

Abstract
Nuclear receptor corepressor (NCoR) and silencing mediator for retinoid and thyroid hormone receptors (SMRT) function as corepressors for diverse transcription factors including nuclear receptors such as estrogen receptors and androgen receptors. Deregulated functions of NCoR and SMRT have been observed in many types of cancers and leukemias. NCoR and SMRT directly bind to transcription factors and nucleate the formation of stable complexes that include histone deacetylase 3, transducin b-like protein 1/TBL1-related protein 1, and G-protein pathway suppressor 2. These NCoR/SMRT-interacting proteins also show deregulated functions in cancers. In this review, we summarize the literature on the mechanism, regulation, and function of the core components of NCoR/SMRT complexes in the context of their involvement in cancers and leukemias. While the current studies support the view that the corepressors are promising targets for cancer treatment, elucidation of the mechanisms of corepressors involved in individual types of cancers is likely required for effective therapy.
Keywords: NCoR, SMRT, HDAC3, TBL1, TBLR1, GPS2, cancer, corepressor, transcriptional repression
Introduction: discovery of NCoR and SMRT as promiscuous nuclear receptor corepressors
The ‘on’-and-‘off’ control of gene expression involves a cascade of transcription factors and is assisted by two classes of cofactors. These cofactors can activate (the coactivators) or repress (the corepressors) gene transcription via modifying specific residues of histones, thereby regulating the accessibility of chromatin to the basal transcription machinery [1]. Two of the first identified corepressors are nuclear receptor corepressors (CoRs), which include NCoR (nuclear receptor corepressor) [2] and SMRT (silencing mediator for retinoid and thyroid hormone receptors) [3]. Prior to the cloning of NCoR and SMRT, several studies have reported that thyroid hormone receptors (TR) and retinoic acid receptors (RAR) have an intrinsic ability to mediate ligand-independent repression, in a manner that is dependent on soluble and titratable factors [4-6]. The search for these factors led to the cloning of NCoR [2] and its homologous protein SMRT [3] (a truncated version) as well as full-length SMRT [7,8]. Binding of TR and RAR to their respective ligands, thyroid hormone (T3) and retinoic acid (RA), disrupts their interactions with NCoR or SMRT, allowing for coactivator recruitment and subsequent activation.
Subsequent studies reveal that NCoR and SMRT also mediate ligand-independent interaction with various other nuclear receptors (NRs), including vitamin D receptors (VDR) [9], peroxisome proliferator-activated receptors (PPAR) [10], liver X receptors (LXR) [11], and with orphan receptors including Rev-Erb [12,13], chicken ovalbumin upstream promoter transcription factor (COUP-TF) [14], and dose-sensitive sex reversal-AHC critical region on the X chromosome, gene 1 (DAX-1) [15]. NCoR and SMRT also have the ability to interact with steroid hormone receptors, namely, estrogen receptor (ER) [16], androgen receptor (AR) [17,18], and progesterone receptor (PR) [19]. Although these interactions are generally weaker than the interactions with non-steroid receptors, the steroid receptor interactions with NCoR/SMRT can, nevertheless, be stabilized by binding to the corresponding antagonists. One of the best-studied examples is tamoxifen, an ERα antagonist used to treat ERα-positive breast cancers. Tamoxifen binding to ERα stabilizes the interaction between ERα and NCoR, which is considered a contributing factor for tamoxifen inhibition of ERα target gene expression [20].
Mechanism of action of NCoR/SMRT
NCoR and SMRT are 270 kDa proteins that share highly homologous domains. The C-terminal region of NCoR and SMRT contains three (NCoR) or two (SMRT) nuclear receptor interaction domains (IDs) that mediate direct interactions with the ligand-binding domain (LBD) of NRs [2] (Figure 1). The functional motifs of IDs contain the so-called CoRNR boxes, which have the consensus sequence L-X-X-I/H-I-X-X-X-L/I (L: leucine, I: isoleucine, X: any amino acids) [21-25]. The binding affinity is predominantly regulated by the conformational change of NR LBD domains that takes place in response to ligands (agonists or antagonists). Binding of agonists or antagonists to NRs induces the movement of the helix 12 region of NRs whose position dictates the binding ability of NCoR/SMRT. In general, agonists destabilize the corepressor interaction, resulting in the dismissal of CoRs and subsequent recruitment of coactivators to drive agonist-dependent transcriptional activation. Antagonists do the opposite by stabilizing NCoR/SMRT interactions. In addition, the specificity and fine tuning of the binding between NRs and N-CoR/SMRT are also shown by the preferential use of different CoRNR boxes within a single CoR molecule and the preferential use of NCoR or SMRT for the binding [26,27].
Figure 1.
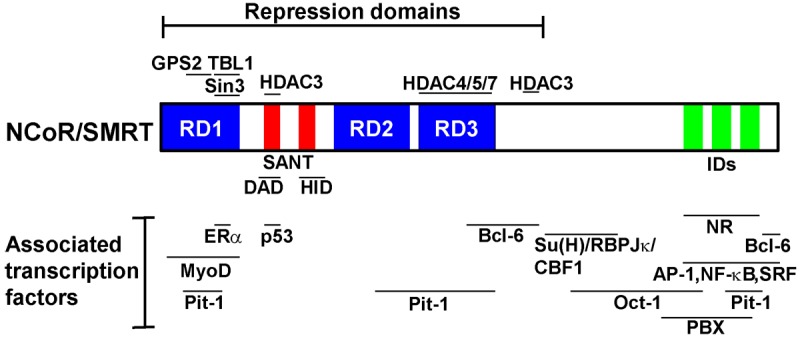
Diagram of NCoR/SMRT corepressors showing their regions involved in various interactions with nuclear receptors, other transcription factors as well as interactions with other components of the core complex.
Biochemical purification of NCoR and SMRT has shown that both NCoR and SMRT are present in large protein complexes (1.6-2 MDa size) that also contain histone deacetylase 3 (HDAC3), transducin β-like protein 1 (TBL1)/TBL1-related protein 1 (TBLR1), and G-protein pathway suppressor 2 (GPS2) [28-30]. It is likely that the core complex contains four subunits, two of which can be either NCoR or SMRT, and either TBL1 or TBLR1. A recent work has shown that TBL1 may exist as a tetramer [31]. The molecular weight of the core complex is about 412 KDa. Multiplication of this by 4 gives rise to ~1.6 MDa, which is in the range of the observed size (1.6-2 MDa). Clearly, the NCoR/SMRT complex should also contain other sub-stoichiometric components, such as Sin3A/HDAC1 [32-37], Class II HDACs [38,39], the subunits of chromatin-remodeling complexes [40], KAP-1 [40], and histone acetyl-transferases such as CBP [1,41-43]. The functional interactions of these sub-stoichiometric components with the core NCoR/SMRT complex may offer additional layers of regulation of target gene transcription.
NCoR/SMRT nucleates the formation of the core complex through interactions provided by the N-terminal portion of the proteins. Historically, the N-terminus of NCoR and SMRT has been characterized to contain three independent repression domains (RD1, RD2 and RD3) [2]. While the function of RD2 and RD3 has yet to be clarified, RD1 has been shown to mediate NCoR/SMRT interactions with both GPS2 and TBL1/TBLR1 [28,44]. Interestingly, the region of RD1 that interacts with TBL1/TBLR1 appears to overlap with the recently-reported interaction with the DNA-binding domain of ERα [45] (Figure 1).
None of the three RD domains interacts with HDAC3. A previously-noted motif in NCoR and SMRT is the SW13/ADA2/NCoR/TFIIB (SANT) domain, two copies of which are located between RD1 and RD2 [46]. The first SANT domain along with a short upstream region is referred to as the deacetylase activation domain (DAD), which directly binds to HDAC3. The binding involves a conformational change of DAD and is critical for the activation of the enzymatic activity of HDAC3 [28,47,48]. Interestingly, a recent work [49] by Adikesavan et al. has shown that the DAD of SMRT can directly bind to p53 (Figure 1). This binding blocks HDAC3 interaction with DAD leading to a net increase in histone acetyltransferase (HAT) activities, which contributes to the activation of p53 target gene in response to DNA damage. SMRT (but not NCoR) has also been shown to function as a coactivator for ERα in MCF-7 breast cancer cells [50], underscoring a specificity between SMRT and NCoR in regulating transcription.
The second SANT domain in CoRs has been shown to function as a histone interaction domain (HID), in line with the reported role of SANT domains in recognizing histones in the context of other proteins [51]. By occupying the nonacetylated histone tails, HID inhibits HAT activities, suggesting a feed-forward mechanism by which the two SANT motifs cooperatively promote histone deacetylation and repression of target genes [52,53]. Similar modes of cooperation have also been reported between HDAC3 and TBL1, which can also directly bind to histones [30,54,55].
Although NCoR and SMRT were originally identified as transcriptional corepressors for NRs, they have emerged as promiscuous corepressors for many other sequence-specific transcription factors that function in different cellular processes [42]. Thus, various studies have shown that SMRT and NCoR interact with Bcl-6/LAZ3, a transcription factor recently shown to play an important role in repressing inflammation [56-59], MyoD [60], HES-related repressor proteins [61]. NCoR/SMRT also interact with the evolutionarily related POU homeodomain factors Pit-1 [62,63], Oct-1 [64], the Notch-activated adapter protein Su(H)/RBP-Jκ/CBF1 [65-67], Pbx [68,69], serum response factor (SRF) [70], NF-κB [71-74], AP-1 [70], and signal transducers and activators of transcription 5 (STAT5). Other factors that are associated with NCoR and SMRT include PLZF [75,76], ETO family proteins involved in acute myeloid leukemias [77-79], SMADs [80], c-Myb [81], aryl hydrocarbon receptor [82], Sharp [83], and Kaiso [84].
Two studies have added new insight into the function and mechanism of CoR-mediated repression. Ebert et al. showed that NCoR can interact with methyl-CpG-binding protein 2 (MeCP2) and phosphorylation of T308 blocks the interaction of the MeCP2 repression domain with NCoR and suppresses the ability of MeCP2 to repress transcription [85], thus linking NCoR to DNA methylation-dependent gene silencing. Another work shows that the NCoR complex binds and deacetylases P-TEFb, an elongation factor important for RNA Pol II-mediated transcription [86], thus revealing a role for corepressors in deacetylating general transcription factors to regulate transcription.
Deregulation of NCoR/SMRT corepressors and other complex subunits in cancer
ER and AR are steroid receptors that play important roles in the initiation and progression of breast and prostate cancers. NCoR/SMRT interact with both ER and AR and serve as potential drug targets in the treatment of these cancers. Recent genome-wide studies have shown that NCoR/SMRT and HDAC3 bind to thousands of genes [56,87], consistent with their use by other transcription factors such as c-Jun and NFκb that impact on fundamental cellular activities. In this section, we summarize the literature on the involvement of NCoR/SMRT and other complex subunits in cancers and leukemias. NCoR and SMRT have been largely studied in the context of ER, AR and leukemia fusion proteins. While previous studies have focused on NCoR, SMRT and HDAC3, some new development has been reported for TBL1/TBLR1 and GPS2, suggesting their important roles in cancer development.
