

Abstract
Transforming Growth Factor-β (TGF-β) regulates the reactive stroma microenvironment associated with most carcinomas and mediates expression of many stromal derived factors important for tumor progression, including FGF-2 and CTGF. TGF-β is over-expressed in most carcinomas, and FGF-2 action is important in tumor-induced angiogenesis. The signaling mechanisms of how TGF-β regulates FGF-2 expression in the reactive stroma microenvironment are not understood. Accordingly, we have assessed key signaling pathways that mediate TGF-β1-induced FGF-2 expression in prostate stromal fibroblasts and mouse embryo fibroblasts (MEFs) null for Smad2 and Smad3. TGF-β1 induced phosphorylation of Smad2, Smad3, p38 and ERK1/2 proteins in both control MEFs and prostate fibroblasts. Of these, Smad3, but not Smad2 was found to be required for TGF-β1 induction of FGF-2 expression in stromal cells. ChIP analysis revealed a Smad3/Smad4 complex was associated with the -1.9 to -2.3 kb upstream proximal promoter of the FGF-2 gene, further suggesting a Smad3-specific regulation. In addition, chemical inhibition of p38 or ERK1/2 MAPK activity also blocked TGF-β1-induced FGF-2 expression in a Smad3-independent manner. Conversely, inhibition of JNK signaling enhanced FGF-2 expression. Together, these data indicate that expression of FGF-2 in fibroblasts in the tumor stromal cell microenvironment is coordinately dependent on both intact Smad3 and MAP kinase signaling pathways. These pathways and key downstream mediators of TGF-β action in the tumor reactive stroma microenvironment, may evolve as putative targets for therapeutic intervention.
Keywords: Tumor microenvironment, TGF-β, signaling pathway, FGF-2, promoter regulation
Introduction
The stromal cell microenvironment is a key regulator of carcinoma progression, however, the biology of how reactive stromal cells influence tumor progression is not well understood. Several growth factors have emerged as regulators of the reactive stroma microenvironment. Among these, TGF-β is perhaps the most multifunctional, exhibiting both direct and indirect effects on carcinoma cells, cancer associated fibroblasts (CAFs), host immune response mechanisms, and angiogenesis [1,2]. TGF-β is overexpressed in most carcinomas and is associated with elevated metastatic potential. The pleiotropic actions of TGF-β regulates wound repair through its cytostatic, chemotactic and fibrotic induction of different cell populations. The fibrotic induction of stroma fibroblasts facilitates granulation tissue formation and tissue remodeling through ECM deposition and growth factor production [3]. Our previous studies have shown that TGF-β action in stromal cells promotes angiogenesis and prostate tumorigenesis in human prostate carcinoma cell/stromal cell recombination xenograft models [4-6]. The actions of TGF-β were mediated, in part, through induced expression of CTGF and FGF-2 in reactive stroma fibroblasts in the tumor microenvironment [6,7]. Elevated stromal expression of CTGF and FGF-2 induced angiogenesis in the tumor microenvironment and elevated rate of tumor growth. Little is understood about specific signaling pathways mediated by TGF-β action in reactive stroma fibroblasts, yet TGF-β action signaling in this compartment of cells appears to be a critical component of tumor progression. Accordingly, these pathways may represent targets of opportunity for novel therapeutic approaches.
FGF-2 is one of the most potent factors regulated by TGF-β in reactive stroma. In stromal tissues, FGF-2 functions as a paracrine and autocrine mitogen, regulates differentiation, induces migration of endothelial cells to promote angiogenesis during wound healing and tumorigenesis, and regulates general homeostasis [8-10]. In both breast and prostate cancer-associated reactive stroma, expression of FGF-2 and cognate receptors are significantly increased and this likely affects rate of tumor progression and metastasis [10-14]. What is not yet understood, are the specific pathways and interaction of these pathways that mediate TGF-β induced FGF-2 expression in the tumor microenvironment fibroblasts. In general, TGF-β action is mediated through phosphorylation of cytoplasmic R-Smads, which subsequently translocate to the nucleus and regulate gene expression by either binding DNA directly or by associating with DNA-binding transcription factors [15-18]. In addition to activation of Smad signaling, TGF-β1 can activate members of the MAPK pathways as well as other kinases. Upon stimulation by TGF-β, crosstalk between ERK1/2, p38, JNK1/2 and Smad pathways is cell type-specific [19-26] although potential crosstalk and integration of pathways in reactive stroma fibroblasts has not been addressed.
In this report we have evaluated critical signaling pathways in order to better understand the TGF-β/FGF-2 signaling axis in prostate cancer reactive stroma and to address specific mechanisms of TGF-β1-induced FGF-2 expression in stromal fibroblasts. Results here show that FGF-2 expression in fibroblasts is upregulated by TGF-β1 and that Smad3, but not Smad2 signaling is required. In addition, FGF-2 expression relied on intact p38 and ERK1/2 signaling, which was induced by TGF-β in a Smad-independent manner. These data show that TGF-β1 regulates FGF-2 via coordinate, yet independent Smad3/4 and MAPK pathways. Key integrated pathways may evolve as potential targets for therapeutic intervention in reactive stroma-related diseases including fibrosis and cancer.
