,47. β-AR stimulation can contribute to arrhythmias by increasing Ca2+ current and increasing in sarcoplasmic reticulum Ca2+ ATPase (SERCA) activity, both of which increase SR Ca2+ load to increase vulnerability to spontaneous Ca2+ release. In HF, although the Ca2+ transient amplitude and SR load is decreased, the prolonged duration of the AP and Ca2+ transient, together with increased Ca2+ leak via the RyR, could favor spontaneous after depolarizations. For example, DeSantiago et. al.48 demonstrated that increased SR Ca2+ by β2-AR activation results in enhanced arrhythmogenesis in myocytes from human and rabbit failing hearts. Interestingly, in the ACi model, the pro-arrhythmic effect of β-AR challenge was not evident during the peak of the response, but was revealed only after recovery from the increased work. We propose that CGP-37157 is protecting against damage induced by mitochondrial ROS accumulation by preserving the pyridine nucleotide/antioxidant capacity during β-AR activation. Among the potential ROS targets, the local oxidation of the RyR by mitochondrial ROS49 could underlie spontaneous Ca2+ release and ectopic activity. RyR channels have multiple cysteine residues that sense the local redox state, and their oxidation is responsible for increased spontaneous SR Ca2+ leak in HF, which can be normalized by dithiothreitol13. In this light, we have previously demonstrated that CGP-37157, by increasing the mitochondrial Ca2+ response and preserving NAD(P)H redox potential during increased work, can prevent DADs triggered by ouabain-induced Na+ overload in myocytes, hearts, and intact animals30. This mechanism is the most likely explanation for the CGP-mediated protection against post-ISO ectopic activity observed in the isolated perfused ACi hearts (Fig. 5).
In addition to ROS-driven dysfunction, impaired mitochondrial energetics in HF also contributes to cardiac decompensation50. For example, limitations of ATP delivery could impair diastolic function by affecting the Ca2+ removal capacity via SERCA when energy demand increases during β-AR stimulation. This has been observed in studies of energetically-deficient animal models. In the creatine kinase deficient mouse, the reduced energy reserve leads to a decreased capacity of SERCA to remove cytosolic Ca2+ 51,52. A peroxisome proliferator-activated receptor gamma coactivator 1-β (PGC-1β) knockout mouse also displays reduced respiration rate and ATP production53; these animals showed increased susceptibility to catecholamine-induced arrhythmias54. Cardiomyocytes from these mice have normal Ca2+ transients at rest, but elevated [Ca2+]c and spontaneous diastolic Ca2+ transients were induced following isoproterenol administration54. By improving energy supply, CGP-37157 treatment could thus help to maintain the activity of SERCA or other energy-dependent processes during increased work to preserve systolic and diastolic function.
The present findings highlight the central role of oxidative stress in both the functional deterioration and arrhythmias associated with HF progression. In addition to direct damage to the myocyte, mounting evidence indicates that ROS modulate various signaling pathways involved in hypertrophic growth, apoptotic and necrotic cell death, and proliferation of cardiac fibroblasts11,55,56. ROS are also downstream mediators of neurohumoral stimuli implicated in HF, such as angiotensin II, endothelin 1, and tumor necrosis factor α. Increased ROS can also directly activate signaling cascades including tyrosine kinases, MAP kinases, PKC, and PI3-kinase57, which are potent regulators of cardiac growth. The effects of CGP on these downstream effectors of ROS signaling remain to be determined, but could also contribute to the observed protection. Nevertheless, given that the hypertrophic response was only partially decreased by CGP treatment, the major beneficial effect of CGP may be to prevent the redox modification of proteins, (e.g. ion channels, myofilament proteins, and mitochondrial enzymes) that lead to contractile dysfunction and deficient energetics. Recent studies confirm the preeminent role of mitochondrial ROS in HF, for example, a mitochondrially-targeted antioxidant peptide (SS-31) protects animals from Ang II-induced cardiac hypertrophy and fibrosis14 and overexpression of mitochondrial, but not cytosolic, catalase prevents changes in the proteome of mice subjected to pressure-overload-induced heart failure15. CGP treatment intervenes in the mechanism leading to the mitochondrial ROS overload, while preserving energy supply and demand balance, which may have advantages over strategies that simply attempt to increase the ROS scavenger capacity.