NCoR and SMRT subunits
Breast cancer
ERα is expressed in approximately 75% of breast cancers where ERα - dependent activation plays an important role in the growth of these cancers. NCoR/SMRT are among the best-characterized ERα corepressors [88]. Since coactivators and corepressors compete for ERα binding, the ratio between coactivators and corepressors is expected to modulate ERα activity in benign and malignant cells. Results from independent studies have supported this model by showing that diminished expression of NCoR/SMRT drives breast cancer initiation and progression. For example, an unbiased pathway analysis has revealed multiple alterations linked to loss of NCoR and SMRT corepressor complexes in luminal A breast tumors [89]. In another study that examines ERα corepressor levels in breast cancer, NCoR levels were found to be downregulated in invasive ductal carcinomas [90].
Downregulation of NCoR/SMRT has also been implicated in endocrine resistance. Selective ER modulators (SERMs), such as tamoxifen, an ERα antagonist, are beneficial in the initial treatment of ERα-positive breast cancer, because these SERMs can inhibit cancer growth by inhibiting the transcriptional activity of ERα. The activity of tamoxifen is in part attributed to its ability to stabilize the binding of ERα to corepressors [91]. Reduced expression of NCoR/SMRT corepressors has been shown to contribute to tamoxifen resistance in breast cancers [92-95]. Two studies have provided mechanistic insights into signal-dependent downregulation of NCoR and SMRT in breast cancer cells at the level of protein degradation, which contributes to cancer progression and acquisition of endocrine resistance. Frasor et al. reported that NCoR level was reduced due to estrogen-dependent upregulation of Siah2, which functions as an E3 ligase for NCoR [96,97]. Interestingly, two recent studies have shown that downregulation of Siah2 is correlated with acquisition of breast cancer tamoxifen resistance [98,99]. In another work, Stanya et al. reported that Pin1 can promote proteasomal degradation of phosphorylated SMRT in response to HER-2 signaling [100].
Several studies have reported alternative mechanisms by which NCoR regulates ERα-dependent transcription in breast cancers. First, NCoR has been shown to regulate ERα expression. More than half of the ERα-positive breast cancers also express PR. Elevated expression levels of PR-B isoform increase the interaction of the receptor with NCoR on the half-PRE site of the ERα promoter, an event incompatible with PR-coactivator interactions. Silencing of NCoR was able to reverse the down-regulation of ERα expression induced by PR-B overexpression [101]. In another work, by using chromatin immunoprecipitation (ChIP) assays, Konduri et al. [102] have demonstrated that ERα represses p53-mediated transcriptional activation in human breast cancer cells by recruiting NCoR/SMRT and HDAC1. Finally, reduction in the ratio of ERβ to ERα expression appears to be correlated with breast tumorigenesis. Bartella et al. [103] showed that ERβ recruits NCoR, leading to hypoacetylation of histones and displacement of RNA-polymerase II at the ERα promoter, providing new mechanistic insight into the antagonism between ERβ and ERα in breast cancer cell growth.
Prostate cancer
AR is the driving force for prostate cancer development and progression. Many patients will benefit from the androgen deprivation therapy in combination with the treatment of AR antagonists (flutamide or bicalutamide). The underlying principle for the use of AR antagonists is that these compounds will compete with agonists for binding to AR, while stabilizing the binding between AR and NCoR/SMRT corepressors, thus preventing coactivator recruitment and AR-dependent activation of target genes [104,105].
The involvement of AR corepressors in prostate cancer has been well documented [106]. Earlier studies have suggested that a reduction of NCoR/SMRT levels may contribute to androgen-independent AR activation and prostate cancer progression [107-109]. Nevertheless, several studies have shown that the corepressor regulation of AR may be more complex than previously thought. It has been shown that overexpression of NCoR/SMRT does not repress AR-dependent gene expression in prostate cancer cell lines, but rather activates it [110]. A caveat is that overexpressed proteins may not faithfully recapitulate the regulation observed under physiological conditions. Another work has shown that the ability of NCoR to enhance antagonist-mediated AR repression is in fact modulated by post-translational modifications of NCoR. Protein kinase A (PKA) is able to directly bind and phosphorylate the Ser-70 residue in the RD1 domain of NCoR. The phosphorylation enhances nuclear localization of NCoR and potentiates the antagonist activity on AR-dependent transcription in prostate cells [111].
Several recent studies have re-affirmed the importance of AR-NCoR/SMRT axis in regulating AR function and prostate cancer progression. It has been shown that reduced recruitment and loss of corepressor SMRT/NCoR alter the ligand response and AR functions in a manner that contributes to prostate cancer progression [112]. In another study, Yoo et al. studied the role of AR corepressors during androgen-independent prostate cancer progression [113]. It was found that casein kinase 2 (CK2)-mediated phosphorylation of NCoR strongly correlates with androgen-independent growth and invasion of prostate cancer cells, and with poor prognosis of prostate cancer patients, suggesting that CK2-NCoR axis is a potential therapeutic target in prostate cancer. Perhaps a more exciting evidence for a direct role of NCoR in antagonizing androgen-independent prostate cancer growth is provided by the study showing that the ubiquitin ligase Siah2 switches AR from repressed to the activated state by selectively targeting NCoR bound, transcriptionally-inactive AR for ubiquitin-dependent degradation. Thus, Siah2 promotes activation of AR target genes, leading to the growth of androgen-independent prostate cancer cells [114]. Taken together, the consensus of the current studies is in line with the model that the efficacy of NCoR/SMRT corepressors in repressing AR activity is directly correlated with the recruitment of these corepressors on AR target genes. Reduction of such recruitment, which would predispose genes for activation, may occur through various mechanisms, such as reduced gene expression, proteasomal degradation or altered modification of the corepressors.
Leukemia
Leukemia is a prototypic type of cancer in which a pro-oncogenic function of NCoR and SMRT was first documented. One form of acute promyelocytic leukemia (APL) is caused by abbrant expression of PML-RARα (promyelocytic leukemia-retinoic acid receptor α) or PLZF-RARα (promyelocytic leukemia zinc finger-retinoic acid receptor α) fusion proteins. As a result of the fusion, the binding affinity between RARα and NCoR/SMRT is increased such that the corepressors cannot be released by the physiological dose of RA [75,115-118]. Accordingly, these fusion proteins promote leukemogenesis due to reduced sensitivity to retinoic acid-dependent transcriptional activation of target genes involved in cell differentiation. The involvement of NCoR/HDAC in the pathogenesis of leukemias is further confirmed by the studies showing that expression of an NCoR fragment that disrupts corepressor interaction restores RA sensitivity in resistant cells [119], and that the NCoR/HDAC complex is a key regulator of the transcriptional repression mediated by PML-RARα in vivo [120]. APL patients that have PML-RARα is sensitive to high doses of RA treatment, whereas APL patients harboring PLZF-RARα translocations do not. This difference is due to the fact that in PLZF-RARα, the PLZF moiety can also bind to NCoR/SMRT corepressors, which cannot be released by high doses of RA, thus explaining the phenotypic difference between PML-RARα and PLZF-RARα. The presence of trichostatin A, a specific inhibitor of histone deacetylases, increases histone acetylation and leads to transcription activation [115].
Two recent studies have provided new insight into the regulation of RARα fusion proteins by NCoR/SMRT. One study shows that PML-RARα and PLZF-RARα have gained the ability to recognize specific spliced variants of NCoR and SMRT variants that are poorly recognized by RARα [121]. Another work showed that failure to dissociate corepressors, or failure to recruit co-activators results in RA-resistant variants of the PML-RARα oncoprotein in APL [122].
The t(8;21) chromosomal translocation is involved in nearly 15% of total acute myeloid leukemia (AML) cases, which generates the AML1-ETO fusion protein between the DNA-binding domain of AML1 and a nearly full-length ETO. The role of NCoR/SMRT corepressors in AML was first revealed by their interaction with ETO, the fusion partner in the t(8;21) AML and NCoR/SMRT [77,123,124]. ETO is a member of a small family of corepressors that can form oligomers and bind to NCoR/SMRT and HDACs [125,126]. Its homologous protein ETO-2 is also involved in AML by fusing to AML1 [127]. The current literature is consistent with the model that the AML1-ETO/ETO-2 promote leukemogenesis in part by recruiting the NCoR/SMRT/HDAC to repress transcription of target genes [77,128-130]. Interestingly, a recent work has shown that another leukemia fusion protein E2A-Pbx1 drives leukemogenesis by escaping ETO-2-mediated repression in order to activate Hox target genes [131]. Thus, the role of corepressors in different types of leukemias is likely context-dependent, emphasizing the need to dissect the individual mechanisms for therapeutic targeting of the corepressors.
Other subunits of the NCoR/SMRT complex
HDAC3
HDACs deacetylate both histones and proteins, and play important roles in the regulation of chromatin biology and gene transcription [77,132-134]. The 18 human HDACs can be divided into 4 classes [28]. Class I, II and IV belong to Zinc-dependent, NAD-independent histone deacetylases, whereas class III belongs to NAD-dependent histone deacetylases. Class I HDACs include HDAC1, 2, 3 and 8 isoforms, which are encoded by separate genes. The Class I HDACs generally show ubiquitous expression, and HDAC1-3 have been well studied. HDAC1 and HDAC2 exist as a dimer in several HDAC1/2-shared complexes, including the NURD chromatin-remodeling complex [135,136], the Sin3A-containing corepressor complex and the CoREST/LSD1-containing histone deacetylase/histone demethylase complex [137,138]. Whereas HDAC1 and HDAC2 generally show constitutive nuclear localization, HDAC3 shows both cytosolic and nuclear localization [139]. Consistent with this, HDAC3 contains both nuclear import and export domains [140]. In the nucleus, HDAC3 specifically associates with NCoR/SMRT corepressor complexes [28].
Several studies have implicated a role for HDAC3 in cancer. Upregulation of HDAC3 has been observed in several types of cancers including colon, lung, prostate and breast cancers, in a manner that correlates with poor survival and prognosis of the patients [141-148]. HDAC3 has served as a potential drug target in the treatment of PML-RARα, PLZF-RARα and AML1-ETO leukemias [77,126,149,150]. Independent studies using cancer and leukemia-derived cell lines have confirmed pro-survival roles for HDAC3. The pro-survival activity of HDAC3 revealed by these studies may be mediated by its roles in transcriptional repression of growth-arrest genes such as p21 [55,151,152], cell cycle progression [153-155], genomic stability [156], or its involvement in apoptosis/cell death/survival pathways [141,142,150,157-159]. In addition to regulating cell proliferation, HDAC3 has also been shown to play an important role in metastasis of cancer cells through its ability to enhance epithelial-mesenchymal transition (EMT) [160]. HIF-1alpha-induced HDAC3 was found to be crucial for hypoxia-induced EMT and metastatic phenotypes. HDAC3 crosstalks with WDR5 to activate the expression of mesenchymal genes expression, while repressing epithelial gene expression. While these studies have supported an anti-tumorigenic function of HDAC3, Bhaskara et al. have shown that mice depleted of HDAC3 in liver develop hepatocellular carcinoma [156,161], suggesting that HDAC3 may have context-dependent function in different cancers.