Material and methods
Cell lines
The control prostate fibroblast cell line TβRIICT was derived from a C57B mouse with floxed TGF-β receptor II alleles as we have described previously [6] and cultured in BFS medium: DMEM (Invitrogen, Carlsbad, CA) supplemented with 5% fetal bovine serum (Hyclone, Logan, UT), 5% Nu Serum (BD Biosciences, Bedford, MA), 0.5 μg/ml testosterone, 5 μg/ml insulin, 100 units/ml penicillin and 100 μg/ml streptomycin (Sigma, St. Louis, MO). The Smad2 wild type control (S2WT), Smad2 null (S2KO), Smad3 wild type control (S3WT) and Smad3 null (S3KO) mouse embryo fibroblasts (MEFs) were generated as described previously [27,28]. These cells were cultured in 20% FBS in DMEM with 100 units/ml penicillin and 100 μg/ml streptomycin. All cell lines were monitored for mycoplasma using the MycoAltert Mycoplasma Detection Kit (Lonza Rockland, Inc., Rockland, ME). During the course of experimentation, the S2WT, S3WT, S2KO, and S3KO cell lines were discovered to be mycoplasma-positive. These cell lines were treated with Mycoplasma Removal Agent (MRA) (MP Biomedicals, Irvine, CA) following recommended procedure. Subsequent screening at regular intervals showed all cell lines were mycoplasma-free. Key experiments were repeated and results were verified using mycoplasma-negative cell lines.
Measurement of FGF-2 protein
To measure induction of FGF-2 protein by TGF-β1, the TβRIICT prostate fibroblasts, or S2WT, S2KO, S3WT and S3KO MEF cells were plated in 6-well plates in full serum for 24 hours, washed with PBS, and serum starved by replacing media with 0.5% FBS in DMEM for 24 hours. Cells were subsequently treated with 2.5 ng/ml (100 pM) porcine TGF-β1 (R&D Systems, Minneapolis, MN) or vehicle control for 24 hours and cell extracts were made in 250 μL RIPA buffer (150 mM NaCl, 50 mM Tris, 0.5% DOC, 0.1% SDS, 1% NP-40 plus protease inhibitors). For chemical inhibitor studies, cells were pretreated with either vehicle control (0.1% DMSO) or UO126 (10 μM), SP600125 (10 μM), or SB203580 (10 μM for prostate fibroblasts or 50 μM for MEFs) (each from Calbiochem, San Diego, CA) for 1-2 h prior to TGF-β1 treatment. FGF-2 protein in 100 μl of cell extracts made with RIPA buffer was measured by an FGF-2-specific ELISA according to the manufacturer’s protocol (R&D Systems). Replicate wells were trypsinized and cells counted using a hemacytometer. FGF-2 levels were standardized to relative cell number. The data was normalized to vehicle control levels and fold changes were determined.
Quantitative PCR
Transcriptional regulation of FGF-2 mRNA levels by TGF-β1 and MAPK proteins was assessed by quantitative PCR (qPCR) analysis as described previously [6]. TβRIICT prostate gland fibroblasts, and S3WT, S3KO, S2WT and S2KO MEFs were grown in 6-well plates in full serum to 70-80% confluence and media was changed to 0.5% FBS in DMEM for 24 hours. Cells were pretreated as described above with the MAPK inhibitors (10 μM for each) and then treated with 2.5 ng/ml (100 pM) porcine TGF-β1 or vehicle control alone for 3, 6, 9, 12, 18 or 24 hours and total RNA was extracted using the RNeasy Miniprep kit (Qiagen, Inc., Valencia, CA). cDNA was prepared and analyzed for FGF-2 and GAPDH levels using qPCR as described previously [6] and relative FGF-2 mRNA levels were determined using the ddCT method. All data was standardized to vehicle control levels.
