

Abstract
A formidable challenge at the forefront of organic synthesis is the control of chemoselectivity to enable the selective formation of diverse structural motifs from a readily available substrate class. Presented herein is a detailed study of chemoselectivity with palladium-based phosphine catalysts and readily available 2-B(pin)-substituted allylic acetates, benzoates, and carbonates. Depending on the choice of reagents, catalysts and reaction conditions, 2-B(pin)-substituted allylic acetates and derivatives can be steered into one of three reaction manifolds: allylic substitution, Suzuki-Miyaura cross-coupling, or elimination to form allenes, all with excellent chemoselectivity. The studies on chemoselectivity of Pd catalysts in their reactivity with boron-bearing allylic acetate derivatives led to the development of diverse and practical reactions with potential utility in synthetic organic chemistry.
Keywords: Chemoselectivity, Palladium, Tsuji-Trost Allylic Substitution, Suzuki-Miyaura cross-coupling, Allene
Introduction
One of the most significant impediments to the efficient synthesis of complex natural and non-natural products is the low level of chemoselectivity exhibited by many reagents and catalysts.[1,2] To compensate for poor chemoselectivity, synthetic organic chemists have been forced to design and implement elaborate protecting group strategies in their construction of complex natural products.[3]
Although considerable effort has been devoted to understanding chemoselectivity with classical reagents, fewer studies have focused on chemoselectivity with transition metal catalysts. Given the increasing importance of transition metal-catalyzed reactions in organic synthesis, [4] we set out to optimize and control catalyst chemoselectivity in some of the most synthetically valuable transition metal-catalyzed reactions, the Tsuji-Trost allylic substitution[5–9] and the Suzuki-Miyaura cross-coupling reactions.[10,11] Palladium phosphine complexes catalyze both reactions even though the mechanistic pathways are distinct (Figure 1). Allylic substitutions begin with coordination of the substrate to palladium(0) to form a π-complex (Figure 1, right-hand cycle). Backside attack by the palladium on the carbon bearing the leaving group in an oxidative ionization generates a π-allyl palladium complex. External nucleophilic addition to the π-allyl palladium complex followed by liberation of the product regenerates the palladium(0). In contrast, in the Suzuki-Miyaura cross-coupling palladium(0) undergoes oxidative addition with an aryl halide to form the palladium(II) aryl halide intermediate (Figure 1, left-hand cycle).[12] Transmetallation with boronic acid derivatives and bases provides the second Pd–C bond.[13] Reductive elimination forms the C–C bond of the product and regenerates palladium(0). More broadly speaking, the palladium does not come into contact with the leaving group or nucleophile in the allylic substitution, [14] whereas in the Suzuki-Miyaura cross-couplings, palladium directly interacts with both coupling partners. The common denominator in these reactions is the ability of the same palladium catalyst to promote both processes.
Figure 1.
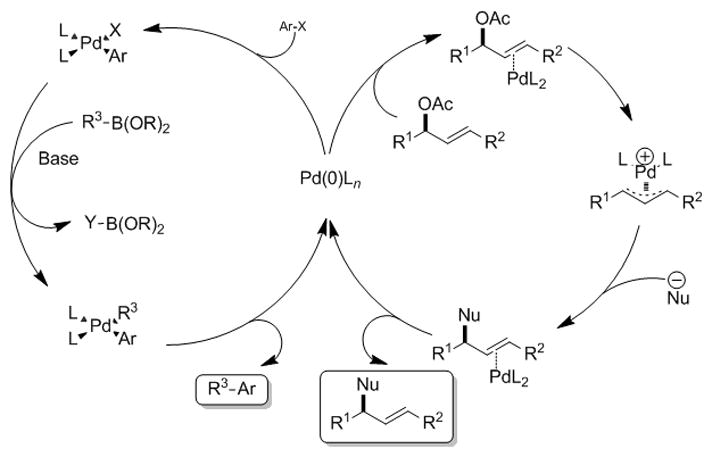
Catalytic cycles of Tsuji-Trost allylic substitution (right-hand cycle) and Suzuki-Miyaura cross-coupling (left-hand cycle).
With this backdrop, we asked a fundamental question: Could the chemoselectivity of a palladium-phosphine-based catalyst toward an allylic acetate, an aryl halide, and a vinyl boronate ester be controlled by careful choice of reagents and conditions? This report answers this question and introduces a third orthogonal reaction pathway of 2-B(pin)-substituted allylic acetates that generates allenes with excellent chemoselectivity. Based on the highly chemoselective reactions developed herein, 2-B(pin)-substituted allylic acetate derivatives are useful “linchpins” that can be utilized for the preparation of an array of small molecules, including functionalized allylic amines and malonates, α-amino ketones, trisubstituted allylic esters and carbonates, and 1,3-disubstituted allenes. A portion of this work related to allylic substitution has been communicated.[15] In the present full report, we disclose the scope of Suzuki-Miyaura cross-coupling and allene formation.
Results and Discussion
The application of multifunctional substrates in tandem reactions enables the rapid construction of diverse chemical arrays. Key to the success of such strategies is the ability to control chemoselectivity by controlling the relative rates of processes that lead into different reaction manifolds. In this study, we have embedded a vinyl boronate ester in an allylic acetate with the goal of controlling chemoselectivity between palladium-catalyzed Tsuji-Trost allylic substitution, Suzuki-Miyaura cross-coupling and allene forming reactions (Figure 2). Given that the same palladium catalysts promote allylic substitution, cross-coupling, and allene formation reactions, we focused on reaction conditions and reagents to control chemoselectivity.
Figure 2.