An active area in HDAC study is the development of isoform-specific HDAC inhibitors [162-164]. While pan-HDAC inhibitors have promising effects in the treatment of cancers, the clinical use of these inhibitors is complicated by their off-target effects [165-168]. It has been proposed that HDAC3 may be responsible for most of the anti-tumor effects of HDAC inhibitors [154,169]. A few studies reported the development and use of HDAC3-specific inhibitors that target the active form of HDAC3 [169,170]. Inhibition of HDAC3 using such a selective inhibitor RGFP966 promotes apoptosis and diminishes cell growth in cutaneous T cell lymphoma cell lines due to DNA damage and compromised S phase progression [169].
An alternative strategy to inhibit HDAC3 function is to prevent its activation by blocking the formation of HDAC3 complexes with corepressors, based on findings that the enzymatic activity of HDAC3 is strictly dependent on its binding to NCoR/SMRT [28,171]. The critical importance of corepressor interaction for HDAC3 activity has been recently demonstrated in vivo [172], which shows that mice harboring loss-of-function mutations in the DAD domain of both NCoR and SMRT have essentially no active HDAC3 in all tested tissues, which correlates with globally-increased histone acetylation. These mice, unlike the embryonic lethality of the total HDAC3 knockout mice [173], are vital, suggesting that free HDAC3 is not non-functional. A follow-up study by the Lazar group [174] recently showed that an HDAC3 mutant that is enzymatically inactive but retains the ability to bind to corepressors can rescue the HDAC3-null defect in liver, indicating that HDAC activity is not required for all HDAC3 functions, but its interaction with corepressors such as NCoR is critical. This would challenge the rationale of using traditionally-developed HDAC inhibitors that target the active site of HDACs to inhibit the function of HDAC3 (and possibly other HDACs) in cancers. Nevertheless, the above-mentioned strategy of using inhibitors to prevent the formation of stable HDAC3-corepressor complexes would continue to be warranted in targeting HDAC3 function in cancers.
It has been shown that the inactive form of HDAC3 is in complexes with Hsc70 and TRiC chaperones [139]. These chaperones are released from HDAC3 in its mature form after corepressor association. A recent work has shown that knockdown of NCoR and SMRT in cultured cells reduces the steady-state protein expression of HDAC3 in all tested cell types including colon, prostate and breast cancer cells [175], indicating that the E3 ligase-driven HDAC3 degradation pathway is still intact in cancer cells. Elucidating the nature of HDAC3-specific E3 ligases may offer a new approach to target HDAC3 in cancer cells. It is noteworthy that the steady-state level of liver HDAC3 in the above-mentioned DAD mutant knock-in mice does not differ significantly from the control mice. The difference between living animals and cultured cell may be caused by the use of different assay conditions. Alternatively, the mice may have been forced to compensate for the loss of HDAC3 given that HDAC3 is required for the early embryonic development.
Previous studies have suggested that the C-terminus of HDAC3 is required for its interaction with CoR [140,171]. Surprisingly, it was shown that if proteasomal activity is blocked, a C-terminus-truncated HDAC3 can still form a complex with CoR in cells [175]. Proteasome-dependent rapid turnover thus may explain earlier failures to detect an interaction between CoR and the C-terminus-truncated HDAC3. Despite the binding to CoR, the C-terminus-truncated HDAC3 remains associated with chaperones and cannot be activated. These data are consistent with a novel function for HDAC3 C-terminus in coupling CoR binding, chaperone dismissal, and subsequent activation and stabilization of HDAC3, suggesting that the C-terminus acts at a step after the binding of corepressors to facilitate the release of chaperones from HDAC3 (Figure 2). A role for C-terminus in regulating HDAC3 activity is also supported by the finding that phosphorylation of the conserved C-terminal residue S424 increases the activity of HDAC3 [176]. A recent work reported the 3D structure of a complex containing a C-terminus-truncated HDAC3 and SMRT-DAD [177]. It is worth noting that the C-terminus is cleaved after the formation of the complex, consistent with its role involved in the complex formation and HDAC3 activation.
Figure 2.
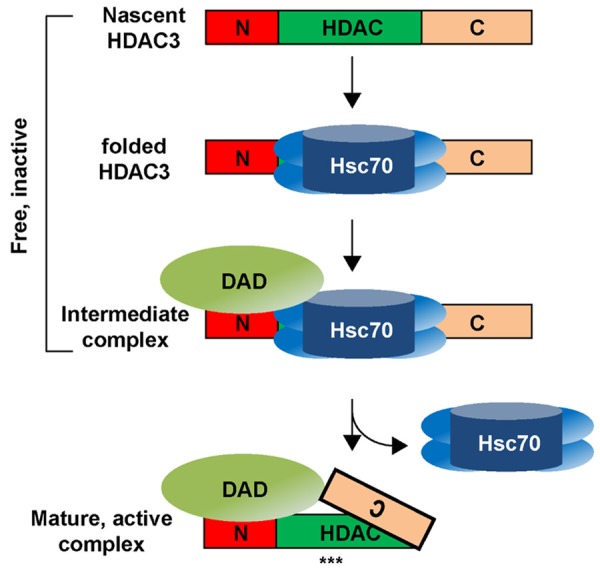
A hypothetical model of HDAC3 activation by corepressors. In the inactive state, HDAC3 is complexed with Hsc70 and TRiC chaperones. Corepressors bind to HDAC3 N-terminus through the DAD domain. The DAD-triggered HDAC3 activation occurs in a stepwise fashion and involves the formation of an intermediate DAD-HDAC3-chaperone complex, whose subsequent conversion into the mature, active state requires a C-terminus dependent conformational change to dissociate chaperones. “***” denotes exposed HDAC3 active site.
TBL1 and TBLR1
TBL1 is a protein initially found to play a role in the X-linked late-onset deafness in human [178]. Its counterpart in fly, ebi, has been shown to play a role in fly eye development by regulating the EGF receptor-mediated signaling pathways [179]. Interestingly, Lazar and Wong groups later discovered that TBL1 is a stoichiometric subunit of NCoR and SMRT nuclear receptor corepressor complexes [30,44]. TBL1 directly interacts with histone tails and facilitates the function of HDAC3 [30,44]. By purifying FLAG-NCoR and FLAG-HDAC3 interacting proteins, Zhang et al. discovered a TBL1-like protein, which they named TBLR1 (TBL1-like protein 1) [28]. TBLR1/TBL1, like NCoR and SMRT, are the alternative subunits of CoR/HDAC3 corepressor complexes. Interestingly, the expression of TBLR1 is reduced in mature hematopoietic cells compared to progenitor cells [180], suggesting a role for TBLR1 in regulating cell differentiation.
Both TBL1 and TBLR1 have been shown to have “coactivator” functions. Although the classical Gal4-fusion assay failed to support an “activation domain” in TBL1/TBLR1, it confirmed that TBL1/TBLR1 harbor transferable repression domains [30,44]. This shows that TBL1/TBLR1 are not classical coactivators. The “coactivator” activity of TBL1/TBLR1 appears to be attributed to their ability to drive proteasomal degradation of NCoR/SMRT and CtBP corepressors in a signal-dependent fashion [181]. Perissi et al. showed that TBL1/TBLR1 promote the clearance of NCoR/SMRT and CtBP corepressors by recruiting 19S proteasome particles to drive the ubiquitination and degradation of NCoR/SMRT and CtBP, thus allowing the recruitment of coactivators. This takes place not only on NRs but also on c-Jun and NF-κB binding sites of affected genes [181,182]. Phosphorylation of TBL1 and TBLR1 on specific residues is required to activate their E3 ligase activities. TBL1 and TBLR1 have imperfect F-boxes. Therefore, an interesting model is that these proteins normally reside in the NCoR/SMRT complex in the inactive state. Phosphorylation and/or possibly other post-translational modifications activate their latent E3 ligase activities, leading to degradation of the corepressors.
Dysregulation of Wnt-β-catenin signaling pathway has been associated with tumorigenesis. Another form of post-translational modifications on TBL1/TBLR1, SUMOylation, has been shown to dissociate TBL1/TBLR1 from the NCoR complex, and subsequently allow them to complex with β-catenin to dock on and activate the promoter of Wnt target genes, which is important for the tumorigenicity of SW480 colon cancer cells [183]. These results suggest that the nature of post-translational modification of TBL1 and TBLR1 determines whether they may function as tumor suppressors or as oncogenes to facilitate repression or activation of context-dependent target genes.
A recent development has implicated TBLR1 involvement in AR regulation and prostate cancer progression. TBLR1 is found to be primarily localized in the nucleus of benign prostate cells and the level is reduced in prostate cancer cells. TBLR1 binds to AR and potentiates AR-dependent transcription of target genes in a phosphorylation- and 19S proteasome-dependent manner. Interestingly, TBLR1 selectively activates genes important for growth suppression and differentiation but not pro-proliferative AR target genes, suggesting a tumor suppressor function of TBLR1 in prostate cancer [184].
GPS2
In search for proteins that can rescue the lethal phenotype of yeast harboring a dominant active yeast Gβγ mutant, Spain et al. have discovered GPS2 as a G-protein pathway suppressor and shown that GPS2 can suppress RAS/MAPK-dependent JNK activation [185]. Another group independently found that GPS2 interacts with Tax, a human T-cell lymphotrophic virus transcription factor, and suppresses TNFalpha-dependent activation of the JNK pathway [186]. Biochemical purification of novel NCoR/HDAC3-interacting proteins identified GPS2 as an integral component of the NCoR/SMRT/HDAC3 complex [28]. A coiled-coil region of GPS2 (amino acids 1-105) directly interacts with the N-terminal regions of both NCoR/SMRT and TBL1/TBLR1 to form a heterotrimeric complex. A recent structural study has confirmed these domain interactions and also shown that TBL1 is organized into a tetrameric structure [31].
Since the discovery of GPS2 as a G-protein pathway suppressor, GPS2 has emerged as a multifunctional protein. GPS2 can act both as a corepressor and a coactivator for various transcription factors including nuclear receptors [187-194], consistent with the presence of distinct repression and activation domains in GPS2 [28,187]. Consistent with its coactivator function, GPS2 has also been shown to facilitate histone demethylation of certain target genes [189,195]. These pro-activation activities of GPS2 may be independent of its interactions with NCoR/SMRT and TBL1/TBLR1. In the context of repression, it has been proposed that GPS2 may, analogous to NCoR/SMRT, tether the corepressor complex to sequence-specific transcription factors. While this idea is still interesting, and in fact is consistent with the reported anti-inflammatory function of GPS2 [196], the nature of transcription factors that use GPS2 to recruit the CoR complex, and the corresponding target genes regulated by the transcription factors, remain to be determined.
Several recent studies have provided evidence that the interaction between GPS2 and CoRs may be a drugable target in cancer. Bi et al. [197] reported that GPS2 is SUMOylated at K45 and K71 within the coiled-coil region that interacts with CoRs. SUMOylation increases the corepressor function of GPS2 for ERα-mediated transcription, initially reported by Cheng et al. [194], in both MCF-7 and T47D breast cancer cells due to enhanced GPS2 incorporation into the TBL1/SMRT complex, which correlates with reduced proliferation of MCF-7 and T47D cells.
Two recent studies reported somatic mutations of GPS2 in a form of aggressive brain tumor, Medulloblastoma [198]. These mutations affect the residues in the coiled-coil region of GPS2 that mediate its interaction with NCoR/SMRT and TBL1/TBLR1. The mutations significantly worsen the prognosis [199]. The impact of these mutations on the GPS2-CoR interaction has yet to be studied. Given the anti-inflammatory function of GPS2, one may speculate that such mutations may reduce the ability of the corepressor complex to suppress cytokine-dependent signaling pathways, which may predispose the affected cells to oncogenesis.