In summary, CGP-37157 preserves cardiac function, attenuates remodeling and fibrosis, and prevents SCD in the guinea pig heart failure and SCD model produced by pressure overload combined with β-AR stimulation. The beneficial effects of CGP-37157 can attributed to the effects of restored [Ca2+]m dynamics by CGP-37157 on mitochondrial energetics and redox balance. Additional studies will be needed to characterize all of the HF-associated modifications impacted by CGP-37157 treatment, including cellular redox status, ROS balance, the electrophysiology of the heart, excitation-contraction coupling, energy metabolism, and post-translational protein modifications. More work will also be required to determine if CGP treatment can reverse established left ventricular hypertrophic remodeling to prevent decompensation and SCD in this model. In addition, given the known variations in cytoplasmic Na+ in different species58, the efficacy of CGP may be different in other models of HF and in humans. Nevertheless, the findings support the hypothesis that [Ca2+]m dynamics plays a critical role in the development of HF and HF-associated SCD and implicate mNCE as a novel therapeutic target for HF.
Supplementary Material
Novelty and Significance.
What Is Known?
Sudden cardiac death (SCD) accounts for up to 50% of deaths in patients with heart failure (HF) and, in many cases, can be attributed to paroxysmal ventricular arrhythmias.
The mechanisms underlying SCD are unknown but ion channel remodeling, action potential prolongation, and abnormal cytoplasmic and mitochondrial Ca2+ handling have been implicated.
Energy metabolism is also known to be altered in HF but the links between impaired energy supply, oxidative stress, and contractile and electrical dysfunction are incompletely understood.
What New Information Does This Article Contribute?
A novel guinea-pig model of HF/SCD is described, combining pressure-overload and daily catecholamine challenge, which displays impaired contractility, long-QT, and a high incidence of SCD due to spontaneous ventricular fibrillation over a 4-8 week time span.
Treatment with an inhibitor of the mitochondrial Na+/Ca2+ exchanger (mNCE) prevents contractile decompensation and SCD in vivo but does not prevent QT prolongation.
mNCE inhibition prevents NAD(P)H oxidation and ROS overload during increased work in myocytes from failing hearts and decreases the incidence of arrhythmias after a catecholamine challenge in perfused failing hearts.
Impaired mitochondrial Ca2+ balance, and the consequent impairment of NAD(P)H availability, plays a key role in heart failure progression and vulnerability to fatal arrhythmias by compromising energy supply and antioxidant capacity in the failing heart.
Sudden cardiac death is a major cause of death in patients with heart failure, but the mechanisms underlying this increased vulnerability are poorly understood. In cardiomyocytes from failing hearts, it was previously shown that insufficient mitochondrial Ca2+ ([Ca2+]m) accumulation, secondary to cytoplasmic Na+ overload, decreases NAD(P)H/NAD(P)+ redox potential, increases oxidative stress, and disrupts Ca2+ handling when workload increases. Here, we show that chronic in vivo treatment with an inhibitor of the mitochondrial Na+/Ca2+ exchanger (CGP-37157) prevents the progression from compensated hypertrophy to heart failure and eliminates the high incidence of spontaneous SCD. The findings highlight the critical role played by mitochondrial Ca2+ dynamics in maintaining redox and energy balance in the failing heart and support the feasibility of targeting mitochondria for therapeutic intervention in heart failure.
Acknowledgments
SOURCES OF FUNDING
This work was supported by NIH grants R01HL101235, R01HL105216 and P01HL077180, and the Fondation Leducq Transatlantic Networks of Excellence Program.