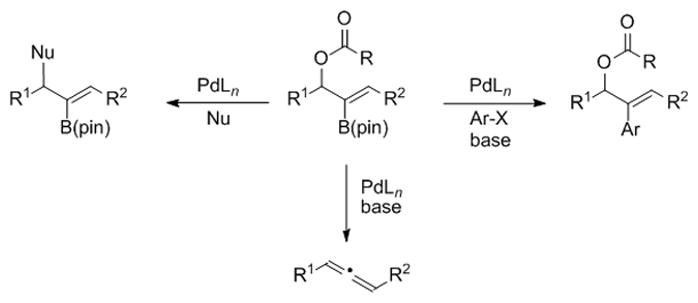
Potential reactions of 2-B(pin)-substituted allylic esters.
Synthesis of 2-B(pin)-substituted allylic acetates
A crucial step toward the development of new synthetic methods is introduction of reliable and scalable procedures to prepare the substrates. Using our stereodefined 1-alkenyl-1,1-heterobimetallic reagents, the 2-B(pin)-substituted allylic acetates were easily prepared in one-pot from readily available alkynyldioxaborolanes (Scheme 1).[15–19] Thus, hydroboration of air-stable alkynyldioxaborolanes with dicyclohexylborane generates 1-alkenyl-1,1-diboro intermediates as the only observable regioisomer (1H NMR).[20–26] Although neither B–C bond is very nucleophilic, the boron centers are electronically quite different due to resonance donation from the pinacolato group of the B(pin). The partially occupied p-orbital of the B(pin) boron dramatically raises the barrier to transmetallation and the vinyl–BCy2 undergoes selective B to Zn transmetallation at −78 °C to generate the heterobimetallic intermediate. Addition of the Zn–C bond to aldehydes and quenching the resulting alkoxides with acetic anhydride provided 2-B(pin)-substituted (E)-allylic acetates 1 in 52–85% yield (Scheme 1). The reaction worked well with linear and branched aliphatic aldehydes (67–85% yield). Likewise, benzaldehyde and its derivatives with electron withdrawing or donating substituents proved to be good reaction partners (52–78% yield). Cinnamaldehyde gave 60% yield of the rearranged dienyl product formed on isomerization of the acetate (see Supporting Information). The alkynyldioxaborolanes can be derived from either aliphatic, aromatic, or functionalized alkynes (R2 = n-Bu, (CH2)4Cl, Ph). The scalability of this method was demonstrated in the synthesis of 2-B(pin)-substituted allylic acetates in gram quantities, positioning us to explore their reactivity and chemoselectivity with palladium phosphine-based catalysts.
Scheme 1.
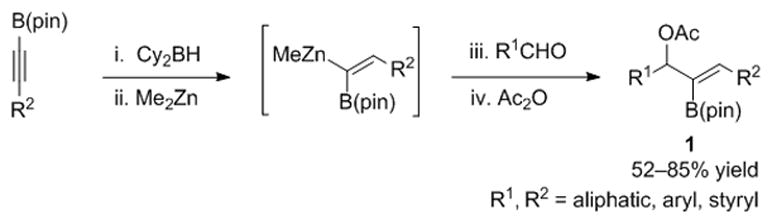
One-pot synthesis of 2-B(pin)-substituted allylic acetates.
Allylic substitution of 2-B(pin)-substituted allylic acetates: Controlling chemoselectivity
At the outset of our studies we were concerned about the ability of 2-B(pin)-substituted allylic acetates to successfully undergo the Tsuji-Trost allylic substitution. Although most acyclic allylic acetate substrates are mono- or disubstituted, [7] leading to mono or 1,3-disubstituted π-allyl intermediates, the reactivity of 1,2,3-trisubstituted allylic derivatives toward allylic ionization with palladium catalysts has been relatively unexplored.[27–29] A few examples of allylic acetate derivatives containing a boron at the 2-position[30,31] had been used in transition metal-catalyzed allylic substitution reactions, [31] although some examples of 3-boron substituted analogs had been generated or proposed as intermediates.[32–44] Despite the lack of clear precedent, the conjugate base of dimethylmalonate and several primary and secondary amines participated in the allylic substitution. For example, 2-B(pin)-substituted allylic acetate 1a underwent substitution with sodium dimethylmalonate (Table 1, entry 1) to furnish 2-B(pin)-substituted allylic alkylation product 2a in 81% yield. Morpholine, N-methylbenzyl amine, and piperidine were good substrates, affording 2-B(pin)-substituted allylic amines 2b–d in 79–83% yield (entries 2–4). Benzyl amine, used as the representative primary amine, also underwent a single allylic substitution to produce 2e in 65% yield (entry 5). The unsymmetrical dialkyl substrate 1c underwent substitution with morpholine catalyzed by [Pd(allyl)Cl]2 and PPh3 (1:4) with nucleophilic attack at the less hindered position of the π-allyl with excellent regioselectivity (>20:1, entry 6). Isomeric 2-B(pin)-substituted allylic acetates 1d and 1f each underwent reaction with morpholine and NaCH(CO2Me)2 using catalyst generated from Pd(OAc)2 (10 mol %) and PPh3 (20 mol %) to afford the same regioisomer of allylic substitution products 2h and 2i with ≥10:1 regioselectivity (entries 7–10). The styryl derivative 1d was found to be less reactive than 1f toward the same palladium catalyst, resulting in lower yields of the substitution products 2g and 2h (entries 7–10, 45 vs. 80% and 51 vs. 79%). In contrast to most π-allyl palladium complexes with a single aryl group at the terminus, allylic substitution took place at the benzylic position in each case. The reversal in regioselectivity is due to formation of the anti-aryl-π-allyl isomer, which exhibits enhanced reactivity at the benzylic position, as outlined in our related study.[19] Analogous reactions with electron withdrawing or donating substituents on the aryl groups (1g–j) provided the benzylic substitution products (2i–l) in 75–92% isolated yield with >10:1 regioselectivity (entries 11–14). The unique regioselectivity observed with aryl substituted substrates enables the synthesis of benzylic amines, which are common structural motifs in medicinal chemistry.[45]
Table 1.
Palladium-catalyzed allylic substitution of 2-B(pin)-substituted allylic acetates.

