

Abstract
Metastasis is the main cause of prostate cancer-associated deaths. While significant progress has been made in the treatment of primary tumors, efficient therapies that target the metastatic spread of prostate cancer are far from clinical reality. To efficiently treat cancer we need be able to impede its spread. Unfortunately, the majority of current therapeutics approved to treat metastatic cancer were originally selected based on their ability to inhibit primary tumor growth. This inherent flaw precludes these therapies from efficiently targeting the development of secondary metastatic lesions, a process that is distinct from that of primary tumor progression. In this review we will summarize the conceptual, cellular and molecular targets that should be considered to design effective anti-metastatic therapies.
Keywords: Metastasis, prostate cancer, molecular targets and biomarkers of metastasis, clinical trial design
Introduction
Metastatic dissemination of the primary tumor is the single major determinant of prostate cancer-associated death. While the five year survival for localized prostate cancer is close to 100%, the survival rate drops to 31.9% for patients with metastatic disease [1]. The introduction of PSA screening has enabled the early detection of many latent prostate cancers, and it is estimated that up to half of new prostate cancer diagnoses detect a tumor that was unlikely to surface clinically in the absence of PSA screening [2]. Despite this success, PSA levels fail to accurately predict prostate cancer metastatic spread resulting on the one hand, missed diagnosis of distant metastases or on the other, overtreatment of patients [2,3]. New therapies and diagnostic and prognostic biomarkers for metastatic disease could substantially improve patient treatment after a diagnosis of prostate cancer. Recent evidence suggests that low risk prostate cancer patients can benefit from “active surveillance” and novel biomarkers would significantly inform cancer intervention protocols. An ideal metastatic biomarker would be easily detectable throughout the entire patient observation period, and additionally serve as a therapeutic target. Active surveillance informed by prognostic biomarker(s) could spare an estimated ~40% of all prostate cancer patients from treatments with significant side effects that are unlikely to improve survival [2-4].
Several unique features of cancer metastasis must be taken into account before designing clinically relevant anti-metastasis therapies and biomarkers. 1) The biology of metastatic dissemination is drastically different from the biology of primary tumor progression. A successful therapeutic agent or biomarker that efficiently inhibits primary tumor growth will not necessarily block cancer metastasis. Therefore, when evaluating an anti-metastatic agent as a potential cancer therapy, its ability to block or delay the formation of new metastatic lesions must serve as a primary readout. Consequently, cancer patients selected for anti-metastatic clinical trials must be carefully selected. Ideal patients should have limited or no metastases at the time of selection, but have a high risk of developing metastatic disease [5,6]. 2) Metastasis can occur early during cancer progression [7-10], therefore in order to develop efficient cancer therapies it will be important to distinguish patients with high risk of metastatic disease from those who are at lower risk. This discrimination could provide a therapeutic window for high-risk patients to receive more aggressive metastasis targeting therapy, while treatment for low risk patients could focus predominantly on treatments targeting the primary tumor. 3) Metastasis is a multistep process that involves complex interactions of metastatic cancer cells with the host microenvironment [9,10]. At any one time, individual cancer cells may exist at all stages of the metastatic cascade and display different sensitivity to treatment. While the steps of metastatic dissemination have distinct features, they rely on overlapping cellular and molecular mechanisms. For this reason, efficient therapeutic agents should simultaneously block multiple steps in the metastatic cascade. For example, cancer cell motility is involved in both the initial (invasion, intravasation) and late steps of metastasis (extravasation, perivascular invasion) [5,11,12]. Furthermore, communication of the cancer cells with the host stroma is critical throughout the whole metastatic cascade,from early metastatic events (invasion and extravasation) to the establishment of the secondary metastatic niche [6,13-16]. 4) Some metastatic steps are rate-limiting and therefore represent more attractive targets then others. Cancer cells in a particular metastatic step(s) may be more accessible to specific treatment(s) and therefore represent ideal candidates for targeted intervention. For example, the initiation of metastatic lesion formation is a bottle-neck step of the metastatic cascade [17-19]. Successful establishment of secondary lesions requires the proper combination of local stromal factors, and the close interaction between the disseminated tumor and preexisting vasculature, making it an optimal target for anti-metastatic therapies [20,21]. Another potential target is the survival of the blood circulating tumor cells (CTCs). The survival of CTCs is dependent on complex interactions with host blood cells which are easily targeted by intravenous delivery strategies [22-24].
Here we discuss the molecular, cellular or conceptual determinants that could be effectively targeted to inhibit metastasis and explore their therapeutic potential to block specific metastatic steps. While the main focus of this review is the metastatic spread of prostate cancer, we will also discuss recent data on breast, pancreatic and other solid cancers, as the molecular mechanisms that control these cancers metastasis are largely overlapping.
Targetable determinants of cancer metastasis
Recent advances in quantitative metastasis assays and intravital imaging techniques has led to the identification of five distinct rate limiting steps of metastasis, and has allowed for the visualization of these key metastatic steps in living tissue [12,25-29]. These steps include: cancer cell survival in the circulation, arrest and extravasation in the small capillaries and formation of overt metastatic lesions [6,10]. While the regulation of these steps is complex, they are controlled only by a few conceptual determinants that can serve as potential therapeutic targets. We define these determinants as 1) Cell autonomous motility; 2) Soluble communication; 3) Cell-cell adhesion; 4) Cell-matrix adhesion (reviewed in [30]). We propose that each metastatic step can be characterized by a unique “heat map” of these determinants that drive cancer cells toward the establishment of overt metastatic lesions (Figure 1). Next, we will summarize the molecular processes that drive the key steps in metastatic progression, with an emphasis on the determinants that can serve as specific anti-metastatic therapeutic targets.
Figure 1.