Nonstandard Abbreviations and Acronyms
- HF
heart failure
- SCD
sudden cardiac death
- ICD
implantable cardiac defibrillator
- ROS
reactive oxygen species
- OXPHOS
oxidative phosphorylation
- GSH
reduced glutathione
- Trx(SH)2
reduced thioredoxin
- [Na+]c
cytoplasmic Na+
- [Ca2+]m
mitochondrial Ca2+
- MCU
mitochondrial Ca2+uniporter
- mNCE
mitochondrial Na+/Ca2+ exchanger
- CGP
CGP-37157 (7-Chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one)
- AC
ascending aortic constriction
- FS
fractional shortening
- LVH
left ventricular hypertrophy
- CM-DCF
5-(and -6)-chloromethyl-2’,7’-dichlorodihydrofluorescein
- LVIDd
diastolic LV internal dimention
- HR
heart rate
- PVB
premature ventricular beat
- HRV
HR variability
- SDRR
standard deviation of RR interval
- rMSSD
root mean square of successive difference
- SD1
short term dispersion
- SD2
long term dispersion
Footnotes
DISCLOSURES
None
References
- 1.Bigger JT., Jr Why patients with congestive heart failure die: arrhythmias and sudden cardiac death. Circulation. 1987;75:IV28–35. [PubMed] [Google Scholar]
- 2.Goldman S, Johnson G, Cohn JN, Cintron G, Smith R, Francis G. Mechanism of death in heart failure. The Vasodilator-Heart Failure Trials The V-HeFT VA Cooperative Studies Group. Circulation. 1993;87:VI24–31. [PubMed] [Google Scholar]
- 3.Abraham WT, Wagoner LE. Medical management of mild-to-moderate heart failure before the advent of beta blockers. Am J Med. 2001;110(Suppl 7A):47S–62S. doi: 10.1016/s0002-9343(98)00386-6. [DOI] [PubMed] [Google Scholar]
- 4.Mann DL, Bristow MR. Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation. 2005;111:2837–2849. doi: 10.1161/CIRCULATIONAHA.104.500546. [DOI] [PubMed] [Google Scholar]
- 5.Gupta A, Akki A, Wang Y, Leppo MK, Chacko VP, Foster DB, Caceres V, Shi S, Kirk JA, Su J, Lai S, Paolocci N, Steenbergen C, Gerstenblith G, Weiss RG. Creatine kinase-mediated improvement of function in failing mouse hearts provides causal evidence the failing heart is energy starved. J Clin Invest. 2012;122:291–302. doi: 10.1172/JCI57426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katz AM. Is the failing heart an energy-starved organ? J Card Fail. 1996;2:267–272. doi: 10.1016/s1071-9164(96)80012-1. [DOI] [PubMed] [Google Scholar]
- 7.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 8.Marin-Garcia J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev. 2008;13:137–150. doi: 10.1007/s10741-007-9079-1. [DOI] [PubMed] [Google Scholar]
- 9.Rosca MG, Hoppel CL. Mitochondria in heart failure. Cardiovasc Res. 2010;88:40–50. doi: 10.1093/cvr/cvq240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ventura-Clapier R, Garnier A, Veksler V, Joubert F. Bioenergetics of the failing heart. Biochim Biophys Acta. 2011;1813:1360–1372. doi: 10.1016/j.bbamcr.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007;93:903–907. doi: 10.1136/hrt.2005.068270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bayeva M, Ardehali H. Mitochondrial dysfunction and oxidative damage to sarcomeric proteins. Curr Hypertens Rep. 2010;12:426–432. doi: 10.1007/s11906-010-0149-8. [DOI] [PubMed] [Google Scholar]
- 13.Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Gyorke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ, Rabinovitch PS. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res. 2012;93:79–88. doi: 10.1093/cvr/cvr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maack C, Bohm M. Targeting mitochondrial oxidative stress in heart failure throttling the afterburner. J Am Coll Cardiol. 2011;58:83–86. doi: 10.1016/j.jacc.2011.01.032. [DOI] [PubMed] [Google Scholar]
- 17.Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–182. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maack C, O’Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. 2007;102:369–392. doi: 10.1007/s00395-007-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–288. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Rourke B, Maack C. The role of Na dysregulation in cardiac disease and how it impacts electrophysiology. Drug Discov Today Dis Models. 2007;4:207–217. doi: 10.1016/j.ddmod.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansford RG, Zorov D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol Cell Biochem. 1998;184:359–369. [PubMed] [Google Scholar]