Western blot
Phosphorylation of MAPK kinase proteins and Smads were evaluated by Western blot of cells treated with 2.5 ng/ml (100 pM) TGF-β1. Cells were seeded in 6-well plates, cultured to 70-80% confluence and serum starved in DMEM for 24 hours. Pretreatment with chemical inhibitors was performed 1 hour before TGF-β1 treatment and cell extracts were made with 400 μl SDS sample buffer containing 62.5 mM Tris-HCL (pH 6.8), 2% w/v SDS, 10% glycerol, 50 mM DTT, and 0.01% bromophenol blue after washing cells 2X with cold PBS. 20 μg of the protein extracts were loaded onto 8% acrylamide gels and transferred onto either nitrocellulose or PVDF membranes for Western blotting. Primary antibodies were as follows: phospho-Smad2 (Ser465/467) (#3101, Cell Signaling, Danvers, MA), Smad2 (#51-1300, Zymed), phospho-Smad3 (Ser433/435) (#9514, Cell Signaling), Smad3 (#51-1500, Zymed), phospho-P38MAPK (Thr180/Tyr182) (#506119, Calbiochem), P38MAPK (#P39520, BD Transduction Laboratory, San Jose, CA), phospho-ERK1 (#E23920, BD Transduction Laboratory), phospho-ERK1/2 (#9106, Cell Signaling), and ERK1/2 (#9102, Cell Signaling). Antibodies were used at recommended concentrations. Western analysis was detected by Western Lightning kit (PerkinElmer Life Science, Inc., Waltham, MA) or SuperSignal West Dura and Femto (Pierce, Rockford, IL) with Kodak Image Station 440. For some experiments, membranes were subsequently stripped for 30 minutes at 50°C in 65 mM Tris-HCl pH6.7, 100 mMβ-ME, 2% SDS and membranes were re-probed with other antibodies.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were carried out with chromatin prepared from Smad2 null MEFs treated with TGF-β1 (2.0 ng/ml, 80 pM) or its vehicle control as reported previously [29]. Briefly, 2.0 μg of anti-Smad4 (B8, Santa Cruz Biotechnology, Santa Cruz, CA) or anti-Smad2/3 (E20, Santa Cruz Biotechnology), and 35 U of chromatin at A260 were used for each ChIP assay. ChIP products were amplified in a PTC-200 Peltier Thermal Cycler by primers designed to the FGF-2 promoter using Primer Express software (Applied Biosystems, Carlsbad, CA) following the guidelines provided by Applied Biosystems and are as follows: (-3542 to -3356) 5’ATGGTGTCCTGTTTCCGAGTCTT and 5’TGTCTCGACTCTGCACATCATCTT; (-2999 to -2593): 5’CCCTCTGCATGCTACCTTACTTC and 5’GCATACACCGTGTTAACTGCCA; (-2353 to -2168): 5’TGTGCTCCTATTGGTAAACATGCA and 5’TCAGCCAGGGATTTGTGGAA; (-1511 to -1384): 5’GGATACAAAGCCCACATTTGAGA and 5’TTGAAGAGGGTGCCAGCTAATG.
Statistical analysis
Mean levels of protein production were compared in control vs. experimental conditions using the unpaired t test (two-tailed). Statistical analyses of relative RNA levels in control vs. experimental conditions over the indicated time course were evaluated using the 2-way ANOVA analysis. Statistical analyses were generated using Prism for Macintosh version 5.0 (GraphPad Software). P < 0.05 was considered statistically significant. Experiments were repeated a minimum of 3 times (independently) and measured in triplicates.
Results
Smad3 signaling is required for TGF-β1 induced FGF2 expression in fibroblasts
Our previous work has shown that TGF-β1 stimulates FGF-2 message and protein expression as well as FGF-2 protein secretion from prostate gland stromal cells [6]. The Smads critical for this regulation and component signaling pathways in fibroblasts are not well defined. To assess the specific role of Smad signaling, mouse embryo fibroblasts (MEFs) from Smad2 null mice and Smad3 null mice along with their individual intact Smad control MEFs [28] were evaluated. As shown in Figure 1A, both wild type Smad2 MEF controls and Smad2 null MEFs exhibited TGF-β1 induced expression of FGF-2 mRNA as assessed by quantitative PCR. In contrast, Smad3 wild type control MEFs exhibited a time-dependent induction of FGF-2 message by TGF-β1, whereas FGF-2 expression remained at near basal levels in Smad3 null MEFs as shown in Figure 1B. In control MEFs, FGF-2 message levels were highest at 9 hr. Figure 1C shows that TGF-β1 induced phosphorylation of both Smad2 and Smad3 through 4 hours of TGF-β1 treatment in control MEFs. As expected, Smad3 protein or phosphorylation was absent in Smad3 null MEFs; however, Smad2 phosphorylation remained intact in these cells. ELISA was used to assess FGF-2 protein induction. Figure 1D shows that Smad3 control MEFs exhibited a significant increase in FGF-2 protein over a 24 hr period, whereas this response was greatly attenuated in Smad3 null MEFs. Together, these data indicate that TGF-β1 induced expression of FGF-2 in fibroblasts is dependent on functional Smad3 signaling but not Smad2.
Figure 1.

Smad3 regulates FGF-2 expression in mouse embryo fibroblasts. A. TGF-β1-induced (100 pM) FGF-2 mRNA expression is equivalent in both Smad2 control MEFs and Smad2 null MEFs; B. A significant upregulation of FGF-2 mRNA by TGF-β1 (100 pM) is evident in Smad3 control MEFs with maximal expression at 9 hr, whereas FGF-2 message remains at near basal levels in TGF-β1 stimulated Smad3 null MEFs; C. TGF-β1 (100 pM) induces Smad2 and Smad3 phosphorylation in Smad3 wild type MEFs cells. In Smad3 null MEFs, Smad3 protein and phosphorylation is absent; however, Smad2 is phosphorylated by TGF-β1; D. A corresponding upregulation of total cellular FGF-2 protein levels by TGF-β1 (100 pM) at 24 hr is observed in Smad3 control MEFs; however, this effect is significantly attenuated in Smad3 null MEFs. All data were normalized to cell number and fold changes were determined relative to control. *P < 0.05, N=3 independent experiments in triplicate.