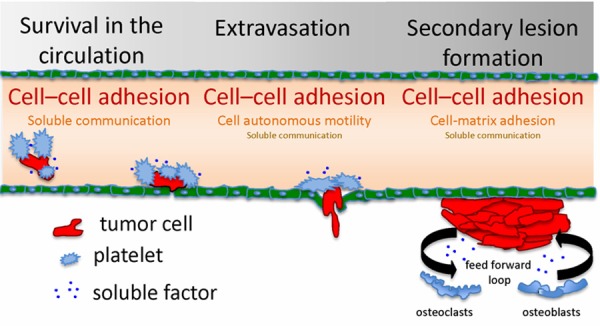
Key targets of the metastatic cascade. Metastatic tumor cells form cytoprotective complexes with host platelets or with each other, while traveling to secondary sites (bone) via the vascular system (Survival in the circulation). Circulating tumor cells arrest in blood vessel constrictions, and transmigrate through the endothelial layer with the assistance of platelets (Extravasation). Within the bone, tumor cells enter a pre-arranged cytoprotective niche assembled with the aid of local stromal cells and matrix bound/soluble factors. Tumor cells may remain dormant or establish proliferative secondary lesions (Secondary lesion formation). Each metastatic step has a unique set of targetable determinants, which are displayed as step specific “heat maps”.
Travel to the metastatic site/survival in circulation
Key targets: cell-cell adhesion; cell-matrix adhesion; soluble communication
The dissemination of metastatic cancer cells through the bloodstream is one of the least studied steps of metastasis. The transient nature of this metastatic step and intricate structure of the vascular system preclude its routine visualization in vivo. Additionally, in vitro models fail to fully recapitulate the cellular and molecular complexity of the bloodstream. Despite these challenges, our understanding of cellular processes involved in the transit of metastatic tumor cells in the bloodstream has significantly increased in the past few years. First, it was discovered that circulating tumor cells (CTCs) do not travel alone, but are typically accompanied by various blood and stromal cells. These interactions protect the cancerous cells from apoptosis, help to avoid immune system detection, and aid in the colonization of the secondary metastatic niche [22-24]. Indeed, it has been long known that cancer patients, especially those in the metastatic stage of disease, have higher risk of blood clots [31]. Recent data suggests that clots arise from circulating tumor cell aggregates that may include platelets, leukocytes or tumor derived fibroblasts. Second, it was demonstrated that the assembly of these complexes involves direct cell-cell or indirect cell-ECM-cell adhesion mechanisms that can serve as therapeutic targets [32-34]. Aggregate formation protects tumor cells from anoikis, the cytotoxic action of host immune cells and blood stream shear force [33]. Recent findings also indicate that co-traveling host cells may promote metastasis by directly enhancing the CTC’s ability to extravasate and establish secondary metastatic lesions [14,32,35]. One of the mechanisms by which platelets increase cancer metastasis is direct shielding of CTCs from cytotoxic NK cell action [14,36]. Additionally, tumor cell-bound platelets may signal to tumor cells in both an adhesion and soluble factor-mediated manner to promote their metastatic potential. For example, platelet-secreted TGFβ synergizes with direct tumor cell-platelet adhesion promoting tumor cell EMT transition, and results in increased lung colonization by colon and breast cancer cells [35]. Furthermore, tumor cell-associated host blood cells may induce local changes in intravascular endothelial adhesion receptor expression, thereby enhancing the tumor cell’s ability to arrest in the vasculature. Tumor cell-bound leukocytes induce endogenous L-selectin ligand expression in endothelial cells adjacent to the intravascular tumor cell emboli to promote tumor cell arrest and extravasation [33]. Tumor cell-associated platelets may also improve the chances of secondary lesion formation. Indeed, activated platelets secrete large amounts of VEGF that may aid extravasated tumor cells by promoting the permeability of the vessel wall and later inducing angiogenic vessel growth [24]. Recently, it has been shown that CTCs may promote their metastatic potential by traveling in clusters or bringing their own “soil”; carcinoma associated fibroblasts. In fact, tumor cells appear to associate with primary tumor derived fibroblasts not only while in the circulation but also within the newly formed metastatic lesions. This implies that the “fibroblast soil” not only promotes tumor cell survival in circulation but also in the metastatic lesion [37]. Suspension culture-induced clustering of Ewing sarcoma cells results in a dramatic upregulation of E-cadherin expression leading to robust activation of the pro-survival ERK/MAPK signaling axis [23]. The survival of CTCs can also be promoted through the upregulation of TrkB, a neurotrophic tyrosine kinase receptor. TrkB overexpression promotes PI3K/PKB pathway activation and the formation of large tumor cell aggregates that survive and proliferate while in the vasculature [38].
Cell-cell and cell-matrix adhesion appears to be main target to consider when designing strategies to target CTCs. Depending on the cancer type, formation of intravascular cell clusters can be mediated by direct or indirect adhesion mechanisms. Specifically, selectin, cadherin, integrin and galectin families of adhesion molecules are implicated in the formation of these cell clusters [23,32,34]. Multiple adhesion-blocking agents such as heparin or integrin blocking peptides/antibodies currently exist, with some of them clinically approved as blood clot inhibitors (see for review [32,34], Table 1). CTCs are a compelling target for anti-metastasis approaches due to the accessibility of CTCs to intravenously delivered therapeutics.
Table 1.