GPS2 turns out to be the first CoR complex subunit that forms fusion proteins with different partners in cancers. Undifferentiated spindle cell sarcoma (UDS) is a type of cancer with poorly defined diagnostic markers. O’Meara et al. identified a novel chromosomal translocation t(17;19)(p13;q13) in a pediatric UDS [200]. Interestingly, this in-frame fusion is between GPS2 and MLL4, which is the active subunit of one of the histone H3K4 methyltransferase complexes [201]. This fusion is oncogenic because it promotes anchorage-independent growth of the cells. It will be interesting to determine whether the fusion protein-containing complexes show multiple histone modification activities conferred by MLL4 (methyltransferase) and GPS2 (deacetylase and/or demethylase), which may be important for deregulation of gene expression in tumors. Besides MLL4, GPS2 has also been reported to fuse to other proteins in a prostate carcinoma cell line [202] and in glioblastoma multiforme [203]. These findings underscore the emerging important role of GPS2 in cancer.
Conclusion and future directions
Since the discovery of NCoR/SMRT nearly 20 years ago, tremendous progress has been made in understanding the mechanisms, regulation and functions of NCoR/SMRT and their interacting proteins in transcription, and their involvement in diverse pathways that regulate metabolism, inflammation and oncogenesis. A major advance in the field is the discovery of the multi-protein complex containing NCoR/SMRT, HDAC3, TBL1/TBLR1 and GPS2. However, to date, we still do not fully understand how these proteins regulate transcription in normal and cancer cells. Future studies may be devoted to understanding (i) the specificity of NCoR, SMRT, and their different isoforms, (ii) how the free form of HDAC3 assembles into the mature NCoR/SMRT-HDAC3 complex, (iii) the mechanism of “coactivator” functions of TBL1/TBLR1 and GPS2 and how these functions may be related to their interactions with NCoR/SMRT, (iv) the regulation of the expression of these proteins in normal and cancer cells, and (v) the genome-wide targets of these proteins. While the literature has supported the concept that deregulated corepressor function facilitates cancer development by disrupting the homeostatic balance between corepressors and coactivators (or other regulatory proteins) in the regulation of chromatin structure, transcription, DNA repair, inflammation and other important biological processes (Figure 3), corepressors may have different mechanisms and play either pro- or anti-tumorigenic roles in different types of cancers and leukemias. It is thus important to fully understand the context-dependent function of corepressors. Given the direct interactions between the various corepressor components and the transcription factors (such as ER, AR and leukemia fusion proteins) involved in cancers and leukemias, these corepressor proteins should serve as important drugable targets for cancer therapy.
Figure 3.
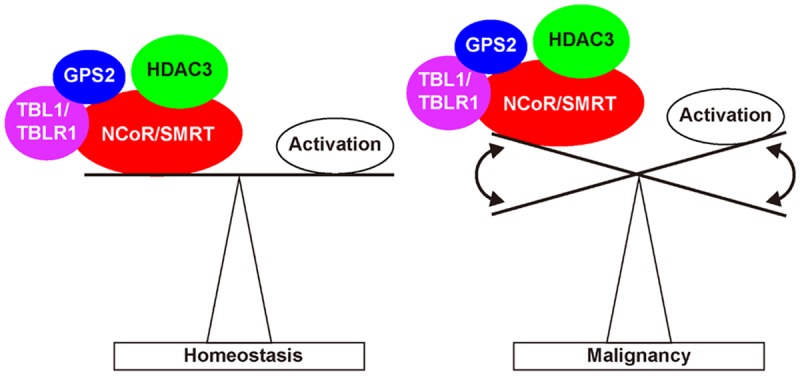
A working model showing that deregulated function of the corepressor complex disrupts homeostatic balance of cellular pathways to promote cancer progression.
Acknowledgements
This work is supported by National Institutes of Health grants R01HL093195 and R21CA178513 (to J.Z.). We apologize for not being able to cite all related references in this review due to length limitations in the main text and references.
Disclosure of conflict of interest
None.
References
- 1.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 2.Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, Rosenfeld MG. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 3.Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 4.Baniahmad A, Köhne AC, Renkawitz R. A transferable silencing domain is present in the thyroid hormone receptor, in the v-erbA oncogene product and in the retinoic acid receptor. EMBO J. 1992;11:1015–1023. doi: 10.1002/j.1460-2075.1992.tb05140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tong GX, Jeyakumar M, Tanen MR, Bagchi MK. Transcriptional silencing by unliganded thyroid hormone receptor beta requires a soluble corepressor that interacts with the ligand-binding domain of the receptor. Mol Cell Biol. 1996;16:1909–1920. doi: 10.1128/mcb.16.5.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J, Lazar MA. The mechanism of action of thyroid hormones. Annu Rev Physiol. 2000;62:439–466. doi: 10.1146/annurev.physiol.62.1.439. [DOI] [PubMed] [Google Scholar]
- 7.Ordentlich P, Downes M, Xie W, Genin A, Spinner NB, Evans RM. Unique forms of human and mouse nuclear receptor corepressor SMRT. Proc Natl Acad Sci U S A. 1999;96:2639–2644. doi: 10.1073/pnas.96.6.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park EJ, Schroen DJ, Yang M, Li H, Li L, Chen JD. SMRTe, a silencing mediator for retinoid and thyroid hormone receptors-extended isoform that is more related to the nuclear receptor corepressor. Proc Natl Acad Sci U S A. 1999;96:3519–3524. doi: 10.1073/pnas.96.7.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dwivedi PP, Muscat GE, Bailey PJ, Omdahl JL, May BK. Repression of basal transcription by vitamin D receptor: evidence for interaction of unliganded vitamin D receptor with two receptor interaction domains in RIP13delta1. J Mol Endocrinol. 1998;20:327–335. doi: 10.1677/jme.0.0200327. [DOI] [PubMed] [Google Scholar]
- 10.Dowell P, Ishmael JE, Avram D, Peterson VJ, Nevrivy DJ, Leid M. Identification of nuclear receptor corepressor as a peroxisome proliferator-activated receptor alpha interacting protein. J Biol Chem. 1999;274:15901–15907. doi: 10.1074/jbc.274.22.15901. [DOI] [PubMed] [Google Scholar]
- 11.Wagner BL, Valledor AF, Shao G, Daige CL, Bischoff ED, Petrowski M, Jepsen K, Baek SH, Heyman RA, Rosenfeld MG, Schulman IG, Glass CK. Promoter-specific roles for liver X receptor/corepressor complexes in the regulation of ABCA1 and SREBP1 gene expression. Mol Cell Biol. 2003;23:5780–5789. doi: 10.1128/MCB.23.16.5780-5789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harding HP, Lazar MA. The monomer-binding orphan receptor Rev-Erb represses transcription as a dimer on a novel direct repeat. Mol Cell Biol. 1995;15:4791–4802. doi: 10.1128/mcb.15.9.4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zamir I, Harding HP, Atkins GB, Horlein A, Glass CK, Rosenfeld MG, Lazar MA. A nuclear hormone receptor corepressor mediates transcriptional silencing by receptors with distinct repression domains. Mol Cell Biol. 1996;16:5458–5465. doi: 10.1128/mcb.16.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata H, Nawaz Z, Tsai SY, O’Malley BW, Tsai MJ. Gene silencing by chicken ovalbumin upstream promoter-transcription factor I (COUP-TFI) is mediated by transcriptional corepressors, nuclear receptor-corepressor (N-CoR) and silencing mediator for retinoic acid receptor and thyroid hormone receptor (SMRT) Mol Endocrinol. 1997;11:714–724. doi: 10.1210/mend.11.6.0002. [DOI] [PubMed] [Google Scholar]
- 15.Crawford PA, Dorn C, Sadovsky Y, Milbrandt J. Nuclear receptor DAX-1 recruits nuclear receptor corepressor N-CoR to steroidogenic factor 1. Mol Cell Biol. 1998;18:2949–2956. doi: 10.1128/mcb.18.5.2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 17.Dotzlaw H, Moehren U, Mink S, Cato AC, Iniguez Lluhi JA, Baniahmad A. The amino terminus of the human AR is target for corepressor action and antihormone agonism. Mol Endocrinol. 2002;16:661–673. doi: 10.1210/mend.16.4.0798. [DOI] [PubMed] [Google Scholar]
- 18.Liao G, Chen LY, Zhang A, Godavarthy A, Xia F, Ghosh JC, Li H, Chen JD. Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. J Biol Chem. 2003;278:5052–5061. doi: 10.1074/jbc.M206374200. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Jeyakumar M, Petukhov S, Bagchi MK. A nuclear receptor corepressor modulates transcriptional activity of antagonist-occupied steroid hormone receptor. Mol Endocrinol. 1998;12:513–524. doi: 10.1210/mend.12.4.0089. [DOI] [PubMed] [Google Scholar]
- 20.Liu XF, Bagchi MK. Recruitment of distinct chromatin-modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J Biol Chem. 2004;279:15050–15058. doi: 10.1074/jbc.M311932200. [DOI] [PubMed] [Google Scholar]
- 21.Hu X, Lazar MA. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature. 1999;402:93–96. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- 22.Perissi V, Staszewski LM, McInerney EM, Kurokawa R, Krones A, Rose DW, Lambert MH, Milburn MV, Glass CK, Rosenfeld MG. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev. 1999;13:3198–3208. doi: 10.1101/gad.13.24.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagy L, Kao HY, Love JD, Li C, Banayo E, Gooch JT, Krishna V, Chatterjee K, Evans RM, Schwabe JW. Mechanism of corepressor binding and release from nuclear hormone receptors. Genes Dev. 1999;13:3209–3216. doi: 10.1101/gad.13.24.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 25.Webb P, Anderson CM, Valentine C, Nguyen P, Marimuthu A, West BL, Baxter JD, Kushner PJ. The nuclear receptor corepressor (N-CoR) contains three isoleucine motifs (I/LXXII) that serve as receptor interaction domains (IDs) Mol Endocrinol. 2000;14:1976–1985. doi: 10.1210/mend.14.12.0566. [DOI] [PubMed] [Google Scholar]
- 26.Cohen RN, Brzostek S, Kim B, Chorev M, Wondisford FE, Hollenberg AN. The specificity of interactions between nuclear hormone receptors and corepressors is mediated by distinct amino acid sequences within the interacting domains. Mol Endocrinol. 2001;15:1049–1061. doi: 10.1210/mend.15.7.0669. [DOI] [PubMed] [Google Scholar]
- 27.Hu X, Li Y, Lazar MA. Determinants of CoRNR-dependent repression complex assembly on nuclear hormone receptors. Mol Cell Biol. 2001;21:1747–1758. doi: 10.1128/MCB.21.5.1747-1758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Kalkum M, Chait BT, Roeder RG. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol Cell. 2002;9:611–623. doi: 10.1016/s1097-2765(02)00468-9. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guenther MG, Lane WS, Fischle W, Verdin E, Lazar MA, Shiekhattar R. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 2000;14:1048–1057. [PMC free article] [PubMed] [Google Scholar]