Smad3/4 interacts directly with the FGF-2 proximal promoter
To assess whether TGF-β1 induction involved direct interaction of Smad3/4 complexes and to pinpoint specific binding regions of the FGF-2 promoter, ChIP assays were conducted. Smad2 null MEFs were used to avoid Smad4 interactions with Smad2 and since they exhibited intact TGF-β1 induced FGF-2 expression. FGF-2 promoter fragments (200-800 bases, Figure 2B) were precipitated using Smad3 and Smad4 antibodies in TGF-β1 treated MEFs and interactions in the first 5 kb upstream of the start site were evaluated with PCR in precipitated material. As shown in Figure 2A, specific interactions were observed with both antibodies using primers directed to amplify the -2353 to -2168 region but not flanking regions (Figure 2A and 2C). These data were consistent with further in silico analysis of DNA sequence in that region. A cluster of 5 potential Smad binding motifs (-1862, -1994, -2143, -2198, and -2560) were found in this expanded region as shown in Figure 2C. Further evaluation using MatInspector by Genomatix confirmed these data, particularly at the -1994 position. Together, these data suggest that TGF-β1 regulates FGF-2 expression directly through interaction of Smad3/Smad4 binding complexes with the FGF-2 proximal promoter approximately 1.9-2.3 kb upstream of the start site, although other potential sites more distal than 5 kb cannot be ruled out.
Figure 2.
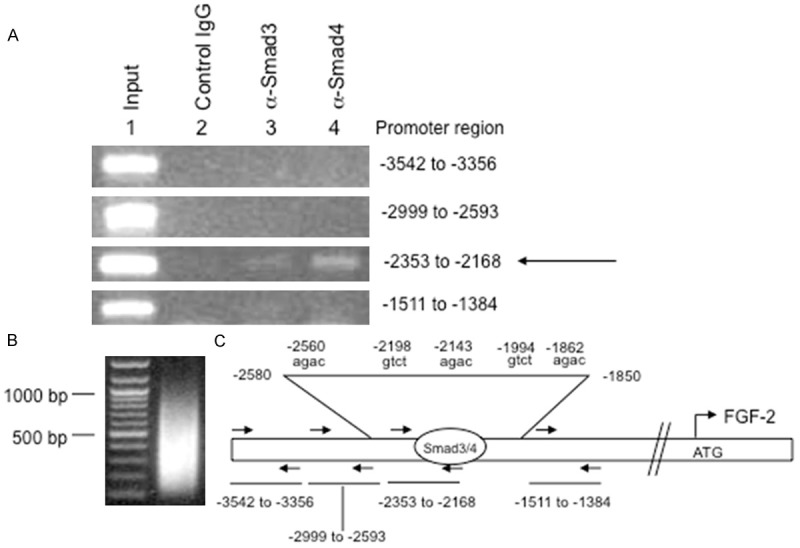
Binding of Smad3/4 complex to the FGF-2 promoter. A. ChIP analysis after TGF-β1 (80 pM) treatment of Smad2 null MEFs showed Smad3 and Smad4 antibodies selectively immunoprecipitated the -2353 to -2168 region (arrow) of the FGF-2 promoter that contains a putative consensus Smad4 binding element; B. Chromatin shearing shows fragments of approximately 200-800 bp; C. Map of regions evaluated by PCR analysis (arrows). Primer sets were designed to scan 5 kb of the FGF-2 promoter, but only amplicons surrounding the potential binding site are shown. Sequence analysis in this region detected five potential SBEs. Subsequent analysis using MatInspector (Genomatix) predicted a gtct SBE motif at -1994 from the start codon as a putative Smad4 binding sequence in the FGF-2 promoter.
TGF-β1-induction of FGF-2 expression in MEFs is regulated by ERK1/2, p38, and JNK signaling
To assess crosstalk mechanisms with MAPK pathways, wild type control MEFs were treated with TGF-β1 (100 pM) in the presence of MAPK inhibitors or vehicle control and FGF-2 mRNA was evaluated by qPCR over a 24 hr. time course. As shown in Figure 3A, both UO126 (a MEK1/2 inhibitor) and SB203580 (a p38 inhibitor) resulted in a significant attenuation of induced FGF-2 mRNA in MEF cells. Of interest, SP600125, a JNK inhibitor, resulted in a significant augmentation of TGF-β1 induction of FGF-2 message. Consistent with these results, Western blots show that TGF-β1 induced phosphorylation of both p38 and ERK 1/2 in wild type control MEFs, although effects on ERK 1/2 exhibited higher basal phosphorylation and TGF-β1 induction of ERK 1/2 was minimal (Figure 3B). Induction of p38 phosporylation was also observed in Smad3 null MEFs, whereas ERK 1/2 is less clear in these cells. Interestingly, TGF-β1 did not affect JNK phosphorylation under any conditions in any cell lines tested (data not shown). Consistent with the mRNA data, use of the p38 and ERK 1/2 MAPK inhibitors resulted in a significant decrease in TGF-β1 induced FGF-2 protein expression at 24 hr as shown by ELISA assay (Figure 3C), although the JNK inhibitor did not seem to affect protein levels at the 24 hr time point.
Figure 3.