Metastatic step-specific anti-cancer therapeutics
| Target | Drug | Company | Metastatic steps targeted | Reference |
|---|---|---|---|---|
| MMP | Marimastat (BB-2516) | British Biotech | Extravasation | Trial NCT00003010 (Metastatic breast cancer) |
| Thrombin | Tinzaparin | Ottawa Hospital Research Institute | Survival in the circulation | Trial NCT01455831 (Metastatic colon cancer) |
| Extravasation | ||||
| Src | AZD0530 | AstraZeneca | Secondary tumor formation | Trial NCT01144481 (Risk factors for skeletal related events in breast cancer) |
| Extravasation | ||||
| EGFR | Herceptin (trastuzumab) | GlaxoSmithKline | Secondary tumor formation | Trial NCT01064349 (Breast cancer metastasis to the brain) |
| VEGFR | Dovitinib | Novartis | Extravasation | Trial NCT01262027 (Metastatic inflammatory breast cancer) |
| Secondary tumor formation | ||||
| α5β1 | Volociximab | Abbott | Extravasation | Trial NCT00100685 (Metastatic renal cell carcinoma) |
| Secondary tumor formation |
Numerous CTC capture and quantification techniques are being developed with the aim to use peripheral blood CTC counts as a quantitative prognostic determinant of risk for metastasis. For example, the CellSearch (Veridex LLC) assay, based on use of EpCAM antibodies conjugated to magnetic beads, has been recently FDA approved for clinical use [39]. It should be noted, however, that even in patients with aggressive metastatic disease, CTCs are often difficult to detect in sufficient numbers in clinically available blood volumes. Other predictive methodologies such as detection of circulating cancer cell exosomes (microparticles) can serve as a more robust approach to assess the tumor’s prognostic features and mutational landscape [40,41]. Circulating cancer microparticles in particular contain cancer-derived surface biomarkers and miRNAs, and are easy to isolate from minute amounts of patient blood [42]. Nevertheless, the detection and enumeration of CTCs is becoming routine procedure [43], which makes it feasible to address them directly as an anti-metastatic target.
Extravasation
Key targets: cell-cell adhesion; cell autonomous motility; soluble communication
During the course of dissemination, circulating tumor cells must adhere to the endothelium and then transit into the surrounding tissue through a process known as extravasation. Originally seen as a rapid and non-rate limiting process, tumor cell extravasation is gaining attention as a highly dynamic step in metastasis that may dictate the organ specificity of metastatic dissemination [44-47].
Tumor cell adhesion to the endothelium presents the first targetable step of extravasation. The initial arrest of tumor cells in the micro-capillaries appears to be relatively non-specific and dictated mainly by tumor cell size. The majority of CTCs that become entrapped within capillaries that are 8-10 μm in diameter, which is comparable with tumor cell diameter [27,32,48]. These entrapped cells can then migrate within the capillaries for extended periods of time using “amoeboid-like” mechanisms to move independently of the blood flow direction [27,48]. Despite being morphologically amoeboid, intravascular migration requires integrin expression by tumor cells [27]. Eventually, vessel-entrapped tumor cells attach closely to the inner side of the vascular wall using selectin, integrin or connexin-dependent mechanisms [27,32,49]. The selectin group of adhesion molecules are believed to be involved in tumor cell adhesion to the vasculature due to similarities between tumor cell extravasation and extravasation of immune cells [32]. Indeed, selectin ligands are often overexpressed by tumor cells and their expression correlates with poor outcomes. Tumor cells that express high levels of selectin ligands better attach to endothelium both in vitro and in vivo [50,51]. Moreover, vessel entrapped tumor cells can induce local upregulation of selectin expression, promoting their attachment to the vascular wall and subsequent extravasation [52]. Similar to selectins, integrin expression by tumor cells negatively correlates with patient prognosis in virtually all types of cancer [34]. While the role of integrin in cancer progression is mainly associated with tumor cell invasion and tumor induced angiogenesis, integrins can play a direct role in tumor cell adhesion to the intravascular endothelium. For example, metastatic breast cancer cells deficient in integrin β1 expression fail to attach and transmigrate through the vessel endothelium [27,53]. Furthermore, over-expression of neuropilin-2 non-kinase receptor (NPR-2) in cancer cells promotes successful attachment in an α5 integrin-dependent manner. Significantly, while NPR-2 expression by cancer cells positively correlates with poor prognosis, it is not required for primary tumor growth or invasion. These data suggest that NPR-2 is specifically involved in extravasation step of metastasis [54]. Recently, the connexin family of cell-cell adhesion and communication proteins has been implicated in circulating tumor cell attachment to the endothelium [49,55]. Clustering of connexin gap junctions was observed upon tumor cell arrest in lung capillaries. Moreover, tumor cell attachment to the endothelium markedly decreases when connexin-mediated cell-cell adhesion is compromised. Similarly, connexins are down-regulated within primary tumors and their over-expression has an inhibitory effect on primary tumor growth and invasion [49,54,55].
While tumor cells may exit the circulation through passive intravascular proliferation, the majority will rely on extravasation, involving the active transmigration of tumor cells through the endothelium and local remodeling of vascular wall structure [27,48,56]. Tumor cells that over-express pro-migratory genes extravasate significantly faster, suggesting that cell autonomous migration mechanisms are directly involved in trans-endothelial passage [27,57]. In addition, tumor cells induce a local angiogenesis-like response that promotes local remodeling and loosening of endothelial cell-cell contacts. For example, secretion of VEGF by extravasating cells induces local vascular leak that alleviates tumor cell escape from the vessel lumen [27,58]. Interestingly, tumor cell extravasation that is mediated through angiogenic factors may be, in part, distally controlled by the primary tumor. Specifically, TGFβ released by the primary tumor up-regulates the secretion of angiopoietin-related protein 4 (ANGPTL4) by metastatic tumor cells, which loosens endothelial cell-cell junctions and promotes tumor cell extravasation in the of lung capillaries [47]. The combination of both pro-adhesion/migratory and pro-angiogenic gene expression signatures may dictate the organ specificity of tumor cell extravasation. The ANGPTL4 or EREG/ COX2/MMP1 and 2 signature is associated with breast cancer metastasis to the lung, while the COX2/HBEGF/ST6GALNAC5 signature specifically mediates breast cancer cell extravasation in the brain [44-47]. Moreover, efficient cancer cell transmigration through the endothelial wall may require the formation of invadopodia since it is dependent on MMP activity. MMPs localize to the invasive edge of cancer cells undergoing extravasation in vitro and this can be blocked by tissue inhibitors of metalloproteinases (TIMPs), however the functional role of these processes in vivo is unclear [59].