- 31.Oberoi J, Fairall L, Watson PJ, Yang JC, Czimmerer Z, Kampmann T, Goult BT, Greenwood JA, Gooch JT, Kallenberger BC, Nagy L, Neuhaus D, Schwabe JW. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat Struct Mol Biol. 2011;18:177–184. doi: 10.1038/nsmb.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Iratni R, Erdjument-Bromage H, Tempst P, Reinberg D. Histone deacetylases and SAP18, a novel polypeptide, are components of a human Sin3 complex. Cell. 1997;89:357–364. doi: 10.1016/s0092-8674(00)80216-0. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Sun ZW, Iratni R, Erdjument-Bromage H, Tempst P, Hampsey M, Reinberg D. SAP30, a novel protein conserved between human and yeast, is a component of a histone deacetylase complex. Mol Cell. 1998;1:1021–1031. doi: 10.1016/s1097-2765(00)80102-1. [DOI] [PubMed] [Google Scholar]
- 34.Jones PL, Sachs LM, Rouse N, Wade PA, Shi YB. Multiple N-CoR complexes contain distinct histone deacetylases. J Biol Chem. 2001;276:8807–8811. doi: 10.1074/jbc.C000879200. [DOI] [PubMed] [Google Scholar]
- 35.Alland L, Muhle R, Hou H Jr, Potes J, Chin L, Schreiber-Agus N, DePinho RA. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387:49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- 36.Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 37.Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373–380. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 38.Huang EY, Zhang J, Miska EA, Guenther MG, Kouzarides T, Lazar MA. Nuclear receptor corepressors partner with class II histone deacetylases in a Sin3-independent repression pathway. Genes Dev. 2000;14:45–54. [PMC free article] [PubMed] [Google Scholar]
- 39.Kao HY, Downes M, Ordentlich P, Evans RM. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 2000;14:55–66. [PMC free article] [PubMed] [Google Scholar]
- 40.Underhill C, Qutob MS, Yee SP, Torchia J. A novel nuclear receptor corepressor complex, N-CoR, contains components of the mammalian SWI/SNF complex and the corepressor KAP-1. J Biol Chem. 2000;275:40463–40470. doi: 10.1074/jbc.M007864200. [DOI] [PubMed] [Google Scholar]
- 41.Cowger JJ, Torchia J. Direct association between the CREB-binding protein (CBP) and nuclear receptor corepressor (N-CoR) Biochemistry (Mosc) 2006;45:13150–13162. doi: 10.1021/bi060562g. [DOI] [PubMed] [Google Scholar]
- 42.Jepsen K, Rosenfeld MG. Biological roles and mechanistic actions of co-repressor complexes. J Cell Sci. 2002;115:689–698. doi: 10.1242/jcs.115.4.689. [DOI] [PubMed] [Google Scholar]
- 43.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 44.Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, Wong J. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J. 2003;22:1336–1346. doi: 10.1093/emboj/cdg120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Varlakhanova N, Snyder C, Jose S, Hahm JB, Privalsky ML. Estrogen receptors recruit SMRT and N-CoR corepressors through newly recognized contacts between the corepressor N terminus and the receptor DNA binding domain. Mol Cell Biol. 2010;30:1434–1445. doi: 10.1128/MCB.01002-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aasland R, Stewart AF, Gibson T. The SANT domain: a putative DNA-binding domain in the SWI-SNF and ADA complexes, the transcriptional co-repressor N-CoR and TFIIIB. Trends Biochem Sci. 1996;21:87–88. [PubMed] [Google Scholar]
- 47.Codina A, Love JD, Li Y, Lazar MA, Neuhaus D, Schwabe JW. Structural insights into the interaction and activation of histone deacetylase 3 by nuclear receptor corepressors. Proc Natl Acad Sci U S A. 2005;102:6009–6014. doi: 10.1073/pnas.0500299102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ishizuka T, Lazar MA. The nuclear receptor corepressor deacetylase activating domain is essential for repression by thyroid hormone receptor. Mol Endocrinol. 2005;19:1443–1451. doi: 10.1210/me.2005-0009. [DOI] [PubMed] [Google Scholar]
- 49.Adikesavan AK, Karmakar S, Pardo P, Wang L, Liu S, Li W, Smith CL. Activation of p53 transcriptional activity by SMRT: a histone deacetylase 3-independent function of a transcriptional corepressor. Mol Cell Biol. 2014;34:1246–1261. doi: 10.1128/MCB.01216-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peterson TJ, Karmakar S, Pace MC, Gao T, Smith CL. The silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) corepressor is required for full estrogen receptor alpha transcriptional activity. Mol Cell Biol. 2007;27:5933–5948. doi: 10.1128/MCB.00237-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boyer LA, Langer MR, Crowley KA, Tan S, Denu JM, Peterson CL. Essential role for the SANT domain in the functioning of multiple chromatin remodeling enzymes. Mol Cell. 2002;10:935–942. doi: 10.1016/s1097-2765(02)00634-2. [DOI] [PubMed] [Google Scholar]
- 52.Yu J, Li Y, Ishizuka T, Guenther MG, Lazar MA. A SANT motif in the SMRT corepressor interprets the histone code and promotes histone deacetylation. EMBO J. 2003;22:3403–3410. doi: 10.1093/emboj/cdg326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hartman HB, Yu J, Alenghat T, Ishizuka T, Lazar MA. The histone-binding code of nuclear receptor co-repressors matches the substrate specificity of histone deacetylase 3. EMBO Rep. 2005;6:445–451. doi: 10.1038/sj.embor.7400391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoon HG, Choi Y, Cole PA, Wong J. Reading and function of a histone code involved in targeting corepressor complexes for repression. Mol Cell Biol. 2005;25:324–335. doi: 10.1128/MCB.25.1.324-335.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karagianni P, Wong J. HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene. 2007;26:5439–5449. doi: 10.1038/sj.onc.1210612. [DOI] [PubMed] [Google Scholar]
- 56.Barish GD, Yu RT, Karunasiri MS, Becerra D, Kim J, Tseng TW, Tai LJ, Leblanc M, Diehl C, Cerchietti L, Miller YI, Witztum JL, Melnick AM, Dent AL, Tangirala RK, Evans RM. The Bcl6-SMRT/NCoR cistrome represses inflammation to attenuate atherosclerosis. Cell Metab. 2012;15:554–562. doi: 10.1016/j.cmet.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang C, Hatzi K, Melnick A. Lineage-specific functions of Bcl-6 in immunity and inflammation are mediated by distinct biochemical mechanisms. Nat Immunol. 2013;14:380–388. doi: 10.1038/ni.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dhordain P, Lin RJ, Quief S, Lantoine D, Kerckaert JP, Evans RM, Albagli O. The LAZ3 (BCL-6) oncoprotein recruits a SMRT/mSIN3A/histone deacetylase containing complex to mediate transcriptional repression. Nucleic Acids Res. 1998;26:4645–4651. doi: 10.1093/nar/26.20.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wong CW, Privalsky ML. Components of the SMRT corepressor complex exhibit distinctive interactions with the POZ domain oncoproteins PLZF, PLZF-RARalpha, and BCL-6. J Biol Chem. 1998;273:27695–27702. doi: 10.1074/jbc.273.42.27695. [DOI] [PubMed] [Google Scholar]
- 60.Bailey P, Downes M, Lau P, Harris J, Chen SL, Hamamori Y, Sartorelli V, Muscat GE. The nuclear receptor corepressor N-CoR regulates differentiation: N-CoR directly interacts with MyoD. Mol Endocrinol. 1999;13:1155–1168. doi: 10.1210/mend.13.7.0305. [DOI] [PubMed] [Google Scholar]
- 61.Iso T, Sartorelli V, Poizat C, Iezzi S, Wu HY, Chung G, Kedes L, Hamamori Y. HERP, a novel heterodimer partner of HES/E(spl) in Notch signaling. Mol Cell Biol. 2001;21:6080–6089. doi: 10.1128/MCB.21.17.6080-6089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scully KM JE, Jepsen K, Lunyak V, Viadiu H, Carrière C, Rose DW, Hooshmand F, Aggarwal AK, Rosenfeld MG. Allosteric effects of Pit-1 DNA sites on long-term repression in cell type specification. Science. 2000;290:1127–1131. doi: 10.1126/science.290.5494.1127. [DOI] [PubMed] [Google Scholar]
- 63.Xu L, Lavinsky RM, Dasen JS, Flynn SE, McInerney EM, Mullen TM, Heinzel T, Szeto D, Korzus E, Kurokawa R, Aggarwal AK, Rose DW, Glass CK, Rosenfeld MG. Signal-specific co-activator domain requirements for Pit-1 activation. Nature. 1998;395:301–306. doi: 10.1038/26270. [DOI] [PubMed] [Google Scholar]
- 64.Kakizawa T, Miyamoto T, Ichikawa K, Takeda T, Suzuki S, Mori J, Kumagai M, Yamashita K, Hashizume K. Silencing mediator for retinoid and thyroid hormone receptors interacts with octamer transcription factor-1 and acts as a transcriptional repressor. J Biol Chem. 2001;276:9720–9725. doi: 10.1074/jbc.M008531200. [DOI] [PubMed] [Google Scholar]
- 65.Kao HY, Ordentlich P, Koyano-Nakagawa N, Tang Z, Downes M, Kintner CR, Evans RM, Kadesch T. A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev. 1998;12:2269–2277. doi: 10.1101/gad.12.15.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oswald F, Kostezka U, Astrahantseff K, Bourteele S, Dillinger K, Zechner U, Ludwig L, Wilda M, Hameister H, Knochel W, Liptay S, Schmid RM. SHARP is a novel component of the Notch/RBP-Jkappa signalling pathway. EMBO J. 2002;21:5417–5426. doi: 10.1093/emboj/cdf549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou S, Hayward SD. Nuclear localization of CBF1 is regulated by interactions with the SMRT corepressor complex. Mol Cell Biol. 2001;21:6222–6232. doi: 10.1128/MCB.21.18.6222-6232.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Asahara H, Dutta S, Kao HY, Evans RM, Montminy M. Pbx-Hox heterodimers recruit coactivator-corepressor complexes in an isoform-specific manner. Mol Cell Biol. 1999;19:8219–8225. doi: 10.1128/mcb.19.12.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saleh M, Rambaldi I, Yang XJ, Featherstone MS. Cell signaling switches HOX-PBX complexes from repressors to activators of transcription mediated by histone deacetylases and histone acetyltransferases. Mol Cell Biol. 2000;20:8623–8633. doi: 10.1128/mcb.20.22.8623-8633.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee SK, Na SY, Jung SY, Choi JE, Jhun BH, Cheong J, Meltzer PS, Lee YC, Lee JW. Activating protein-1, nuclear factor-kappaB, and serum response factor as novel target molecules of the cancer-amplified transcription coactivator ASC-2. Mol Endocrinol. 2000;14:915–925. doi: 10.1210/mend.14.6.0471. [DOI] [PubMed] [Google Scholar]
- 71.Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell. 2002;110:55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- 72.Espinosa L, Santos S, Ingles-Esteve J, Munoz-Canoves P, Bigas A. p65-NFkappaB synergizes with Notch to activate transcription by triggering cytoplasmic translocation of the nuclear receptor corepressor N-CoR. J Cell Sci. 2002;115:1295–1303. doi: 10.1242/jcs.115.6.1295. [DOI] [PubMed] [Google Scholar]