| ||||||
|---|---|---|---|---|---|---|
| Entry | Acetate | Nucleophile | Product | Yield (%)[c] | ||
| 1 |

|
1a | NaCH(CO2Me)2 |

|
2a | 81[a] |
| 2 | 1a |
|

|
2b | 83[a] | |
| 3 | 1a | HNMeBn |

|
2c | 80[a] | |
| 4 | 1a |
|

|
2d | 79[a] | |
| 5 | 1a | BnNH2 |

|
2e | 65[a] | |
| 6 |

|
1c |
|

|
2f | 60[a],[d] |
| 7 |

|
1d |
|

|
2g | 45[b],[d],[e] |
| 8 |

|
1f |
|

|
2g | 80[b],[d] |
| 9 |

|
1d | NaCH(CO2Me)2 |

|
2h | 51[a],[d] |
| 10 |

|
1f | NaCH(CO2Me)2 | 2h | 79[a],[d] | |
| 11 |

|
1g |
|

|
2i | 75[b],[d] |
| 12 | 1g | CH2(CO2Me)2 |

|
2j | 90[b],[d],[f] | |
| 13 |

|
1h | CH2(CO2Me)2 |

|
2k | 92[b],[d],[f] |
| 14 |

|
1j | CH2(CO2Me)2 |

|
2l | 80[b],[d],[f] |
[Pd(allyl)Cl]2.
Pd(OAc)2.
Yield of purified and isolated products.
Regioselectivities ≥10:1 (except entry 6) and were determined by 1H NMR spectroscopy of unpurified reaction mixtures.
BSA and catalytic KOAc were used.
30% recovered starting material.
Development of tandem reactions initiated with allylic substitution
Tandem reactions are important in streamlining organic synthesis, enabling a rapid increase in molecular complexity while minimizing synthetic steps and isolations.[46–51] We, therefore, undertook development of tandem processes with 2-B(pin) allylic acetates beginning with allylic substitutions and followed by reactions of the vinyl boronate ester.[52]
Tandem allylic substitution/oxidation sequences
One of the most utilized transformations of the B–C bond is its oxidation. In the case of vinyl boronate esters, the oxidation products are ketones, and allylic substitution/oxidation will lead to α-functionalized ketones. For the tandem allylic substitution/oxidation process, the allylic substitution was performed as outlined in Table 1.
After the allylic substitution, the crude reaction mixture was treated with alkaline hydrogen peroxide to oxidize the B–C bond. When NaCH(CO2Me)2 was employed as the nucleophile in the substitution, subsequent oxidation generated substituted ketone 3a in 85% yield in this one-pot procedure (Table 2, entry 1). Secondary amines performed well in the tandem allylic substitution/oxidation sequence to form α-amino ketones 3b–3e in 65–82% yield (entries 2–5). Both aliphatic (entries 1–4) and 1-phenyl-2-B(pin)-substituted allylic acetates (entries 5–6) were good substrates for the tandem process. A milder oxidation with NaBO3·H2O[53,54] resulted in an increased yield of 3e (compare entries 5 and 6).
Table 2.
One-pot synthesis of α-substituted ketones via tandem allylic substitution/oxidation.