Substantial evidence suggests that, similar to CTCs, extravasating tumor cells may co-opt host platelets and macrophages to aid them in their escape from the bloodstream. Adherence to platelets may enhance tumor cell arrest in the small capillaries due to passive (size restriction) and active mechanisms (selectin-mediated adhesion) [60]. Breast cancer cells extravasate in the lung with the help of a subpopulation of host CD11b+Gr1− macrophages that directly bind to lung arrested tumor cells and promote their transendothelial migration through a currently unknown mechanism [61].
Multiple selectin, integrin and connexin-blocking agents are available, however their effect on circulating tumor cell extravasation in the clinical setting remain to be evaluated [34,51,62,63]. Heparin has been widely used clinically to prevent blood clotting in cancer patients, however its clinical efficiency as a tumor cell extravasation inhibitor has not been evaluated [56,62]. Tumor cell transendothelial migration and/or tumor cell-blood cell communications represent important clinical targets as there are multiple therapeutic compounds presently available that regulate these processes (see Table 1).
Formation of secondary metastatic lesions
Key targets: cell-matrix adhesion; soluble communication; cell-cell adhesion
Cancer cells begin to disseminate from the primary tumor often long before they can be detected by conventional methods. However, only a minute fraction of these disseminating cells establish overt secondary lesions [10]. Formation of proliferative metastatic colonies is often seen as a rate-limiting step in the metastatic cascade, with only an estimated 0.01% of disseminated cancer cells able to complete this step. To successfully disseminate, tumor cells must pass several checkpoints, including the establishment of adhesive contacts with local vasculature and extracellular matrix, the initiation of feed-forward communication loops with local stromal and immune cells, and the induction of angiogenic responses from the adjacent vasculature [10,18].
A critical aspect of secondary lesion formation is the establishment of strong tumor cell contacts with the basal lamina. Metastatic cancer cells that invade away from the vasculature post-extravasation rapidly die over time [12], and those that successfully establish metastatic lesions maintain close contact with the vasculature [12,21]. Remarkably, metastatic cells can survive in close contact with the vasculature for extended periods of time without proliferating . This highlights the key role that vascular wall lamina adhesion has in the dormancy of metastatic tumor cells [12,64]. Secretion of the metastasis suppressor thrombospondin-1 (TSP1) by mature vasculature is associated with the dormant state of extravasated cancer cells, while even brief contact with growing, angiogenic vessel tips triggers cancer cell proliferation and leads to neovessel formation [65]. For their part, integrins are believed to be central players that control tumor cell adhesion to the vascular wall. Indeed, blocking integrin function significantly diminishes the formation of perivascular metastatic lesions and tumor cell invasion along the vasculature [21,66]. Activation of β1 integrin signaling is necessary for the transition from dormant tumor cells to proliferative metastatic lesions [67]. Moreover, engagement of integrin receptor-mediated adhesion is associated with the tumor cell’s ability to initiate an angiogenic switch at the secondary tumor sites [68]. In fact, the expression signature of integrin adhesion protein machinery in cancer cells can serve as a prognostic indicator of a cancer patient’s metastasis risk. For example, integrin-binding tetraspanin CD151 was shown to be required for efficient cancer cell invasion out of the primary tumor and formation of invasive metastatic lesions. Furthermore, a population of CD151 that is not bound to integrins (CD151free) may independently play a role in cancer cell invasion. Depletion of CD151free from the cancer cell surface blocks its migratory and metastatic potential. Correspondingly, histological detection of CD151free in prostate cancer tissue correlates with poor patient outcome and increased risk of metastasis [26,69]. Recently, the connexin group of cell-cell adhesion and communication proteins were implicated in the establishment of metastatic lesions. Connexin expression is elevated in perivascular regions of invasive metastatic lesions, implying a direct role in communications between tumor and endothelial cells [70,71]. Tumor cell adhesion to the extracellular matrix provides more than just engagement and activation of adhesion receptors. Recent findings indicate that metastatic tumor cell interaction with ECM component Tenascin C (TNC) specifically supports the survival of a stem cell-like population that promotes the formation of proliferative metastatic lesions [72]. Furthermore, the TNC-interacting ECM factor periostin was found to potentiate Wnt pathway signaling in metastatic stem cell-like breast cancer cells (CD24+Thy-1+). These signals promoted the survival of these disseminated cells and their ability to initiate proliferative metastatic lesions [20,73,74].
Soluble communications between the primary tumor and stromal cells within the metastatic niche play a crucial role in the formation of the metastatic lesion. For example, tumor-derived soluble factors can activate and direct bone marrow-derived VEGFR1+ progenitor cells to metastatic niche sites in an organ-specific manner [16,75]. Activated progenitor cells home to the metastatic niches in bone, lungs and other organs where they induce local ECM remodeling through the action of MMP9/LOX, preparing these sites for the arrival of tumor cells. Moreover, tumor-derived factors also stimulate fibroblasts to deposit excess fibronectin that promotes metastatic tumor cell adhesion [74,76]. Primary tumors may communicate with the metastatic niche not only by means of single soluble factors but also through the release of cancer microparticles. Prostate cancer microparticles activate fibroblasts in the metastatic niche through Erk1/2 phosphorylation and MMP-9 upregulation. Fibroblast activation also results in the shedding of fibroblast-derived microparticles, which in turn induce CX3CL1 dependent chemotaxis of prostate cancer cells [77].