- 73.Espinosa L, Ingles-Esteve J, Robert-Moreno A, Bigas A. IkappaBalpha and p65 regulate the cytoplasmic shuttling of nuclear corepressors: cross-talk between Notch and NFkappaB pathways. Mol Biol Cell. 2003;14:491–502. doi: 10.1091/mbc.E02-07-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jang MK, Goo YH, Sohn YC, Kim YS, Lee SK, Kang H, Cheong J, Lee JW. Ca2+/calmodulin-dependent protein kinase IV stimulates nuclear factor-kappa B transactivation via phosphorylation of the p65 subunit. J Biol Chem. 2001;276:20005–20010. doi: 10.1074/jbc.M010211200. [DOI] [PubMed] [Google Scholar]
- 75.Hong SH, David G, Wong CW, Dejean A, Privalsky ML. SMRT corepressor interacts with PLZF and with the PML-retinoic acid receptor alpha (RARalpha) and PLZF-RARalpha oncoproteins associated with acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 1997;94:9028–9033. doi: 10.1073/pnas.94.17.9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.He LZ, Guidez F, Tribioli C, Peruzzi D, Ruthardt M, Zelent A, Pandolfi PP. Distinct interactions of PML-RARalpha and PLZF-RARalpha with co-repressors determine differential responses to RA in APL. Nat Genet. 1998;18:126–135. doi: 10.1038/ng0298-126. [DOI] [PubMed] [Google Scholar]
- 77.Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci PG, Lazar MA. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol. 1998;18:7185–7191. doi: 10.1128/mcb.18.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lutterbach B, Westendorf JJ, Linggi B, Patten A, Moniwa M, Davie JR, Huynh KD, Bardwell VJ, Lavinsky RM, Rosenfeld MG, Glass C, Seto E, Hiebert SW. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol Cell Biol. 1998;18:7176–7184. doi: 10.1128/mcb.18.12.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc Natl Acad Sci U S A. 1998;95:10860–10865. doi: 10.1073/pnas.95.18.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luo K, Stroschein SL, Wang W, Chen D, Martens E, Zhou S, Zhou Q. The Ski oncoprotein interacts with the Smad proteins to repress TGFbeta signaling. Genes Dev. 1999;13:2196–2206. doi: 10.1101/gad.13.17.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li X, McDonnell DP. The transcription factor B-Myb is maintained in an inhibited state in target cells through its interaction with the nuclear corepressors N-CoR and SMRT. Mol Cell Biol. 2002;22:3663–3673. doi: 10.1128/MCB.22.11.3663-3673.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nguyen TA, Hoivik D, Lee JE, Safe S. Interactions of nuclear receptor coactivator/corepressor proteins with the aryl hydrocarbon receptor complex. Arch Biochem Biophys. 1999;367:250–257. doi: 10.1006/abbi.1999.1282. [DOI] [PubMed] [Google Scholar]
- 83.Shi Y, Downes M, Xie W, Kao HY, Ordentlich P, Tsai CC, Hon M, Evans RM. Sharp, an inducible cofactor that integrates nuclear receptor repression and activation. Genes Dev. 2001;15:1140–1151. doi: 10.1101/gad.871201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoon HG, Chan DW, Reynolds AB, Qin J, Wong J. N-CoR Mediates DNA Methylation-Dependent Repression through a Methyl CpG Binding Protein Kaiso. Mol Cell. 2003;12:723–734. doi: 10.1016/j.molcel.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 85.Ebert DH, Gabel HW, Robinson ND, Kastan NR, Hu LS, Cohen S, Navarro AJ, Lyst MJ, Ekiert R, Bird AP, Greenberg ME. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature. 2013;499:341–345. doi: 10.1038/nature12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fu J, Yoon HG, Qin J, Wong J. Regulation of P-TEFb elongation complex activity by CDK9 acetylation. Mol Cell Biol. 2007;27:4641–4651. doi: 10.1128/MCB.00857-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dobrzycka KM, Townson SM, Jiang S, Oesterreich S. Estrogen receptor corepressors -- a role in human breast cancer? Endocr Relat Cancer. 2003;10:517–536. doi: 10.1677/erc.0.0100517. [DOI] [PubMed] [Google Scholar]
- 89.Ciriello G, Sinha R, Hoadley KA, Jacobsen AS, Reva B, Perou CM, Sander C, Schultz N. The molecular diversity of Luminal A breast tumors. Breast Cancer Res Treat. 2013;141:409–420. doi: 10.1007/s10549-013-2699-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kurebayashi J, Otsuki T, Kunisue H, Tanaka K, Yamamoto S, Sonoo H. Expression levels of estrogen receptor-alpha, estrogen receptor-beta, coactivators, and corepressors in breast cancer. Clin Cancer Res. 2000;6:512–518. [PubMed] [Google Scholar]
- 91.Heldring N, Pawson T, McDonnell D, Treuter E, Gustafsson JA, Pike AC. Structural insights into corepressor recognition by antagonist-bound estrogen receptors. J Biol Chem. 2007;282:10449–10455. doi: 10.1074/jbc.M611424200. [DOI] [PubMed] [Google Scholar]
- 92.Smith CL, Nawaz Z, O’Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997;11:657–666. doi: 10.1210/mend.11.6.0009. [DOI] [PubMed] [Google Scholar]
- 93.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 94.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 95.Graham JD, Bain DL, Richer JK, Jackson TA, Tung L, Horwitz KB. Nuclear receptor conformation, coregulators, and tamoxifen-resistant breast cancer. Steroids. 2000;65:579–584. doi: 10.1016/s0039-128x(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 96.Zhang J, Guenther MG, Carthew RW, Lazar MA. Proteasomal regulation of nuclear receptor corepressor-mediated repression. Genes Dev. 1998;12:1775–1780. doi: 10.1101/gad.12.12.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Frasor J, Danes JM, Funk CC, Katzenellenbogen BS. Estrogen down-regulation of the corepressor N-CoR: mechanism and implications for estrogen derepression of N-CoR-regulated genes. Proc Natl Acad Sci U S A. 2005;102:13153–13157. doi: 10.1073/pnas.0502782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jansen MP, Ruigrok-Ritstier K, Dorssers LC, van Staveren IL, Look MP, Meijer-van Gelder ME, Sieuwerts AM, Helleman J, Sleijfer S, Klijn JG, Foekens JA, Berns EM. Downregulation of SIAH2, an ubiquitin E3 ligase, is associated with resistance to endocrine therapy in breast cancer. Breast Cancer Res Treat. 2009;116:263–271. doi: 10.1007/s10549-008-0125-z. [DOI] [PubMed] [Google Scholar]
- 99.Interiano RB, Yang J, Harris AL, Davidoff AM. Seven In Absentia Homolog 2 (SIAH2) downregulation is associated with tamoxifen resistance in MCF-7 breast cancer cells. J Surg Res. 2014;190:203–209. doi: 10.1016/j.jss.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stanya KJ, Liu Y, Means AR, Kao HY. Cdk2 and Pin1 negatively regulate the transcriptional corepressor SMRT. J Cell Biol. 2008;183:49–61. doi: 10.1083/jcb.200806172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.De Amicis F, Zupo S, Panno ML, Malivindi R, Giordano F, Barone I, Mauro L, Fuqua SA, Ando S. Progesterone receptor B recruits a repressor complex to a half-PRE site of the estrogen receptor alpha gene promoter. Mol Endocrinol. 2009;23:454–465. doi: 10.1210/me.2008-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Konduri SD, Medisetty R, Liu W, Kaipparettu BA, Srivastava P, Brauch H, Fritz P, Swetzig WM, Gardner AE, Khan SA, Das GM. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc Natl Acad Sci U S A. 2010;107:15081–15086. doi: 10.1073/pnas.1009575107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bartella V, Rizza P, Barone I, Zito D, Giordano F, Giordano C, Catalano S, Mauro L, Sisci D, Panno ML, Fuqua SA, Andò S. Estrogen receptor beta binds Sp1 and recruits a corepressor complex to the estrogen receptor alpha gene promoter. Breast Cancer Res Treat. 2012;134:569–581. doi: 10.1007/s10549-012-2090-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nat Rev Cancer. 2002;2:389–396. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Salesi N, Carlini P, Ruggeri EM, Ferretti G, Bria E, Cognetti F. Prostate cancer: the role of hormonal therapy. J Exp Clin Cancer Res. 2005;24:175–180. [PubMed] [Google Scholar]
- 106.Burd CJ, Morey LM, Knudsen KE. Androgen receptor corepressors and prostate cancer. Endocr Relat Cancer. 2006;13:979–994. doi: 10.1677/erc.1.01115. [DOI] [PubMed] [Google Scholar]
- 107.Yoon HG, Wong J. The corepressors silencing mediator of retinoid and thyroid hormone receptor and nuclear receptor corepressor are involved in agonist- and antagonist-regulated transcription by androgen receptor. Mol Endocrinol. 2006;20:1048–1060. doi: 10.1210/me.2005-0324. [DOI] [PubMed] [Google Scholar]
- 108.Hodgson MC, Astapova I, Cheng S, Lee LJ, Verhoeven MC, Choi E, Balk SP, Hollenberg AN. The androgen receptor recruits nuclear receptor CoRepressor (N-CoR) in the presence of mifepristone via its N and C termini revealing a novel molecular mechanism for androgen receptor antagonists. J Biol Chem. 2005;280:6511–6519. doi: 10.1074/jbc.M408972200. [DOI] [PubMed] [Google Scholar]
- 109.Berrevoets CA, Umar A, Trapman J, Brinkmann AO. Differential modulation of androgen receptor transcriptional activity by the nuclear receptor co-repressor (N-CoR) Biochem J. 2004;379:731–738. doi: 10.1042/BJ20031456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Laschak M, Bechtel M, Spindler KD, Hessenauer A. Inability of NCoR/SMRT to repress androgen receptor transcriptional activity in prostate cancer cell lines. Int J Mol Med. 2011;28:645–651. doi: 10.3892/ijmm.2011.735. [DOI] [PubMed] [Google Scholar]
- 111.Choi HK, Yoo JY, Jeong MH, Park SY, Shin DM, Jang SW, Yoon HG, Choi KC. Protein kinase A phosphorylates NCoR to enhance its nuclear translocation and repressive function in human prostate cancer cells. J Cell Physiol. 2013;228:1159–1165. doi: 10.1002/jcp.24269. [DOI] [PubMed] [Google Scholar]
- 112.Godoy AS, Sotomayor PC, Villagran M, Yacoub R, Montecinos VP, McNerney EM, Moser M, Foster BA, Onate SA. Altered corepressor SMRT expression and recruitment to target genes as a mechanism that change the response to androgens in prostate cancer progression. Biochem Biophys Res Commun. 2012;423:564–570. doi: 10.1016/j.bbrc.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 113.Yoo JY, Lim BJ, Choi HK, Hong SW, Jang HS, Kim C, Chun KH, Choi KC, Yoon HG. CK2-NCoR signaling cascade promotes prostate tumorigenesis. Oncotarget. 2013;4:972–983. doi: 10.18632/oncotarget.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Qi T, Chen Y, Zhu Y, Jiang J, Wang L, Qi J. Contrast-enhanced transrectal ultrasonography for detection and localization of prostate index tumor: correlation with radical prostatectomy findings. Urology. 2014;84:138–143. doi: 10.1016/j.urology.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 115.Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara FF, Zamir I, Seiser C, Lazar MA, Minucci S, Pelicci PG. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391:815–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- 116.Lin RJ, Nagy L, Inoue S, Shao W, Miller WH Jr, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 1998;391:811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- 117.Lin RJ, Sternsdorf T, Tini M, Evans RM. Transcriptional regulation in acute promyelocytic leukemia. Oncogene. 2001;20:7204–7215. doi: 10.1038/sj.onc.1204853. [DOI] [PubMed] [Google Scholar]