- 22.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 23.Moreno-Sanchez R. Contribution of the translocator of adenine nucleotides and the ATP synthase to the control of oxidative phosphorylation and arsenylation in liver mitochondria. J Biol Chem. 1985;260:12554–12560. [PubMed] [Google Scholar]
- 24.Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol Cell Physiol. 2000;278:C423–435. doi: 10.1152/ajpcell.2000.278.2.C423. [DOI] [PubMed] [Google Scholar]
- 25.Denton RM, McCormack JG. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annual Review of Physiology. 1990;52:451–466. doi: 10.1146/annurev.ph.52.030190.002315. [DOI] [PubMed] [Google Scholar]
- 26.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 27.Murphy E, Eisner DA. Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res. 2009;104:292–303. doi: 10.1161/CIRCRESAHA.108.189050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Babsky A, Doliba N, Doliba N, Savchenko A, Wehrli S, Osbakken M. Na+ effects on mitochondrial respiration and oxidative phosphorylation in diabetic hearts. Exp Biol Med (Maywood) 2001;226:543–551. doi: 10.1177/153537020122600606. [DOI] [PubMed] [Google Scholar]
- 29.Cox DA, Matlib MA. A role for the mitochondrial Na(+)-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J Biol Chem. 1993;268:938–947. [PubMed] [Google Scholar]
- 30.Liu T, Brown DA, O’Rourke B. Role of mitochondrial dysfunction in cardiac glycoside toxicity. J Mol Cell Cardiol. 2010;49:728–736. doi: 10.1016/j.yjmcc.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O’Rourke B, Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hasking GJ, Esler MD, Jennings GL, Burton D, Johns JA, Korner PI. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation. 1986;73:615–621. doi: 10.1161/01.cir.73.4.615. [DOI] [PubMed] [Google Scholar]
- 33.Messerli FH, Ventura HO, Elizardi DJ, Dunn FG, Frohlich ED. Hypertension and sudden death. Increased ventricular ectopic activity in left ventricular hypertrophy. Am J Med. 1984;77:18–22. doi: 10.1016/0002-9343(84)90430-3. [DOI] [PubMed] [Google Scholar]
- 34.Breidthardt T, Christ M, Matti M, Schrafl D, Laule K, Noveanu M, Boldanova T, Klima T, Hochholzer W, Perruchoud AP, Mueller C. QRS and QTc interval prolongation in the prediction of long-term mortality of patients with acute destabilised heart failure. Heart. 2007;93:1093–1097. doi: 10.1136/hrt.2006.102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brendorp B, Elming H, Jun L, Kober L, Malik M, Jensen GB, Torp-Pedersen C, Group DS. Qtc interval as a guide to select those patients with congestive heart failure and reduced left ventricular systolic function who will benefit from antiarrhythmic treatment with dofetilide. Circulation. 2001;103:1422–1427. doi: 10.1161/01.cir.103.10.1422. [DOI] [PubMed] [Google Scholar]
- 36.Moss AJ. Measurement of the QT interval and the risk associated with QTc interval prolongation: a review. Am J Cardiol. 1993;72:23B–25B. doi: 10.1016/0002-9149(93)90036-c. [DOI] [PubMed] [Google Scholar]
- 37.Piccirillo G, Magri D, Matera S, Magnanti M, Torrini A, Pasquazzi E, Schifano E, Velitti S, Marigliano V, Quaglione R, Barilla F. QT variability strongly predicts sudden cardiac death in asymptomatic subjects with mild or moderate left ventricular systolic dysfunction: a prospective study. Eur Heart J. 2007;28:1344–1350. doi: 10.1093/eurheartj/ehl367. [DOI] [PubMed] [Google Scholar]
- 38.Antzelevitch C. Cardiac repolarization. The long and short of it. Europace. 2005;7(Suppl 2):3–9. doi: 10.1016/j.eupc.2005.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stengl M. Experimental models of spontaneous ventricular arrhythmias and of sudden cardiac death. Physiol Res. 2010;59(Suppl 1):S25–31. doi: 10.33549/physiolres.932001. [DOI] [PubMed] [Google Scholar]
- 40.Bristow MR, Shakar SF, Linseman JV, Lowes BD. Inotropes and beta-blockers: is there a need for new guidelines? J Card Fail. 2001;7:8–12. doi: 10.1054/jcaf.2001.26655. [DOI] [PubMed] [Google Scholar]
- 41.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 42.Osadchii OE. Cardiac hypertrophy induced by sustained beta-adrenoreceptor activation: pathophysiological aspects. Heart Fail Rev. 2007;12:66–86. doi: 10.1007/s10741-007-9007-4. [DOI] [PubMed] [Google Scholar]