| |||||
|---|---|---|---|---|---|
| Entry | Allyl acetate | α-Substituted ketones | Yield (%)[a] | ||
| 1 | 1a |

|
3a | 85 | |
| 2 | 1a |

|
3b | 82 | |
| 3 | 1a |

|
3c | 81 | |
| 4 | 1a |

|
3d | 75 | |
| 5 | 1f |
|

|
3e | 65 |
| 6 | 1f | 3e | 78[b] | ||
Yield of purified and isolated products.
Allylic substitution carried out with Pd(OAc)2 (5 mol %) and PPh3 (10 mol %) at rt, and oxidation carried out with 3 equiv of NaBO3·H2O in THF/H2O (1:1) at rt.
It is noteworthy that the tandem allylic substitution/oxidation sequence is equivalent to an α-carbonyl cation synthon and represents an umpolung ketone (Scheme 2).[55–59] α-Substituted ketones are difficult to synthesize with standard π-allyl chemistry. Trost and Gowland demonstrated that 2-alkoxy allylic acetates underwent allylic substitution to afford enol ethers that were hydrolyzed to afford ketones (Scheme 3A).[55] Organ and coworkers performed similar chemistry with allylic phenoxides (Scheme 3B).[59]
Scheme 2.
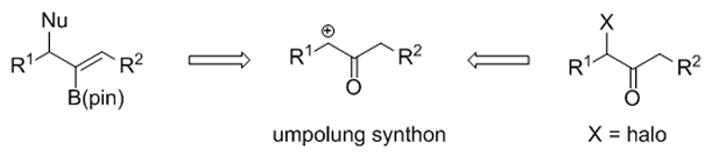
2-B(pin)-substituted allylic substitution products as synthons for α-electrophilic carbonyl substrates.
Scheme 3.
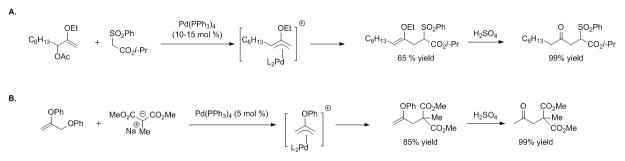
2-B(pin)-substituted allylic substitution products as synthons for α-electrophilic carbonyl substrates.
Enantioenriched allylic amines are valuable synthetic intermediates and are common in natural products and compounds with biological activity.[45,60] One possible approach to access such compounds is via allylic substitutions of enantioenriched 2-B(pin)-substituted allylic acetates. With this in mind, 2-B(pin)-substituted allylic acetate 1f of 80% ee was prepared by Sharpless-Katsuki kinetic resolution[61] of 2-B(pin) allylic alcohol 4a[16] and subsequent acetylation with acetic anhydride (Scheme 4).[15] We then conducted the allylic substitution of 1f and morpholine to form the 2-B(pin)-substituted benzyl amine 2b in 82% yield without loss of enantiomeric excess (Scheme 5). A tandem allylic substitution/B–C bond oxidation with NaBO3·H2O generated the α-amino ketone with 77% ee in 80% yield (Scheme 5). Unfortunately, employing alkaline H2O2 in place of NaBO3·H2O for the oxidation led to racemization of the amino ketone product.
Scheme 4.
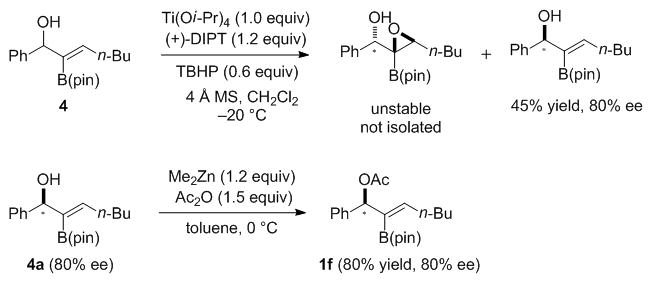
Kinetic resolution of 2-B(pin)-substituted allylic alcohol 4 to prepare enantioenriched 2-B(pin)-substituted allylic acetate 1f.
Scheme 5.
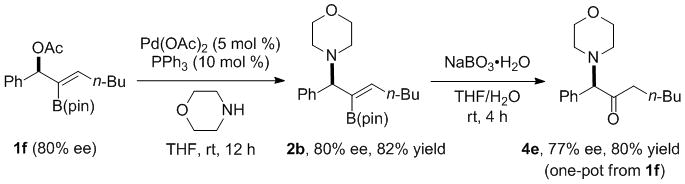
Allylic substitution of enantioenriched 1f with morpholine followed by oxidation with sodium perborate.