- 118.He LZ, Guidez F, Tribioli C, Peruzzi D, Ruthardt M, Zelent A, Pandolfi PP. Distinct interactions of PML-RARalpha and PLZF-RARalpha with co-repressors determine differential responses to RA in APL. Nat Genet. 1998;18:126–135. doi: 10.1038/ng0298-126. [DOI] [PubMed] [Google Scholar]
- 119.Racanicchi S, Maccherani C, Liberatore C, Billi M, Gelmetti V, Panigada M, Rizzo G, Nervi C, Grignani F. Targeting fusion protein/corepressor contact restores differentiation response in leukemia cells. EMBO J. 2005;24:1232–1242. doi: 10.1038/sj.emboj.7600593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Atsumi A, Tomita A, Kiyoi H, Naoe T. Histone deacetylase 3 (HDAC3) is recruited to target promoters by PML-RARalpha as a component of the N-CoR co-repressor complex to repress transcription in vivo. Biochem Biophys Res Commun. 2006;345:1471–1480. doi: 10.1016/j.bbrc.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 121.Mengeling BJ, Phan TQ, Goodson ML, Privalsky ML. Aberrant corepressor interactions implicated in PML-RAR(alpha) and PLZF-RAR(alpha) leukemogenesis reflect an altered recruitment and release of specific NCoR and SMRT splice variants. J Biol Chem. 2011;286:4236–4247. doi: 10.1074/jbc.M110.200964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Farris M, Lague A, Manuelyan Z, Statnekov J, Francklyn C. Altered nuclear cofactor switching in retinoic-resistant variants of the PML-RARα oncoprotein of acute promyelocytic leukemia. Proteins. 2012;80:1095–1099. doi: 10.1002/prot.24010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lutterbach B, Westendorf JJ, Linggi B, Patten A, Moniwa M, Davie JR, Huynh KD, Bardwell VJ, Lavinsky RM, Rosenfeld MG, Glass C, Seto E, Hiebert SW. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol Cell Biol. 1998;18:7176–7184. doi: 10.1128/mcb.18.12.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yan M, Burel SA, Peterson LF, Kanbe E, Iwasaki H, Boyapati A, Hines R, Akashi K, Zhang DE. Deletion of an AML1-ETO C-terminal NcoR/SMRT-interacting region strongly induces leukemia development. Proc Natl Acad Sci U S A. 2004;101:17186–17191. doi: 10.1073/pnas.0406702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang J, Hug BA, Huang EY, Chen CW, Gelmetti V, Maccarana M, Minucci S, Pelicci PG, Lazar MA. Oligomerization of ETO is obligatory for corepressor interaction. Mol Cell Biol. 2001;21:156–163. doi: 10.1128/MCB.21.1.156-163.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hug BA, Lazar MA. ETO interacting proteins. Oncogene. 2004;23:4270–4274. doi: 10.1038/sj.onc.1207674. [DOI] [PubMed] [Google Scholar]
- 127.Gamou T, Kitamura E, Hosoda F, Shimizu K, Shinohara K, Hayashi Y, Nagase T, Yokoyama Y, Ohki M. The partner gene of AML1 in t(16;21) myeloid malignancies is a novel member of the MTG8(ETO) family. Blood. 1998;91:4028–4037. [PubMed] [Google Scholar]
- 128.Melnick AM, Westendorf JJ, Polinger A, Carlile GW, Arai S, Ball HJ, Lutterbach B, Hiebert SW, Licht JD. The ETO protein disrupted in t(8;21)-associated acute myeloid leukemia is a corepressor for the promyelocytic leukemia zinc finger protein. Mol Cell Biol. 2000;20:2075–2086. doi: 10.1128/mcb.20.6.2075-2086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Linggi B, Muller-Tidow C, van de Locht L, Hu M, Nip J, Serve H, Berdel WE, van der Reijden B, Quelle DE, Rowley JD, Cleveland J, Jansen JH, Pandolfi PP, Hiebert SW. The t(8;21) fusion protein, AML1 ETO, specifically represses the transcription of the p14(ARF) tumor suppressor in acute myeloid leukemia. Nat Med. 2002;8:743–750. doi: 10.1038/nm726. [DOI] [PubMed] [Google Scholar]
- 130.Zhang J, Kalkum M, Yamamura S, Chait BT, Roeder RG. E protein silencing by the leukemogenic AML1-ETO fusion protein. Science. 2004;305:1286–1289. doi: 10.1126/science.1097937. [DOI] [PubMed] [Google Scholar]
- 131.Gow CH, Guo C, Wang D, Hu Q, Zhang J. Differential involvement of E2A-corepressor interactions in distinct leukemogenic pathways. Nucleic Acids Res. 2014;42:137–152. doi: 10.1093/nar/gkt855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Secrist JP, Zhou X, Richon VM. HDAC inhibitors for the treatment of cancer. Curr Opin Investig Drugs. 2003;4:1422–1427. [PubMed] [Google Scholar]
- 133.Cress WD, Seto E. Histone deacetylases, transcriptional control, and cancer. J Cell Physiol. 2000;184:1–16. doi: 10.1002/(SICI)1097-4652(200007)184:1<1::AID-JCP1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 134.Kramer OH, Gottlicher M, Heinzel T. Histone deacetylase as a therapeutic target. Trends Endocrinol Metab. 2001;12:294–300. doi: 10.1016/s1043-2760(01)00438-6. [DOI] [PubMed] [Google Scholar]
- 135.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–1935. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–861. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 137.Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 138.Humphrey GW, Wang Y, Russanova VR, Hirai T, Qin J, Nakatani Y, Howard BH. Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1. J Biol Chem. 2001;276:6817–6824. doi: 10.1074/jbc.M007372200. [DOI] [PubMed] [Google Scholar]
- 139.Guenther MG, Yu J, Kao GD, Yen TJ, Lazar MA. Assembly of the SMRT-histone deacetylase 3 repression complex requires the TCP-1 ring complex. Genes Dev. 2002;16:3130–3135. doi: 10.1101/gad.1037502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yang WM, Tsai SC, Wen YD, Fejer G, Seto E. Functional domains of histone deacetylase-3. J Biol Chem. 2002;277:9447–9454. doi: 10.1074/jbc.M105993200. [DOI] [PubMed] [Google Scholar]
- 141.Godman CA, Joshi R, Tierney BR, Greenspan E, Rasmussen TP, Wang HW, Shin DG, Rosenberg DW, Giardina C. HDAC3 impacts multiple oncogenic pathways in colon cancer cells with effects on Wnt and vitamin D signaling. Cancer Biol Ther. 2008;7:1570–1580. doi: 10.4161/cbt.7.10.6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Spurling CC, Godman CA, Noonan EJ, Rasmussen TP, Rosenberg DW, Giardina C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol Carcinog. 2008;47:137–147. doi: 10.1002/mc.20373. [DOI] [PubMed] [Google Scholar]
- 143.Weichert W, Roske A, Gekeler V, Beckers T, Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M, Kristiansen G. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 2008;98:604–610. doi: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Giannini R, Cavallini A. Expression analysis of a subset of coregulators and three nuclear receptors in human colorectal carcinoma. Anticancer Res. 2005;25:4287–4292. [PubMed] [Google Scholar]
- 145.Chen JS, Faller DV, Spanjaard RA. Short-chain fatty acid inhibitors of histone deacetylases: promising anticancer therapeutics? Curr Cancer Drug Targets. 2003;3:219–236. doi: 10.2174/1568009033481994. [DOI] [PubMed] [Google Scholar]
- 146.Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, Tsuneyoshi M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18:769–774. [PubMed] [Google Scholar]
- 147.Bartling B, Hofmann HS, Boettger T, Hansen G, Burdach S, Silber RE, Simm A. Comparative application of antibody and gene array for expression profiling in human squamous cell lung carcinoma. Lung Cancer. 2005;49:145–154. doi: 10.1016/j.lungcan.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 148.Krusche CA, Wulfing P, Kersting C, Vloet A, Bocker W, Kiesel L, Beier HM, Alfer J. Histone deacetylase-1 and -3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Res Treat. 2005;90:15–23. doi: 10.1007/s10549-004-1668-2. [DOI] [PubMed] [Google Scholar]
- 149.Wang J, Saunthararajah Y, Redner RL, Liu JM. Inhibitors of histone deacetylase relieve ETO-mediated repression and induce differentiation of AML1-ETO leukemia cells. Cancer Res. 1999;59:2766–2769. [PubMed] [Google Scholar]
- 150.Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 151.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wolffe AP. Histone deacetylase: a regulator of transcription. Science. 1996;272:371–372. doi: 10.1126/science.272.5260.371. [DOI] [PubMed] [Google Scholar]
- 153.Schneider G, Reichert M, Saur D, Hamacher R, Fritsch R, Schmid RM. HDAC3 is linked to cell cycle machinery in MiaPaCa2 cells by regulating transcription of skp2. Cell Prolif. 2007;40:522–531. doi: 10.1111/j.1365-2184.2007.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Li Y, Kao GD, Garcia BA, Shabanowitz J, Hunt DF, Qin J, Phelan C, Lazar MA. A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes Dev. 2006;20:2566–2579. doi: 10.1101/gad.1455006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ishii S, Kurasawa Y, Wong J, Yu-Lee LY. Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proc Natl Acad Sci U S A. 2008;105:4179–4184. doi: 10.1073/pnas.0710140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, Yenamandra A, Locke K, Yuan JL, Bonine-Summers AR, Wells CE, Kaiser JF, Washington MK, Zhao Z, Wagner FF, Sun ZW, Xia F, Holson EB, Khabele D, Hiebert SW. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436–447. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jin JS, Tsao TY, Sun PC, Yu CP, Tzao C. SAHA Inhibits the Growth of Colon Tumors by Decreasing Histone Deacetylase and the Expression of Cyclin D1 and Survivin. Pathol Oncol Res. 2012;18:713–20. doi: 10.1007/s12253-012-9499-7. [DOI] [PubMed] [Google Scholar]
- 158.Mariadason JM. Dissecting HDAC3-mediated tumor progression. Cancer Biol Ther. 2008;7:1581–1583. doi: 10.4161/cbt.7.10.6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Takami Y, Nakayama T. N-terminal region, C-terminal region, nuclear export signal, and deacetylation activity of histone deacetylase-3 are essential for the viability of the DT40 chicken B cell line. J Biol Chem. 2000;275:16191–16201. doi: 10.1074/jbc.M908066199. [DOI] [PubMed] [Google Scholar]
- 160.Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, Teng SC, Wu KJ. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell. 2011;43:811–822. doi: 10.1016/j.molcel.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 161.Knutson SK, Chyla BJ, Amann JM, Bhaskara S, Huppert SS, Hiebert SW. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008;27:1017–1028. doi: 10.1038/emboj.2008.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Balasubramanian S, Verner E, Buggy JJ. Isoform-specific histone deacetylase inhibitors: the next step? Cancer Lett. 2009;280:211–221. doi: 10.1016/j.canlet.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 163.Thaler F, Minucci S. Next generation histone deacetylase inhibitors: the answer to the search for optimized epigenetic therapies? Expert Opin Drug Discov. 2011;6:393–404. doi: 10.1517/17460441.2011.557660. [DOI] [PubMed] [Google Scholar]