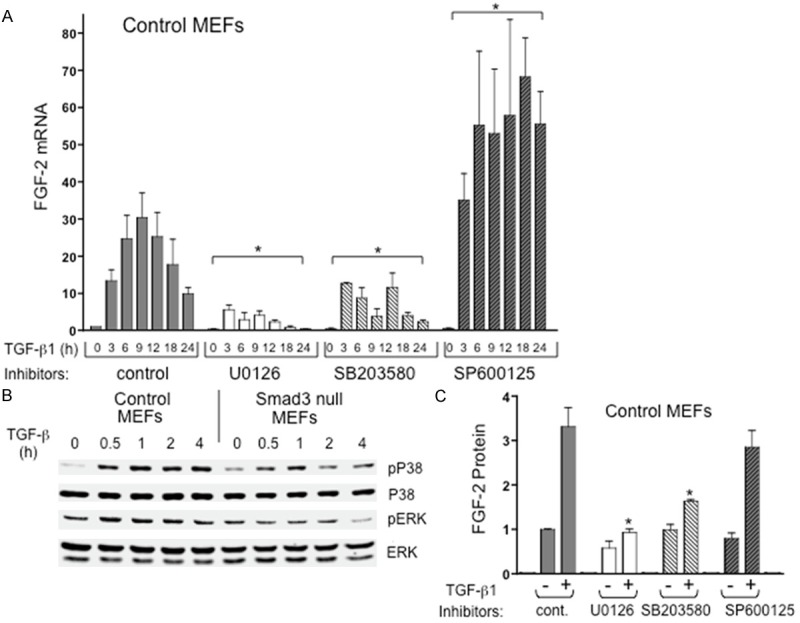
TGF-β1-induced FGF-2 mRNA and protein expression in mouse embryo fibroblasts is regulated by ERK1/2, p38 and JNK signaling pathways. A. Chemical inhibition of either ERK1/2 phosphorylation (UO126) or p38 activity (SB203580) significantly attenuated TGF-β1-induced FGF-2 mRNA expression relative to TGF-β1 control, whereas inhibition of JNK signaling (SP600125) significantly elevated FGF-2 mRNA induction by TGF-β1 (asterisks). *P < 0.05, N ≥ 3 independent experiments in triplicate; B. Western blots show TGF-β1 induced phosphorylation of p38 in mouse embryo fibroblasts with less obvious induction in ERK 1/2. C. FGF-2 protein in MEF cell extracts from cells treated with either TGF-β1 (+) or vehicle control (-), with or without chemical inhibitors. TGF-β1 induction of FGF-2 protein was significantly inhibited in MEFs treated with ERK1/2 and p38 inhibitors (asterisks). *P < 0.05, N ≥ 3 independent experiments with replicate wells/experiment.
TGF-β1 activates MAPKs in prostate stromal cells
C57BL/6 mouse prostate fibroblasts (TβRIICT) were generated as described previously [6] and, as expected, exhibited TGF-β1 induced Smad3 phosphorylation (Figure 4A). Although a basal level of phosphorylation of p38 was noted at the zero time point, TGF-β1 induced elevated phosphorylation of p38 in prostate fibroblasts evident at the 1-4 hr time points (Figure 4A), similar to control MEF cells. Basal level of ERK 1/2 phosphorylation was also observed at the 0-2 hr time points. TGF-β1 induced an elevated phosphorylation of ERK 1/2 that was apparent at the 4 hr time point (Figure 4B). This induction of ERK 1/2 phosphorylation was specific, since pretreatment of cells with UO126 resulted in attenuated ERK1/2 phosphorylation in the presence of TGF-β1 (Figure 4B).
Figure 4.
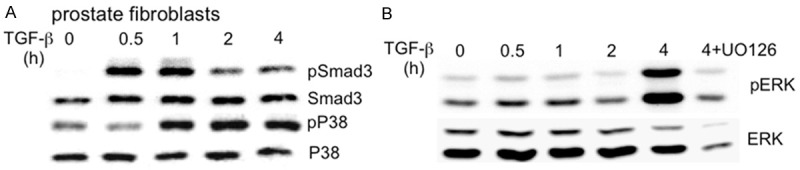
TGF-β1 induces phosphorylation of Smad3, p38, and ERK 1/2 in prostate gland fibroblasts. A. Western blots show induction of Smad3, p38 phosphorylation by TGF-β1 treatment in prostate gland fibroblasts; B. TGF-β1 induced ERK 1/2 phosphorylation in prostate gland fibroblasts. Basal phosphorylation was evident and TGF-β1 induced phosphorylation was most apparent at 4 hr, which was inhibited by pretreatment with MEK inhibitor U0126 (4 + U0126) as a control.
TGF-β1-induction of FGF-2 expression in prostate stromal cells is regulated by ERK1/2, p38, and JNK signaling
In TβRIICT prostate fibroblasts, TGF-β1 induced FGF-2 messages following a similar time course as compared to MEFs as shown in Figure 5A. Maximum FGF-2 message was observed at 6-18 hours post-TGF-β1 treatment. Similar to regulation in MEFs, both the ERK 1/2 and p38 inhibitors (U0126 and SB203580 respectively) resulted in a significant attenuation of FGF-2 message induction. Consistent with results using MEFs, the JNK inhibitor SP600125 also tends to elevate FGF-2 message in prostate stromal cells treated with TGF-β1 but this did not achieve statistical significance as in MEFs. Protein levels exhibited a similar trend with the ERK 1/2 and p38 inhibitors resulting in a significant attenuation of TGF-β1 induced FGF-2 protein at the 24 hr time point (Figure 5B). As suggested with induced FGF-2 message, use of the JNK inhibitor SP600125 did result in a significant elevation of FGF-2 protein in prostate stromal cells (Figure 5B).