Tandem allylic substitution/Suzuki-Miyaura cross-coupling
Allylic amines are present in molecules with diverse pharmacological properties such as Acrivastine (Semprex), [45,62] Flunarizine (Sibelium, Ca channel blocker), [63,64] and several BA uptake inhibitors.[65] In the synthesis of allylic amines, it is usually important that the geometry about the C=C bond be controlled to provide a single product. With that in mind, we sought to achieve a one-pot allylic substitution/Suzuki-Miyaura cross-coupling reaction of 2-B(pin)-substituted allylic acetates. As alluded to in Figure 1, it is conceivable that the allylic substitution and Suzuki-Miyaura cross-coupling could be catalyzed using the same palladium source. One of the key factors to turn on the Suzuki-Miyaura cross-coupling will be the activation of the B–C bond, initiated by hydrolysis to the boronic acid. It is noteworthy that a successful allylic substitution/cross-coupling sequence enables the synthesis of a variety of 2-arylated allylic amines and malonates from easily accessible 2-B(pin)-substituted allylic acetates, aryl halides, amines, and malonates. The allylic substitution of 2-B(pin) allylic acetate 1a and morpholine was performed following the conditions in Table 1 entry 8. After completion of the substitution, as judged by TLC, the reaction mixture was diluted with THF/water (10:1), Cs2CO3 and aryl halide were added, and the reaction mixture was heated (Table 3). After workup, 2-arylated trisubstituted allylic amine derivatives 5a–c were isolated in 52–70% yield (entries 1–3), including the 3-pyridyl derivative. Likewise, unsymmetrical 2-B(pin)-substituted allylic acetate 1f underwent the tandem allylic substitution with morpholine/cross-coupling to yield trisubstituted benzylic amines 5d–f in 50–70% yield (entries 4–7). Aryl bromide coupling partners with electron donating or withdrawing substituents were well tolerated. Iodobenzene gave lower yield than the aryl bromides (entries 4 and 5). Tandem Suzuki-Miyaura cross-coupling of allylic substitution product 2h with sodium malonate failed with iodobenzene, recovering allylic substitution product 2h (43% yield, entry 8). In contrast, 1f and 1h underwent tandem allylic substitution with NaCH(CO2Me)2/cross-couplings with bromobenzene in 51 and 61% yield (entries 9 and 10). Importantly, in all cases, the geometry of the double bond was maintained.
Table 3.
Tandem allylic substitution/Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic acetates.