To overcome the hostile environments of secondary sites, metastatic cancer cells harness local stromal and immune cells to promote their survival upon arrival in these secondary sites and to generate the metastatic niche. The best-studied examples of these interactions are the soluble and adhesive communications established between breast and prostate cancer cells within the bone metastatic niche stroma. Upon arrival to the bone metastatic niche, breast and prostate cancer cells begin to secrete an array of factors that induce differentiation and activation of bone tissue housekeeping cells-osteoclasts and osteoblasts. Abnormal osteoblast/osteoclast activity induces bone resorption leading to the release of growth factors (such as TGFβ) from the bone tissue. This provides a positive signal to tumor cell that promotes their continued proliferation [15,78,79]. In addition to soluble communications, disseminated cancer cells can also directly interact with bone stromal cells. First, dormant metastatic breast cancer cells in bone can recruit osteoclast progenitors and promote local osteoclast activity in a VCAM-1 adhesive-dependent mechanism. Engagement of tumor cell expressed VCAM-1 induces tumor cell proliferation and transition into an overt metastatic lesion [79].
The complex biology of the metastatic lesion formation step makes it one of the most promising therapeutic targets within the metastatic cascade. The involvement of tumor cell-matrix adhesion in the initiation and development of metastatic tumors is a key target within this step. Many agents that target tumor cell adhesion to the matrix are already showing promising results in preclinical and early clinical studies (also see Table 1 for examples of current therapeutics that may target metastatic tumor formation) [34]. Blocking soluble communication or tumor cell-stromal cell adhesion are also potential targets, with promise in preclinical studies [75,76]. Monitoring new metastatic lesion formation provides a direct readout for the efficacy of therapies that target this metastatic step.
Conclusions and future directions
By the time of diagnosis, many cancer patients will already have sub-clinical disseminated tumor cells. While targeting of the primary tumor can delay disease progression, many cancer patients will eventually succumb from the development of metastatic lesions. Despite this, the majority of current anti-cancer therapies target the proliferation and survival of primary tumors. Moreover, the current design of anti-cancer agent clinical trials specifically selects those treatments that target primary tumor development and largely ignores those that may block the metastatic spread of cancer. We believe that several major initiatives need to be taken both at preclinical and clinical levels to address this (Figure 2).
Figure 2.
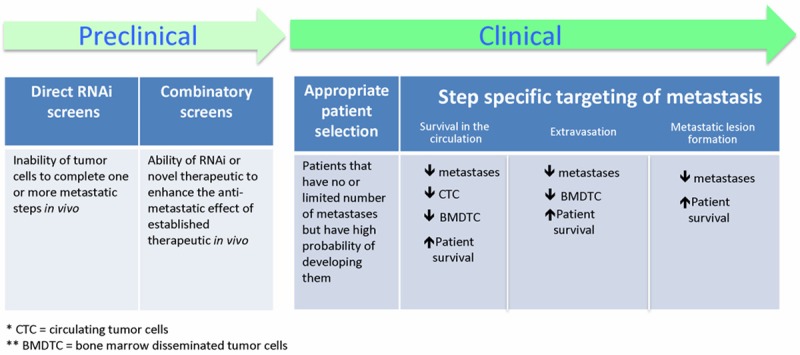
Steps towards anti-metastatic therapies. At the preclinical level, efforts should be directed towards the identification of novel anti-metastatic targets. At the clinical level, the current anti-cancer therapeutic clinical trial design should be altered to assure appropriate patient selection and proper step specific therapy readouts.
At the preclinical level, priority should be given to research that aims to identify and validate novel therapeutic targets that block cancer metastasis in vivo. With modern intravital imaging techniques, each step of the metastatic cascade can be quantitatively visualized in vivo [25,32,34,80]. Modern gene silencing techniques continue to allow researchers to conduct genome wide screens to identify the genes that control metastatic dissemination of cancer from the primary tumor [81-84]. There is a distinct opportunity for imaging-based siRNA screens using animal models of human cancer to discover novel, clinically relevant anti-metastasis targets. Additionally, the identification of anti-metastatic agents that complement currently approved anti-cancer agents must be a priority.
An inherent flaw in the current design of clinical trails is that they fail to identify novel anti-metastatic agents [6,85,86]. In order to evaluate these agents properly in the clinical trial setting, novel endpoints must be devised.Standard phase I-III clinical trail design selects for therapeutic agents that have low toxicity and can shrink or stabilize primary tumors. It is highly probable that therapeutic compounds that block tumor cell dissemination will have no effect on primary tumor growth, and therefore they are likely to be discarded early in development. Clearly, the selection of patients and endpoints for the evaluation of potential anti-metastatic agents must be drastically different from those that target primary tumor progression. Patients with aggressive primary tumors but no or limited numbers of distant metastases should be selected, perhaps on the basis of CTC or circulating cancer microparticle status. Second, endpoints for anti-metastatic clinical trials must be selected to properly assess the distinct steps of metastasis. We provide a list of potential metastasis step-specific readouts in Figure 2.
The advent of powerful high-throughput screening and clinical detection technologies has made this an ideal time to undertake a comprehensive search for novel anti-metastatic targets, to clinically validate existing metastasis step-specific therapies, and to integrate measures of tumor cell dissemination in the design and rationale of clinical trials in cancer. It has become wholly feasible to design and validate clinically effective anti-metastatic therapies, and this will be absolutely necessary if we are to make a tangible impact on this most deadly aspect of cancer.
Acknowledgements
Dr. Lewis holds the Frank and Carla Sojonky Chair in Prostate Cancer Research funded by the Alberta Cancer Foundation. Dr. Zijlstra and Ms. Hebron received funding from the National Institutes of Health (CA143081 and CA009592 respectively).
Disclosure of conflict of interest
None.