Figure 5.
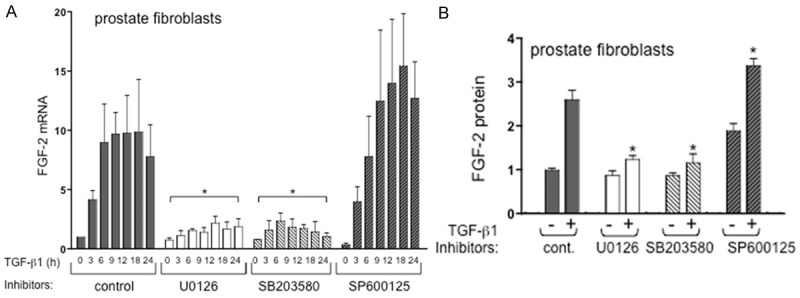
TGF-β1-induced FGF-2 expression in prostate gland fibroblasts requires ERK1/2 and p38 activity. A. Chemical inhibition of either ERK1/2 phosphorylation (UO126) or p38 activity (SB203580) significantly attenuated TGF-β1-induced FGF-2 mRNA expression in prostate gland fibroblasts relative to control (asterisks). *P < 0.05, N ≥ 3 independent experiments in triplicate. Inhibition of JNK activity (SP600125) resulted in a trend toward elevated mRNA that did not reach statistical significance; B. FGF-2 protein in prostate gland fibroblast cell extracts from cells treated with either TGF-β1 (+) or vehicle controls (-) with or without chemical inhibitors. TGF-β1 induced FGF-2 protein was significantly attenuated (asterisks) in the presence of ERK1/2 and p38 kinase inhibitors, whereas inhibition of JNK resulted in a significant increase in FGF-2 protein (asterisks). *P < 0.05, N=2-4 independent experiments with replicate wells per experiment.
Discussion
TGF-β is a key regulator of the reactive stroma microenvironment that is typified by an induced reactive stroma composed primarily of activated fibroblasts and myofibroblasts. FGF-2 may mediate much of the pro-fibrosis biology induced by TGF-β1 at sites of reactive stroma [6,12]. Moreover, FGF-2 may mediate metastatic spread of prostate cancer, as we have shown recently that FGF receptor 1 is critical for metastasis in a mouse model [30]. In this report, we examined TGF-β1 regulation of FGF-2 expression in prostate fibroblasts and mouse embryo fibroblasts by potential mediators of TGF-β1 signaling, in order to define key pathways. Our data shows that FGF-2 mRNA and protein expression are regulated by Smads3/4 and the MAPK proteins p38, ERK1/2, and JNK. These data show that functional signaling of the Smad3, p38 and ERK 1/2, but not Smad2, are each required for TGF-β1 induced FGF-2 mRNA and protein expression in fibroblasts. Our data also shows that the -1.9 to -2.3 region of FGF-2 promoter is a potential site for Smad3/4 complex regulation of FGF-2 expression, although other sites of interaction in the more distal promoter cannot be ruled out. However, our data also suggests that functional Smad3 signaling is not necessarily required for TGF-β1 induced phosphorylation of the selected MAPK proteins.
The role of Smad3 and exclusion of Smad2 in regulating FGF-2 expression is consistent with other pro-fibrotic roles assigned to functional Smad3 signaling. Smad3 has been shown to regulate pro-fibrotic, pro-angiogenic proteins including collagens I and III, VEGF-A [31] and CTGF [32]. Consistent with these findings, wounding studies have shown an inhibited fibrotic response in Smad3 null mice [27]. It is likely that attenuated FGF-2 may have accounted for this decreased fibrotic, wound repair response. Based on data reported here, FGF-2 can be categorized as a partner in the TGF-β-induced and Smad3 dependent fibrotic tissue response. Of interest is the Smad3 independent activation of MAPK signaling in fibroblasts. These data suggest that even in attenuated or blocked Smad3 signaling conditions, some fibroblasts cell responses to TGF-β1 can be generated by MAPK signaling.
In contrast to the pro-fibrotic activities of p38 and ERK 1/2 signaling, JNK activation is suggested to be generally anti-fibrotic due to its inhibition of Smad3-driven transcription [33]. Furthermore, activation of the JNK1/2 SAPK signaling pathway by pro-inflammatory cytokines like TNF-β acts as an antagonist of Smad3-driven expression of pro-fibrotic and pro-angiogenic genes by preventing Smad3 transcriptional activity by a mechanism that is incompletely understood [34,35]. Consistent with this, transplantation of bone marrow from a TNF-α knockout mouse in a wounding model potentiated the fibrotic response suggesting that cytokines expressed by inflammatory cells at sites of wound repair may inhibit the fibrotic response [36]. In alignment with these possibilities, data reported here shows that the JNK pathway may function to inhibit TGF-β1 induced expression of FGF-2 in stromal fibroblasts and MEFs and that TGF-β1 inhibits JNK activity. Accordingly, it is likely that pro-fibrotic and anti-inflammatory response generated concurrently at sites of repair by TGF-β1 are mediated via differential activation of ERK 1/2 and p38, along with repression of JNK pathways in activated fibroblasts. This is important since activated fibroblasts are the central cell type in any type of wound repair, reactive stroma, granulation tissue, or fibrotic response.