| ||||||
|---|---|---|---|---|---|---|
| Entry | Allylic acetate | Nu−/NuH | Ar-X | Product | Yield (%)[a] | |
| 1 |
 1a |
|
4-Me-C6H4-Br |

|
5a | 70 |
| 2 | 1a |
|
4-MeO-C6H4-Br | 5b | 65 | |
| 3 | 1a |
|
3-C5H3N-Br | 5c | 52 | |
| 4 |
 1f |
|
Ph-I |

|
5d | 50 |
| 5 | 1f |
|
Ph-Br | 5d | 70 | |
| 6 | 1f |
|
4-MeO-C6H4-Br | 5e | 69 | |
| 7 | 1f |
|
4-O2N-C6H4-Br | 5f | 59 | |
| 8 | 1f | NaCH(CO2Me)2 | Ph-I | –[b] | ||
| 9 | 1f | NaCH(CO2Me)2 | Ph-Br |

|
5g | 51 |
| 10 |
 1h |
NaCH(CO2Me)2 | Ph-Br |

|
5h | 61 |
Yield of purified and isolated products.
No cross-coupling product was observed, but 43% of 2h was isolated.
Related palladium-catalyzed tandem reactions have been reported by the groups of Kazmaier and Pucheault. Kazmaier and co-workers established one-pot allylic amination/Stille couplings of β-stannylated allylic carbonates to provide allylic amine derivatives (Scheme 6A).[66,67] This approach provides disubstituted allylic amines and is complementary to the chemistry in Table 3, which generates trisubstituted products. After publication of our initial communication, [15] Pucheault and co-workers followed with a tandem palladium-catalyzed Tsuji-Trost allylic substitution/Suzuki-Miyaura cross-couplings of γ-borylated allylic acetates (Scheme 6B).[42] This reaction provides traditional allylic substitution products that can be generated in an efficient tandem fashion. With respect to the synthesis of compound libraries, our approach furnishes 1,2,3-trisubstituted allyl products, which contain an additional point of diversity over those in Scheme 6. To further expand the synthetic utility of 2-B(pin)-substituted allylic ester derivatives, we next set out to reverse the chemoselectivity by performing the Suzuki-Miyaura cross-coupling in the presence of the allylic ester derivatives.
Scheme 6.
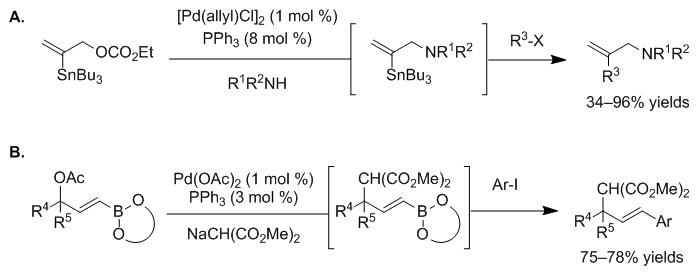
A. Kazmaier’s one-pot allylic amination/Stille couplings. B. Pucheault’s tandem Tsuji-Trost allylic substitution/Suzuki-Miyaura cross-couplings.
Chemoselective Suzuki-Miyaura cross-coupling in the presence of allylic leaving groups
As demonstrated in the tables above, allylic substitutions of 2-B(pin)-substituted allylic acetates proceed readily despite the large size of the B(pin) group. We then asked the question whether the Suzuki-Miyaura cross-coupling reaction could be conducted in the presence of the allylic ester derivatives. As outlined in Figures 1 and 2, the determining factor controlling bifurcation between the two reaction manifolds is the relative rate of oxidative ionization of the allylic ester vs. oxidative addition of the aryl halide. Although there have been many studies on oxidative addition of aryl and vinyl halides and numerous investigations into oxidative ionization of allylic acetates, [12,14] very few studies compare the relative rates of these two processes.[68–71] As pointed out by Organ, [59,72] in cases where a comparison was made, the substrates employed were frequently biased by either steric effects or conformational restrictions that disfavor proper orbital alignment needed for the allylic ionization to proceed.
Precedents for oxidative addition of aryl halides in the presence of allylic acetates
The chemoselectivity of palladium-based catalysts has been investigated by several groups.[73–76] Lautens and co-workers developed a reductive coupling of allylic acetates that involves β-OAc elimination (Scheme 7A).[77,78] Jiao and co-workers demonstrated a complementary Heck reaction of allylic acetates that undergo traditional β-hydride elimination rather than β-acetate elimination (Scheme 7B).[79] Genet and co-workers conducted a chemoselective Suzuki-Miyaura cross-coupling reaction of γ-borylated allylic acetates with activated alkenyl iodides (Scheme 7C).[80] Kočovský and co-workers investigated Suzuki-Miyaura cross-coupling reaction of simple γ-borylated allylic carbonates with aryl iodides (Scheme 7D).[39] The cross-coupling products were isolated in low to moderate yields (32% average yield for 15 substrates) and the reactions failed with aryl bromides. Although the conditions and catalysts used in Scheme 7A–D are different, the trend suggests that oxidative addition of sp2 C–I bonds is faster than ionization of allylic acetates, but the increased reactivity of carbonates results in low cross-coupling yields (Scheme 7D).