References
- 1.Schroder FH, Carter HB, Wolters T, van den Bergh RC, Gosselaar C, Bangma CH, Roobol MJ. Early detection of prostate cancer in 2007. Part 1: PSA and PSA kinetics. Eur Urol. 2008;53:468–477. doi: 10.1016/j.eururo.2007.10.047. [DOI] [PubMed] [Google Scholar]
- 2.Klotz L. Active surveillance: the Canadian experience. Curr Opin Urol. 2012;22:222–230. doi: 10.1097/MOU.0b013e328352598c. [DOI] [PubMed] [Google Scholar]
- 3.Carroll PR. Early stage prostate cancer--do we have a problem with over-detection, overtreatment or both? J Urol. 2005;173:1061–1062. doi: 10.1097/01.ju.0000156838.67623.10. [DOI] [PubMed] [Google Scholar]
- 4.Cooperberg MR, Lubeck DP, Meng MV, Mehta SS, Carroll PR. The changing face of low-risk prostate cancer: trends in clinical presentation and primary management. J. Clin. Oncol. 2004;22:2141–2149. doi: 10.1200/JCO.2004.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steeg PS, Camphausen KA, Smith QR. Brain metastases as preventive and therapeutic targets. Nature reviews. Cancer. 2011;11:352–363. doi: 10.1038/nrc3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steeg PS. Perspective: The right trials. Nature. 2012;485:S58–59. doi: 10.1038/485S58a. [DOI] [PubMed] [Google Scholar]
- 7.Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321:1841–1844. doi: 10.1126/science.1161621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmuller G, Klein CA. Systemic spread is an early step in breast cancer. Cancer cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nature reviews. Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 11.Comen E, Norton L, Massague J. Clinical implications of cancer self-seeding. Nat Rev Clin Oncol. 2011;8:369–377. doi: 10.1038/nrclinonc.2011.64. [DOI] [PubMed] [Google Scholar]
- 12.Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, Winkler F. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010;16:116–122. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- 13.Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 2009;69:9498–9506. doi: 10.1158/0008-5472.CAN-09-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576–590. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castellana D, Zobairi F, Martinez MC, Panaro MA, Mitolo V, Freyssinet JM, Kunzelmann C. Membrane microvesicles as actors in the establishment of a favorable prostatic tumoral niche: a role for activated fibroblasts and CX3CL1-CX3CR1 axis. Cancer Res. 2009;69:785–793. doi: 10.1158/0008-5472.CAN-08-1946. [DOI] [PubMed] [Google Scholar]
- 16.Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the ‘pre-metastatic niche’: within bone and beyond. Cancer Metastasis Rev. 2006;25:521–529. doi: 10.1007/s10555-006-9036-9. [DOI] [PubMed] [Google Scholar]
- 17.McGowan PM, Kirstein JM, Chambers AF. Micrometastatic disease and metastatic outgrowth: clinical issues and experimental approaches. Future Oncol. 2009;5:1083–1098. doi: 10.2217/fon.09.73. [DOI] [PubMed] [Google Scholar]
- 18.Chambers AF, Naumov GN, Varghese HJ, Nadkarni KV, MacDonald IC, Groom AC. Critical steps in hematogenous metastasis: an overview. Surg Oncol Clin N Am. 2001;10:243–255. vii. [PubMed] [Google Scholar]
- 19.Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153:865–873. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, Huelsken J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2012;481:85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 21.Carbonell WS, Ansorge O, Sibson N, Muschel R. The vascular basement membrane as “soil” in brain metastasis. PLoS One. 2009;4:e5857. doi: 10.1371/journal.pone.0005857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11:123–134. doi: 10.1038/nrc3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duda DG, Duyverman AM, Kohno M, Snuderl M, Steller EJ, Fukumura D, Jain RK. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci U S A. 2010;107:21677–21682. doi: 10.1073/pnas.1016234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nieswandt B, Hafner M, Echtenacher B, Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999;59:1295–1300. [PubMed] [Google Scholar]
- 25.Sahai E. Illuminating the metastatic process. Nature reviews. Cancer. 2007;7:737–749. doi: 10.1038/nrc2229. [DOI] [PubMed] [Google Scholar]
- 26.Beerling E, Ritsma L, Vrisekoop N, Derksen PW, van Rheenen J. Intravital microscopy: new insights into metastasis of tumors. J Cell Sci. 2011;124:299–310. doi: 10.1242/jcs.072728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zijlstra A, Lewis J, Degryse B, Stuhlmann H, Quigley JP. The inhibition of tumor cell intravasation and subsequent metastasis via regulation of in vivo tumor cell motility by the tetraspanin CD151. Cancer Cell. 2008;13:221–234. doi: 10.1016/j.ccr.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leong HS, Chambers AF, Lewis JD. Assessing cancer cell migration and metastatic growth in vivo in the chick embryo using fluorescence intravital imaging. Methods Mol Biol. 2012;872:1–14. doi: 10.1007/978-1-61779-797-2_1. [DOI] [PubMed] [Google Scholar]
- 29.Palmer TD, Lewis J, Zijlstra A. Quantitative analysis of cancer metastasis using an avian embryo model. J Vis Exp. 2011 doi: 10.3791/2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer TD, Ashby WJ, Lewis JD, Zijlstra A. Targeting tumor cell motility to prevent metastasis. Adv Drug Deliv Rev. 2011;63:568–581. doi: 10.1016/j.addr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, Klemke R. Visualizing extravasation dynamics of metastatic tumor cells. J Cell Sci. 2010;123:2332–2341. doi: 10.1242/jcs.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lyman GH, Khorana AA. Cancer, clots and consensus: new understanding of an old problem. J. Clin. Oncol. 2009;27:4821–4826. doi: 10.1200/JCO.2009.22.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konstantopoulos K, Thomas SN. Cancer cells in transit: the vascular interactions of tumor cells. Annu Rev Biomed Eng. 2009;11:177–202. doi: 10.1146/annurev-bioeng-061008-124949. [DOI] [PubMed] [Google Scholar]