The further identification of signaling pathways and genes regulated directly or indirectly by TGF-β1 in fibroblasts at sites of tissue repair and in carcinoma-associated fibroblasts or myofibroblasts at reactive stroma sites in the tumor microenvironment will facilitate our understanding of the common signaling schemes utilized by TGF-β1 in its regulation of downstream genes in cancer, wound repair, and pro-fibrotic diseases, including vessel wall disease. In addition, it is equally imperative to further understand the pathways utilized by pro-inflammatory cytokines in negatively regulating Smad3-driven pro-fibrotic biology in order to identify additional potential therapeutic targets.
Acknowledgements
We thank Dr. Rainer Lanz for expert assistance with qPCR. This work was supported by NIH grants, RO1-CA058093, U54-CA126568, RO1-DK045909, SPORE P50-CA58204, UO1-CA84296, and Department of Defense grant W81XWH-04-1-0189.
Disclosure of conflict of interest
None.
References
- 1.Barcellos-Hoff MH, Akhurst RJ. Transforming growth factor-beta in breast cancer: too much, too late. Breast Cancer Res. 2009;11:202. doi: 10.1186/bcr2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flanders KC, Wakefield LM. Transforming growth factor-(beta)s and mammary gland involution; functional roles and implications for cancer progression. J Mammary Gland Biol Neoplasia. 2009;14:131–144. doi: 10.1007/s10911-009-9122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 4.Tuxhorn JA, McAlhany SJ, Dang TD, Ayala GE, Rowley DR. Stromal cells promote angiogenesis and growth of human prostate tumors in a differential reactive stroma (DRS) xenograft model. Cancer Res. 2002;62:3298–3307. [PubMed] [Google Scholar]
- 5.Tuxhorn JA, McAlhany SJ, Yang F, Dang TD, Rowley DR. Inhibition of TGF-β activity decreases angiogenesis in a human prostate cancer reactive stroma xenograft model. Cancer Res. 2002;62:6021–6025. [PubMed] [Google Scholar]
- 6.Yang F, Strand DW, Rowley DR. Fibroblast growth factor-2 mediates transforming growth factor-beta action in prostate cancer reactive stroma. Oncogene. 2008;27:450–459. doi: 10.1038/sj.onc.1210663. [DOI] [PubMed] [Google Scholar]
- 7.Yang F, Tuxhorn JA, Ressler SJ, McAlhany SJ, Dang TD, Rowley DR. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res. 2005;65:8887–8895. doi: 10.1158/0008-5472.CAN-05-1702. [DOI] [PubMed] [Google Scholar]
- 8.Dow JK, deVere White RW. Fibroblast growth factor 2: its structure and property, paracrine function, tumor angiogenesis, and prostate-related mitogenic and oncogenic functions. Urology. 2000;55:800–806. doi: 10.1016/s0090-4295(00)00457-x. [DOI] [PubMed] [Google Scholar]
- 9.Bikfalvi A, Klein S, Pintucci G, Rifkin DB. Biological roles of fibroblast growth factor-2. Endocr Rev. 1997;18:26–45. doi: 10.1210/edrv.18.1.0292. [DOI] [PubMed] [Google Scholar]
- 10.Kwabi-Addo B, Ozen M, Ittmann M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr Relat Cancer. 2004;11:709–724. doi: 10.1677/erc.1.00535. [DOI] [PubMed] [Google Scholar]
- 11.Su G, Blaine SA, Qiao D, Friedl A. Shedding of syndecan-1 by stromal fibroblasts stimulates human breast cancer cell proliferation via FGF2 activation. J Biol Chem. 2007;282:14906–14915. doi: 10.1074/jbc.M611739200. [DOI] [PubMed] [Google Scholar]
- 12.Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin Cancer Res. 1999;5:1063–1071. [PubMed] [Google Scholar]
- 13.Giulianelli S, Cerliani JP, Lamb CA, Fabris VT, Bottino MC, Gorostiaga MA, Novaro V, Gongora A, Baldi A, Molinolo A, Lanari C. Carcinoma-associated fibroblasts activate progesterone receptors and induce hormone independent mammary tumor growth: A role for the FGF-2/FGFR-2 axis. Int J Cancer. 2008;123:2518–2531. doi: 10.1002/ijc.23802. [DOI] [PubMed] [Google Scholar]
- 14.Feng S, Wang F, Matsubara A, Kan M, McKeehan WL. Fibroblast growth factor receptor 2 limits and receptor 1 accelerates tumorigenicity of prostate epithelial cells. Cancer Res. 1997;57:5369–5378. [PubMed] [Google Scholar]
- 15.Barrett-Lee P, Travers M, Luqmani Y, Coombes RC. Transcripts for transforming growth factors in human breast cancer: clinical correlates. Br J Cancer. 1990;61:612–617. doi: 10.1038/bjc.1990.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coffey RJ Jr, Shipley GD, Moses HL. Production of transforming growth factors by human colon cancer lines. Cancer Res. 1986;46:1164–1169. [PubMed] [Google Scholar]