Scheme 7.
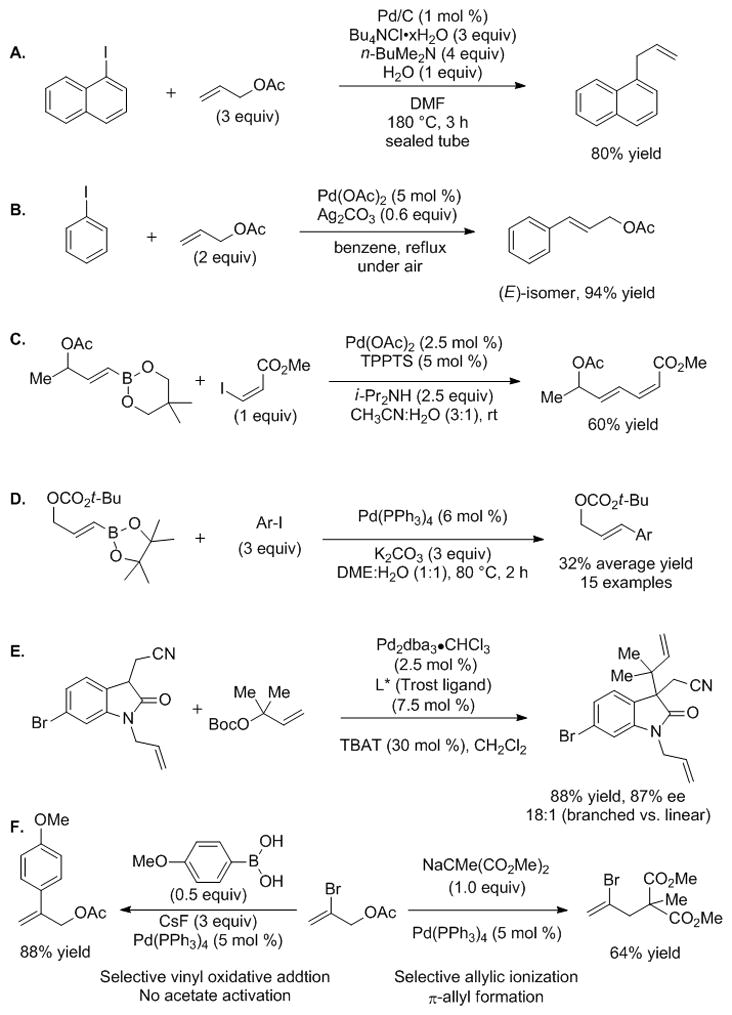
Precedents for oxidative addition of aryl halides in the presence of allylic acetates. See text for discussion.
In the case of aryl bromides, Trost, Malhotra, and Chan recently found that Boc activated allylic alcohols undergo allylic substitution in the presence of an aryl bromide (Scheme 7E).[81] Interestingly, Organ demonstrated that the same palladium catalyst can be used to selectively afford either the allylic substitution or vinylic cross-coupling products from polyfunctionalized olefin building blocks (Scheme 7F).[59,72] Due to the diverse substrates, catalysts, and conditions employed in Scheme 7, only a fragmented picture of the reactivity landscape is provided. A systematic study is necessary to map the relative reactivity of aryl halides in the Suzuki-Miyaura cross-coupling in the presence of allylic ester derivatives of varying reactivity.
Development of orthogonal reaction conditions for the palladium-catalyzed allylic substitution and Suzuki-Miyaura cross-coupling
To favor the Suzuki-Miyaura cross-coupling manifold, the palladium(0) form of the catalyst would need to be rapidly captured by the aryl halide. It is well known that the relative rate of oxidative addition of a homologous series of aryl halides is Ar–I > Ar–Br > Ar–Cl.[12,13] We envisioned that an aryl iodide would rapidly undergo oxidative addition to Pd(0) to generate a Pd(II) aryl iodide intermediate that is unreactive toward oxidative ionization of the allylic acetate (Figure 1). To explore the possibility of conducting the Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic acetates, the di-n-butyl substrate 1a was combined with iodobenzene, Pd(OAc)2 (15 mol %), PPh3 (30 mol %), and Cs2CO3 (3 equiv) in a 10:1 THF/water reaction mixture (Scheme 8). At 70 °C a clean reaction occurred to generate the 2-aryl-substituted allylic acetate 6a in 90% isolated yield. At lower catalyst loading, the cross-coupling was incomplete and gave diminished yields. With the result in Scheme 8, we set out to optimize the Suzuki-Miyaura cross-coupling reaction of 2-B(pin)-substituted allylic acetates to lower the catalyst loading, increase the yields, and develop a broad substrate scope.
Scheme 8.
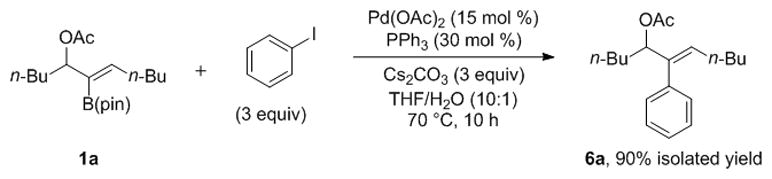
Suzuki-Miyaura Cross-Coupling of 2-B(pin)-substituted Allylic Acetate 1a.
Optimization of Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic acetates
Lowering the catalyst loading from 15 mol % (Scheme 8 and Table 4, entry 1) to 10 mol % Pd resulted in decreased yield of the cross-coupled product 6a (Table 4, entry 2). To optimize the coupling, different phosphine ligands were employed. The bulky P(o-Tol)3 formed the product in only 20% yield (entry 3). Bidentate phosphine ligands gave poor yields of the arylated allylic acetates (27–62% yield, entries 4–6). Subjecting the di-n-butyl allylic acetate 1a to various Pd precursors showed that Pd(OAc)2/P(t-Bu)3, Pd(PPh3)4, and PdCl2(PPh3)2/PPh3 all formed suitable catalysts for chemoselective activation of iodobenzene vs. oxidative ionization of the allylic acetate (entries 7–9). Several bases were screened (Cs2CO3, CsF, and K3PO4), and Cs2CO3 was found to give the highest yield (entries 10–12). When toluene was used in place of THF or dioxane, the catalyst loading could be lowered to 5 mol % with shorter reaction times and up to 96% yield (entries 13–16). Further decreasing the catalyst loading, however, led to incomplete reactions and lower yields. We found a 1:3 ratio of Pd(OAc)2 to PPh3 to be optimal for catalyst formation (entries 13 and 14, 95–96% yield) and suitable for evaluation of the scope of the cross-coupling.
Table 4.
Optimization of Pd-catalyzed Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic acetate 1a.