- 43.Badenhorst D, Veliotes D, Maseko M, Tsotetsi OJ, Brooksbank R, Naidoo A, Woodiwiss AJ, Norton GR. Beta-adrenergic activation initiates chamber dilatation in concentric hypertrophy. Hypertension. 2003;41:499–504. doi: 10.1161/01.HYP.0000056601.29613.DD. [DOI] [PubMed] [Google Scholar]
- 44.Du XJ, Autelitano DJ, Dilley RJ, Wang B, Dart AM, Woodcock EA. beta(2)-adrenergic receptor overexpression exacerbates development of heart failure after aortic stenosis. Circulation. 2000;101:71–77. doi: 10.1161/01.cir.101.1.71. [DOI] [PubMed] [Google Scholar]
- 45.Grote-Wessels S, Baba HA, Boknik P, El-Armouche A, Fabritz L, Gillmann HJ, Kucerova D, Matus M, Muller FU, Neumann J, Schmitz M, Stumpel F, Theilmeier G, Wohlschlaeger J, Schmitz W, Kirchhefer U. Inhibition of protein phosphatase 1 by inhibitor-2 exacerbates progression of cardiac failure in a model with pressure overload. Cardiovasc Res. 2008;79:464–471. doi: 10.1093/cvr/cvn113. [DOI] [PubMed] [Google Scholar]
- 46.Viatchenko-Karpinski S, Terentyev D, Gyorke I, Terentyeva R, Volpe P, Priori SG, Napolitano C, Nori A, Williams SC, Gyorke S. Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ Res. 2004;94:471–477. doi: 10.1161/01.RES.0000115944.10681.EB. [DOI] [PubMed] [Google Scholar]
- 47.Wehrens XH, Marks AR. Altered function and regulation of cardiac ryanodine receptors in cardiac disease. Trends Biochem Sci. 2003;28:671–678. doi: 10.1016/j.tibs.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 48.Desantiago J, Ai X, Islam M, Acuna G, Ziolo MT, Bers DM, Pogwizd SM. Arrhythmogenic effects of beta2-adrenergic stimulation in the failing heart are attributable to enhanced sarcoplasmic reticulum Ca load. Circ Res. 2008;102:1389–1397. doi: 10.1161/CIRCRESAHA.107.169011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou L, Aon MA, Liu T, O’Rourke B. Dynamic modulation of Ca2+ sparks by mitochondrial oscillations in isolated guinea pig cardiomyocytes under oxidative stress. J Mol Cell Cardiol. 2011;51:632–639. doi: 10.1016/j.yjmcc.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci U S A. 2005;102:808–813. doi: 10.1073/pnas.0408962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crozatier B, Badoual T, Boehm E, Ennezat PV, Guenoun T, Su J, Veksler V, Hittinger L, Ventura-Clapier R. Role of creatine kinase in cardiac excitation-contraction coupling: studies in creatine kinase-deficient mice. FASEB J. 2002;16:653–660. doi: 10.1096/fj.01-0652com. [DOI] [PubMed] [Google Scholar]
- 52.Spindler M, Meyer K, Stromer H, Leupold A, Boehm E, Wagner H, Neubauer S. Creatine kinase-deficient hearts exhibit increased susceptibility to ischemia-reperfusion injury and impaired calcium homeostasis. Am J Physiol Heart Circ Physiol. 2004;287:H1039–1045. doi: 10.1152/ajpheart.01016.2003. [DOI] [PubMed] [Google Scholar]
- 53.Lelliott CJ, Medina-Gomez G, Petrovic N, Kis A, Feldmann HM, Bjursell M, Parker N, Curtis K, Campbell M, Hu P, Zhang D, Litwin SE, Zaha VG, Fountain KT, Boudina S, Jimenez-Linan M, Blount M, Lopez M, Meirhaeghe A, Bohlooly YM, Storlien L, Stromstedt M, Snaith M, Oresic M, Abel ED, Cannon B, Vidal-Puig A. Ablation of PGC-1beta results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 2006;4:e369. doi: 10.1371/journal.pbio.0040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gurung IS, Medina-Gomez G, Kis A, Baker M, Velagapudi V, Neogi SG, Campbell M, Rodriguez-Cuenca S, Lelliott C, McFarlane I, Oresic M, Grace AA, Vidal-Puig A, Huang CL. Deletion of the metabolic transcriptional coactivator PGC1beta induces cardiac arrhythmia. Cardiovasc Res. 2011;92:29–38. doi: 10.1093/cvr/cvr155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS. Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol. 2002;34:379–388. doi: 10.1006/jmcc.2002.1526. [DOI] [PubMed] [Google Scholar]
- 57.Sabri A, Hughie HH, Lucchesi PA. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid Redox Signal. 2003;5:731–740. doi: 10.1089/152308603770380034. [DOI] [PubMed] [Google Scholar]
- 58.Pieske B, Houser SR. [Na+]i handling in the failing human heart. Cardiovasc Res. 2003;57:874–886. doi: 10.1016/s0008-6363(02)00841-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.