- 34.Laubli H, Stevenson JL, Varki A, Varki NM, Borsig L. L-selectin facilitation of metastasis involves temporal induction of Fut7-dependent ligands at sites of tumor cell arrest. Cancer Res. 2006;66:1536–1542. doi: 10.1158/0008-5472.CAN-05-3121. [DOI] [PubMed] [Google Scholar]
- 35.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nature reviews. Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, Jirouskova M, Degen JL. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. 2005;105:178–185. doi: 10.1182/blood-2004-06-2272. [DOI] [PubMed] [Google Scholar]
- 37.Pinedo HM, Verheul HM, D’Amato RJ, Folkman J. Involvement of platelets in tumour angiogenesis? Lancet. 1998;352:1775–1777. doi: 10.1016/s0140-6736(98)05095-8. [DOI] [PubMed] [Google Scholar]
- 38.Zhong X, Rescorla FJ. Cell surface adhesion molecules and adhesion-initiated signaling: understanding of anoikis resistance mechanisms and therapeutic opportunities. Cell Signal. 2012;24:393–401. doi: 10.1016/j.cellsig.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 39.Miller MC, Doyle GV, Terstappen LW. Significance of Circulating Tumor Cells Detected by the CellSearch System in Patients with Metastatic Breast Colorectal and Prostate Cancer. J Oncol. 2010;2010:617421. doi: 10.1155/2010/617421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hosseini-Beheshti E, Pham S, Adomat H, Li N, Tomlinson Guns ES. Exosomes as biomarker enriched microvesicles: characterization of exosomal proteins derived from a panel of prostate cell lines with distinct AR phenotypes. Mol Cell Proteomics. 2012;11:863–885. doi: 10.1074/mcp.M111.014845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leong HS, Podor TJ, Manocha B, Lewis JD. Validation of flow cytometric detection of platelet microparticles and liposomes by atomic force microscopy. J Thromb Haemost. 2011;9:2466–2476. doi: 10.1111/j.1538-7836.2011.04528.x. [DOI] [PubMed] [Google Scholar]
- 42.Dijkstra S, Mulders PF, Schalken JA. Clinical use of novel urine and blood based prostate cancer biomarkers: A review. Clin Biochem. 2013 doi: 10.1016/j.clinbiochem.2013.10.023. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 43.Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430:1034–1039. doi: 10.1038/nature02765. [DOI] [PubMed] [Google Scholar]
- 44.Pantel K, Alix-Panabieres C. Circulating tumour cells in cancer patients: challenges and perspectives. Trends Mol Med. 2010;16:398–406. doi: 10.1016/j.molmed.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 45.Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, Massague J. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li F, Glinskii OV, Zhou J, Wilson LS, Barnes S, Anthony DC, Glinsky VV. Identification and analysis of signaling networks potentially involved in breast carcinoma metastasis to the brain. PLoS One. 2011;6:e21977. doi: 10.1371/journal.pone.0021977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, Gomis RR, Manova-Todorova K, Massague J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446:765–770. doi: 10.1038/nature05760. [DOI] [PubMed] [Google Scholar]
- 48.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, Massague J. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoffman RM. Real-time subcellular imaging in live animals: new visible targets for cancer drug discovery. IDrugs. 2006;9:632–635. [PubMed] [Google Scholar]
- 50.Elzarrad MK, Haroon A, Willecke K, Dobrowolski R, Gillespie MN, Al-Mehdi AB. Connexin-43 upregulation in micrometastases and tumor vasculature and its role in tumor cell attachment to pulmonary endothelium. BMC Med. 2008;6:20. doi: 10.1186/1741-7015-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sato M, Narita T, Kimura N, Zenita K, Hashimoto T, Manabe T, Kannagi R. The association of sialyl Lewis(a) antigen with the metastatic potential of human colon cancer cells. Anticancer Res. 1997;17:3505–3511. [PubMed] [Google Scholar]
- 52.Bendas G, Borsig L. Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol. 2012;2012:676731. doi: 10.1155/2012/676731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Auguste P, Fallavollita L, Wang N, Burnier J, Bikfalvi A, Brodt P. The host inflammatory response promotes liver metastasis by increasing tumor cell arrest and extravasation. Am J Pathol. 2007;170:1781–1792. doi: 10.2353/ajpath.2007.060886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pollmann MA, Shao Q, Laird DW, Sandig M. Connexin 43 mediated gap junctional communication enhances breast tumor cell diapedesis in culture. Breast Cancer Res. 2005;7:R522–534. doi: 10.1186/bcr1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klemke M, Weschenfelder T, Konstandin MH, Samstag Y. High affinity interaction of integrin alpha4beta1 (VLA-4) and vascular cell adhesion molecule 1 (VCAM-1) enhances migration of human melanoma cells across activated endothelial cell layers. J Cell Physiol. 2007;212:368–374. doi: 10.1002/jcp.21029. [DOI] [PubMed] [Google Scholar]
- 56.Stoletov K, Strnadel J, Zardouzian E, Momiyama M, Park FD, Kelber JA, Pizzo DP, Hoffman R, VandenBerg SR, Klemke RL. Role of connexins in metastatic breast cancer and melanoma brain colonization. J Cell Sci. 2013;126:904–913. doi: 10.1242/jcs.112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miles FL, Pruitt FL, van Golen KL, Cooper CR. Stepping out of the flow: capillary extravasation in cancer metastasis. Clin Exp Metastasis. 2008;25:305–324. doi: 10.1007/s10585-007-9098-2. [DOI] [PubMed] [Google Scholar]