- 164.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 165.McKinsey TA. Isoform-selective HDAC inhibitors: closing in on translational medicine for the heart. J Mol Cell Cardiol. 2011;51:491–496. doi: 10.1016/j.yjmcc.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 166.Bieliauskas AV, Pflum MK. Isoform-selective histone deacetylase inhibitors. Chem Soc Rev. 2008;37:1402–1413. doi: 10.1039/b703830p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rasheed WK, Johnstone RW, Prince HM. Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig Drugs. 2007;16:659–678. doi: 10.1517/13543784.16.5.659. [DOI] [PubMed] [Google Scholar]
- 168.Garber K. HDAC inhibitors overcome first hurdle. Nat Biotechnol. 2007;25:17–19. doi: 10.1038/nbt0107-17. [DOI] [PubMed] [Google Scholar]
- 169.Wells CE, Bhaskara S, Stengel KR, Zhao Y, Sirbu B, Chagot B, Cortez D, Khabele D, Chazin WJ, Cooper A, Jacques V, Rusche J, Eischen CM, McGirt LY, Hiebert SW. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS One. 2013;8:e68915. doi: 10.1371/journal.pone.0068915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR, Wood MA. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci U S A. 2013;110:2647–2652. doi: 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. 2001;21:6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013;20:182–187. doi: 10.1038/nsmb.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, Hiebert SW. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008;30:61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sun Z, Feng D, Fang B, Mullican SE, You SH, Lim HW, Everett LJ, Nabel CS, Li Y, Selvakumaran V, Won KJ, Lazar MA. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell. 2013;52:769–782. doi: 10.1016/j.molcel.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Guo C, Gow CH, Li Y, Gardner A, Khan S, Zhang J. Regulated clearance of histone deacetylase 3 protects independent formation of nuclear receptor corepressor complexes. J Biol Chem. 2012;287:12111–12120. doi: 10.1074/jbc.M111.327023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang X, Ozawa Y, Lee H, Wen YD, Tan TH, Wadzinski BE, Seto E. Histone deacetylase 3 (HDAC3) activity is regulated by interaction with protein serine/threonine phosphatase 4. Genes Dev. 2005;19:827–839. doi: 10.1101/gad.1286005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Watson PJ, Fairall L, Santos GM, Schwabe JW. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bassi MT, Ramesar RS, Caciotti B, Winship IM, De Grandi A, Riboni M, Townes PL, Beighton P, Ballabio A, Borsani G. X-linked late-onset sensorineural deafness caused by a deletion involving OA1 and a novel gene containing WD-40 repeats. Am J Hum Genet. 1999;64:1604–1616. doi: 10.1086/302408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dong X, Tsuda L, Zavitz KH, Lin M, Li S, Carthew RW, Zipursky SL. ebi regulates epidermal growth factor receptor signaling pathways in Drosophila. Genes Dev. 1999;13:954–965. doi: 10.1101/gad.13.8.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhang X, Dormady SP, Basch RS. Identification of four human cDNAs that are differentially expressed by early hematopoietic progenitors. Exp Hematol. 2000;28:1286–1296. doi: 10.1016/s0301-472x(00)00539-7. [DOI] [PubMed] [Google Scholar]
- 181.Perissi V, Scafoglio C, Zhang J, Ohgi KA, Rose DW, Glass CK, Rosenfeld MG. TBL1 and TBLR1 phosphorylation on regulated gene promoters overcomes dual CtBP and NCoR/SMRT transcriptional repression checkpoints. Mol Cell. 2008;29:755–766. doi: 10.1016/j.molcel.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
- 183.Choi HK, Choi KC, Yoo JY, Song M, Ko SJ, Kim CH, Ahn JH, Chun KH, Yook JI, Yoon HG. Reversible SUMOylation of TBL1-TBLR1 regulates β-catenin-mediated Wnt signaling. Mol Cell. 2011;43:203–216. doi: 10.1016/j.molcel.2011.05.027. [DOI] [PubMed] [Google Scholar]
- 184.Daniels G, Li Y, Gellert LL, Zhou A, Melamed J, Wu X, Zhang X, Zhang D, Meruelo D, Logan SK, Basch R, Lee P. TBLR1 as an androgen receptor (AR) coactivator selectively activates AR target genes to inhibit prostate cancer growth. Endocr Relat Cancer. 2014;21:127–142. doi: 10.1530/ERC-13-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Spain BH, Bowdish KS, Pacal AR, Staub SF, Koo D, Chang CY, Xie W, Colicelli J. Two human cDNAs, including a homolog of Arabidopsis FUS6 (COP11), suppress G-protein- and mitogen-activated protein kinase-mediated signal transduction in yeast and mammalian cells. Mol Cell Biol. 1996;16:6698–6706. doi: 10.1128/mcb.16.12.6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jin DY, Teramoto H, Giam CZ, Chun RF, Gutkind JS, Jeang KT. A human suppressor of c-Jun N-terminal kinase 1 activation by tumor necrosis factor alpha. J Biol Chem. 1997;272:25816–25823. doi: 10.1074/jbc.272.41.25816. [DOI] [PubMed] [Google Scholar]
- 187.Peng YC, Breiding DE, Sverdrup F, Richard J, Androphy EJ. AMF-1/Gps2 binds p300 and enhances its interaction with papillomavirus E2 proteins. J Virol. 2000;74:5872–5879. doi: 10.1128/jvi.74.13.5872-5879.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cardamone MD, Krones A, Tanasa B, Taylor H, Ricci L, Ohgi KA, Glass CK, Rosenfeld MG, Perissi V. A protective strategy against hyperinflammatory responses requiring the nontranscriptional actions of GPS2. Mol Cell. 2012;46:91–104. doi: 10.1016/j.molcel.2012.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jakobsson T, Venteclef N, Toresson G, Damdimopoulos AE, Ehrlund A, Lou X, Sanyal S, Steffensen KR, Gustafsson JA, Treuter E. GPS2 is required for cholesterol efflux by triggering histone demethylation, LXR recruitment, and coregulator assembly at the ABCG1 locus. Mol Cell. 2009;34:510–518. doi: 10.1016/j.molcel.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 190.Peng YC, Kuo F, Breiding DE, Wang YF, Mansur CP, Androphy EJ. AMF1 (GPS2) modulates p53 transactivation. Mol Cell Biol. 2001;21:5913–5924. doi: 10.1128/MCB.21.17.5913-5924.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sanyal S, Bavner A, Haroniti A, Nilsson LM, Lundasen T, Rehnmark S, Witt MR, Einarsson C, Talianidis I, Gustafsson JA, Treuter E. Involvement of corepressor complex subunit GPS2 in transcriptional pathways governing human bile acid biosynthesis. Proc Natl Acad Sci U S A. 2007;104:15665–15670. doi: 10.1073/pnas.0706736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Venteclef N, Jakobsson T, Ehrlund A, Damdimopoulos A, Mikkonen L, Ellis E, Nilsson LM, Parini P, Janne OA, Gustafsson JA, Steffensen KR, Treuter E. GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRbeta in the hepatic acute phase response. Genes Dev. 2010;24:381–395. doi: 10.1101/gad.545110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhang D, Harry GJ, Blackshear PJ, Zeldin DC. G-protein pathway suppressor 2 (GPS2) interacts with the regulatory factor X4 variant 3 (RFX4_v3) and functions as a transcriptional co-activator. J Biol Chem. 2008;283:8580–8590. doi: 10.1074/jbc.M708209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cheng X, Kao HY. G protein pathway suppressor 2 (GPS2) is a transcriptional corepressor important for estrogen receptor alpha-mediated transcriptional regulation. J Biol Chem. 2009;284:36395–36404. doi: 10.1074/jbc.M109.062109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cardamone MD, Tanasa B, Chan M, Cederquist CT, Andricovich J, Rosenfeld MG, Perissi V. GPS2/KDM4A Pioneering Activity Regulates Promoter-Specific Recruitment of PPARgamma. Cell Rep. 2014;8:163–176. doi: 10.1016/j.celrep.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Toubal A, Clement K, Fan R, Ancel P, Pelloux V, Rouault C, Veyrie N, Hartemann A, Treuter E, Venteclef N. SMRT-GPS2 corepressor pathway dysregulation coincides with obesity-linked adipocyte inflammation. J Clin Invest. 2013;123:362–379. doi: 10.1172/JCI64052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bi H, Li S, Wang M, Jia Z, Chang AK, Pang P, Wu H. SUMOylation of GPS2 protein regulates its transcription-suppressing function. Mol Biol Cell. 2014;25:2499–508. doi: 10.1091/mbc.E13-12-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, Greulich H, Lawrence MS, Lennon NJ, McKenna A, Meldrim J, Ramos AH, Ross MG, Russ C, Shefler E, Sivachenko A, Sogoloff B, Stojanov P, Tamayo P, Mesirov JP, Amani V, Teider N, Sengupta S, Francois JP, Northcott PA, Taylor MD, Yu F, Crabtree GR, Kautzman AG, Gabriel SB, Getz G, Jager N, Jones DT, Lichter P, Pfister SM, Roberts TM, Meyerson M, Pomeroy SL, Cho YJ. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bien-Willner GA, Mitra RD. Mutation and expression analysis in medulloblastoma yields prognostic variants and a putative mechanism of disease for i17q tumors. Acta Neuropathol Commun. 2014;2:74. doi: 10.1186/s40478-014-0074-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.O’Meara E, Stack D, Phelan S, McDonagh N, Kelly L, Sciot R, Debiec-Rychter M, Morris T, Cochrane D, Sorensen P, O’Sullivan MJ. Identification of an MLL4-GPS2 fusion as an oncogenic driver of undifferentiated spindle cell sarcoma in a child. Genes Chromosomes Cancer. 2014 doi: 10.1002/gcc.22208. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 201.Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, Ge K. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem. 2007;282:20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.McPherson A, Wu C, Hajirasouliha I, Hormozdiari F, Hach F, Lapuk A, Volik S, Shah S, Collins C, Sahinalp SC. Comrad: detection of expressed rearrangements by integrated analysis of RNA-Seq and low coverage genome sequence data. Bioinformatics. 2011;27:1481–1488. doi: 10.1093/bioinformatics/btr184. [DOI] [PubMed] [Google Scholar]
- 203.Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, Danussi C, Dolgalev I, Porrati P, Pellegatta S, Heguy A, Gupta G, Pisapia DJ, Canoll P, Bruce JN, McLendon RE, Yan H, Aldape K, Finocchiaro G, Mikkelsen T, Prive GG, Bigner DD, Lasorella A, Rabadan R, Iavarone A. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45:1141–1149. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]