- 17.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 18.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 19.Blanchette F, Rivard N, Rudd P, Grondin F, Attisano L, Dubois CM. Cross-talk between the p42/p44 MAP kinase and Smad pathways in transforming growth factor beta 1-induced furin gene transactivation. J Biol Chem. 2001;276:33986–33994. doi: 10.1074/jbc.M100093200. [DOI] [PubMed] [Google Scholar]
- 20.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 21.Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005;24:5742–5750. doi: 10.1038/sj.onc.1208928. [DOI] [PubMed] [Google Scholar]
- 22.Sowa H, Kaji H, Yamaguchi T, Sugimoto T, Chihara K. Activations of ERK1/2 and JNK by transforming growth factor beta negatively regulate Smad3-induced alkaline phosphatase activity and mineralization in mouse osteoblastic cells. J Biol Chem. 2002;277:36024–36031. doi: 10.1074/jbc.M206030200. [DOI] [PubMed] [Google Scholar]
- 23.Ungefroren H, Lenschow W, Chen WB, Faendrich F, Kalthoff H. Regulation of biglycan gene expression by transforming growth factor-beta requires MKK6-p38 mitogen-activated protein Kinase signaling downstream of Smad signaling. J Biol Chem. 2003;278:11041–11049. doi: 10.1074/jbc.M300035200. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe H, de Caestecker MP, Yamada Y. Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J Biol Chem. 2001;276:14466–14473. doi: 10.1074/jbc.M005724200. [DOI] [PubMed] [Google Scholar]
- 25.Xiao YQ, Malcolm K, Worthen GS, Gardai S, Schiemann WP, Fadok VA, Bratton DL, Henson PM. Cross-talk between ERK and p38 MAPK mediates selective suppression of pro-inflammatory cytokines by transforming growth factor-beta. J Biol Chem. 2002;277:14884–14893. doi: 10.1074/jbc.M111718200. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Derynck R. Regulation of Smad signalling by protein associations and signalling crosstalk. Trends Cell Biol. 1999;9:274–279. doi: 10.1016/s0962-8924(99)01579-2. [DOI] [PubMed] [Google Scholar]
- 27.Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C, Roberts AB. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1:260–266. doi: 10.1038/12971. [DOI] [PubMed] [Google Scholar]
- 28.Piek E, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL, Weinstein M, Deng C, Kucherlapati R, Bottinger EP, Roberts AB. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. 2001;276:19945–19953. doi: 10.1074/jbc.M102382200. [DOI] [PubMed] [Google Scholar]
- 29.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 30.Yang F, Zhang Y, Ressler SJ, Ittmann MM, Ayala GE, Dang TD, Wang F, Rowley DR. FGFR1 is essential for prostate cancer progression and metastasis. Cancer Res. 2013;73:3716–3724. doi: 10.1158/0008-5472.CAN-12-3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakagawa T, Li JH, Garcia G, Mu W, Piek E, Bottinger EP, Chen Y, Zhu HJ, Kang DH, Schreiner GF, Lan HY, Johnson RJ. TGF-beta induces proangiogenic and antiangiogenic factors via parallel but distinct Smad pathways. Kidney Int. 2004;66:605–613. doi: 10.1111/j.1523-1755.2004.00780.x. [DOI] [PubMed] [Google Scholar]
- 32.Leivonen SK, Hakkinen L, Liu D, Kahari VM. Smad3 and extracellular signal-regulated kinase 1/2 coordinately mediate transforming growth factor-beta-induced expression of connective tissue growth factor in human fibroblasts. J Invest Dermatol. 2005;124:1162–1169. doi: 10.1111/j.0022-202X.2005.23750.x. [DOI] [PubMed] [Google Scholar]
- 33.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 34.Abraham DJ, Shiwen X, Black CM, Sa S, Xu Y, Leask A. Tumor necrosis factor alpha suppresses the induction of connective tissue growth factor by transforming growth factor-beta in normal and scleroderma fibroblasts. J Biol Chem. 2000;275:15220–15225. doi: 10.1074/jbc.275.20.15220. [DOI] [PubMed] [Google Scholar]
- 35.Verrecchia F, Tacheau C, Wagner EF, Mauviel A. A central role for the JNK pathway in mediating the antagonistic activity of pro-inflammatory cytokines against transforming growth factor-beta-driven SMAD3/4-specific gene expression. J Biol Chem. 2003;278:1585–1593. doi: 10.1074/jbc.M206927200. [DOI] [PubMed] [Google Scholar]
- 36.Saika S, Ikeda K, Yamanaka O, Flanders KC, Okada Y, Miyamoto T, Kitano A, Ooshima A, Nakajima Y, Ohnishi Y, Kao WW. Loss of tumor necrosis factor alpha potentiates transforming growth factor beta-mediated pathogenic tissue response during wound healing. Am J Pathol. 2006;168:1848–1860. doi: 10.2353/ajpath.2006.050980. [DOI] [PMC free article] [PubMed] [Google Scholar]