- 58.Chen MB, Whisler JA, Jeon JS, Kamm RD. Mechanisms of tumor cell extravasation in an in vitro microvascular network platform. Integr Biol (Camb) 2013;5:1262–1271. doi: 10.1039/c3ib40149a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weis S, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol. 2004;167:223–229. doi: 10.1083/jcb.200408130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Voura EB, English JL, Yu HY, Ho AT, Subarsky P, Hill RP, Hojilla CV, Khokha R. Proteolysis during Tumor Cell Extravasation In Vitro: Metalloproteinase Involvement across Tumor Cell Types. PLoS One. 2013;8:e78413. doi: 10.1371/journal.pone.0078413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karpatkin S, Pearlstein E. Role of platelets in tumor cell metastases. Ann Intern Med. 1981;95:636–641. doi: 10.7326/0003-4819-95-5-636. [DOI] [PubMed] [Google Scholar]
- 62.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fritzsche J, Simonis D, Bendas G. Melanoma cell adhesion can be blocked by heparin in vitro: suggestion of VLA-4 as a novel target for antimetastatic approaches. Thromb Haemost. 2008;100:1166–1175. [PubMed] [Google Scholar]
- 64.Evans WH, Leybaert L. Mimetic peptides as blockers of connexin channel-facilitated intercellular communication. Cell Commun Adhes. 2007;14:265–273. doi: 10.1080/15419060801891034. [DOI] [PubMed] [Google Scholar]
- 65.Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nature reviews. Cancer. 2010;10:871–877. doi: 10.1038/nrc2933. [DOI] [PubMed] [Google Scholar]
- 66.Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DY, Chen EI, Lyden D, Bissell MJ. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15:807–817. doi: 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Piao Y, Lu L, de Groot J. AMPA receptors promote perivascular glioma invasion via beta1 integrin-dependent adhesion to the extracellular matrix. Neuro Oncol. 2009;11:260–273. doi: 10.1215/15228517-2008-094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barkan D, Chambers AF. beta1-integrin: a potential therapeutic target in the battle against cancer recurrence. Clin Cancer Res. 2011;17:7219–7223. doi: 10.1158/1078-0432.CCR-11-0642. [DOI] [PubMed] [Google Scholar]
- 69.Lorger M, Krueger JS, O’Neal M, Staflin K, Felding-Habermann B. Activation of tumor cell integrin alphavbeta3 controls angiogenesis and metastatic growth in the brain. Proc Natl Acad Sci U S A. 2009;106:10666–10671. doi: 10.1073/pnas.0903035106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Palmer TD, Martinez CH, Vasquez C, Hebron K, Jones-Paris C, Arnold SA, Chan SM, Chalasani V, Gomez-Lemus JA, Williams AK, Chin JL, Giannico GA, Ketova T, Lewis JD, Zijlstra A. Integrin-free tetraspanin CD151 can inhibit tumor cell motility upon clustering and is a clinical indicator of prostate cancer progression. Cancer Res. 2014;74:173–87. doi: 10.1158/0008-5472.CAN-13-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chao Y, Wu Q, Acquafondata M, Dhir R, Wells A. Partial mesenchymal to epithelial reverting transition in breast and prostate cancer metastases. Cancer Microenviron. 2012;5:19–28. doi: 10.1007/s12307-011-0085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saito-Katsuragi M, Asada H, Niizeki H, Katoh F, Masuzawa M, Tsutsumi M, Kuniyasu H, Ito A, Nojima H, Miyagawa S. Role for connexin 26 in metastasis of human malignant melanoma: communication between melanoma and endothelial cells via connexin 26. Cancer. 2007;110:1162–1172. doi: 10.1002/cncr.22894. [DOI] [PubMed] [Google Scholar]
- 73.Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, Massague J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17:867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kii I, Nishiyama T, Li M, Matsumoto K, Saito M, Amizuka N, Kudo A. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J Biol Chem. 2010;285:2028–2039. doi: 10.1074/jbc.M109.051961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, Le QT, Giaccia AJ. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nature reviews. Cancer. 2011;11:411–425. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Joseph J, Shiozawa Y, Jung Y, Kim JK, Pedersen E, Mishra A, Zalucha JL, Wang J, Keller ET, Pienta KJ, Taichman RS. Disseminated prostate cancer cells can instruct hematopoietic stem and progenitor cells to regulate bone phenotype. Mol Cancer Res. 2012;10:282–292. doi: 10.1158/1541-7786.MCR-11-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, Yan J, Hua Y, Tiede BJ, Haffty BG, Pantel K, Massague J, Kang Y. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell. 2011;20:701–714. doi: 10.1016/j.ccr.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zijlstra A, Mellor R, Panzarella G, Aimes RT, Hooper JD, Marchenko ND, Quigley JP. A quantitative analysis of rate-limiting steps in the metastatic cascade using human-specific real-time polymerase chain reaction. Cancer Res. 2002;62:7083–7092. [PubMed] [Google Scholar]
- 82.Cronan MR, Nakamura K, Johnson NL, Granger DA, Cuevas BD, Wang JG, Mackman N, Scott JE, Dohlman HG, Johnson GL. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene. 2012;31:3889–3900. doi: 10.1038/onc.2011.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lara R, Mauri FA, Taylor H, Derua R, Shia A, Gray C, Nicols A, Shiner RJ, Schofield E, Bates PA, Waelkens E, Dallman M, Lamb J, Zicha D, Downward J, Seckl MJ, Pardo OE. An siRNA screen identifies RSK1 as a key modulator of lung cancer metastasis. Oncogene. 2011;30:3513–3521. doi: 10.1038/onc.2011.61. [DOI] [PubMed] [Google Scholar]
- 84.Simpson CD, Hurren R, Kasimer D, MacLean N, Eberhard Y, Ketela T, Moffat J, Schimmer AD. A genome wide shRNA screen identifies alpha/beta hydrolase domain containing 4 (ABHD4) as a novel regulator of anoikis resistance. Apoptosis. 2012;17:666–678. doi: 10.1007/s10495-012-0723-4. [DOI] [PubMed] [Google Scholar]
- 85.Gobeil S, Zhu X, Doillon CJ, Green MR. A genome-wide shRNA screen identifies GAS1 as a novel melanoma metastasis suppressor gene. Genes Dev. 2008;22:2932–2940. doi: 10.1101/gad.1714608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rubin EH, Gilliland DG. Drug development and clinical trials--the path to an approved cancer drug. Nat Rev Clin Oncol. 2012;9:215–222. doi: 10.1038/nrclinonc.2012.22. [DOI] [PubMed] [Google Scholar]