| ||||||
|---|---|---|---|---|---|---|
| Entry | Pd/ligand[a] | Base[b] | Solvent[c] | Conc.(M)[d] | Time (h) | yield (%)[e] |
| 1 | Pd(OAc)2/PPh3 (15 mol %, 1:2) | Cs2CO3 | THF | 0.022 | 24 | 90[f] |
| 2 | Pd(OAc)2/PPh3 (10 mol %, 1:2) | Cs2CO3 | THF | 0.022 | 48 | 62 |
| 3 | Pd(OAc)2/P(o-Tol)3 (10 mol %, 1:2) | Cs2CO3 | THF | 0.022 | 48 | 20 |
| 4 | Pd(OAc)2/DPPP (10 mol %, 1:1) | Cs2CO3 | THF | 0.022 | 48 | 32 |
| 5 | Pd(OAc)2/DPPE (10 mol %, 1:1) | Cs2CO3 | THF | 0.022 | 48 | 27 |
| 6 | Pd(OAc)2/DPPF (10 mol %, 1:1) | Cs2CO3 | THF | 0.022 | 48 | 62 |
| 7 | Pd(OAc)2/P(t-Bu)3 (10 mol %, 1:1) | Cs2CO3 | THF | 0.022 | 48 | 80 |
| 8 | Pd(PPh3)4 (10 mol %) | Cs2CO3 | THF | 0.022 | 36 | 87 |
| 9 | PdCl2(PPh3)2/PPh3 (10 mol %, 1:1) | Cs2CO3 | THF | 0.08 | 24 | 71 |
| 10 | PdCl2(PPh3)2/PPh3 (10 mol %, 1:1) | Cs2CO3 | Dioxane | 0.08 | 18 | 61 |
| 11 | PdCl2(PPh3)2/PPh3 (10 mol %, 1:1) | CsF | Dioxane | 0.08 | 18 | 25 |
| 12 | PdCl2(PPh3)2/PPh3 (10 mol %, 1:1) | K3PO4 | toluene | 0.08 | 24 | 52 |
| 13 | Pd(OAc)2/PPh3 (10 mol %, 1:3) | Cs2CO3 | toluene | 0.17 | 4 | 96[f] |
| 14 | Pd(OAc)2/PPh3 (5 mol %, 1:3) | Cs2CO3 | toluene | 0.17 | 18 | 95[f],[g] |
| 15 | PdCl2(PPh3)2/PPh3 (5 mol %, 1:1) | Cs2CO3 | toluene | 0.17 | 18 | 93[f],[g] |
| 16 | Pd(PPh3)4 (10 mol %) | Cs2CO3 | toluene | 0.17 | 18 | 96[f],[g] |
mol % of Pd and molar ratio of Pd to phosphine provided in parenthesis.
3 equiv of base and iodobenzene used unless otherwise noted.
10:1 ratio of solvent to H2O.
Ratio of 1a (mmol)/solvent (mL).
Yield determined by 1H NMR integration of the crude reaction mixture using 1,4-dimethoxybenzene as the internal standard.
Yield of isolated and purified products.
2 equiv of Cs2CO3 and iodobenzene used.
Substrate scope of Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic acetates with aryl iodides
In order to determine the substrate scope for Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic acetates, we decided to vary the allylic acetate (R1 and R2) as well as the coupling partner (Ar–I). Aliphatic 2-B(pin)-substituted allylic acetate 1a cleanly provided the arylated allylic acetates 6a–c in good to excellent yields with several aryl iodides bearing electron withdrawing or electron donating groups (60–96% yield, Table 5, entries 1–3). α–Branching is tolerated in the allylic acetate as shown from the reaction of the allylic acetate 1b with iodobenzene (67% yield, entry 4). The styryl derived allylic acetate 1d gave 92–99% yield of the cross-coupled products 6e and 6f (entries 5 and 6). With the isomeric allylic acetate 1f, 70–75% yield of the Suzuki-Miyaura cross-coupled products 6g and 6h were obtained (entries 7 and 8). The isomeric pairs in entries 5 and 7 and entries 6 and 8 each undergo Suzuki-Miyaura cross-coupling with no scrambling of the acetate position. These results indicate that the oxidative ionization of acetate is not reversible or does not disrupt a tight ion pair under these conditions.[14]
Table 5.
Chemoselective Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic acetates with aryl iodides.

| |||||
|---|---|---|---|---|---|
| Entry | Acetate | Ar-I | Product | Yield (%)[a] | |
| 1 | 1a | Ph-I |

|
6a | 96 |
| 2 | 1a | 4-MeO-C6H4-I |

|
6b | 60 |
| 3 | 1a | 4-O2N-C6H4-I |

|
6c | 92 |
| 4 | 1b | Ph-I |

|
6d | 67 |
| 5 | 1d | Ph-I |

|
6e | 99 |
| 6 | 1d | 4-O2N-C6H4-I |

|
6f | 92 |
| 7 | 1f | Ph-I |

|
6g | 70[b] |
| 8 | 1f | 4-O2N-C6H4-I |

|
6h | 75[b] |
Yield of isolated and purified products.
The reaction was conducted at 75°C in THF/H2O (10:1).
To demonstrate scalability of the Suzuki-Miyaura cross-coupling 2-B(pin)-substituted allylic acetates 1e was subjected to cross-coupling conditions on a 5 mmol scale to give 1.3 g of the 2-arylated allylic acetate 6i in 80% isolated yield (Scheme 9). Given that aryl iodides react faster with palladium(0) than allylic acetates, we next examined less reactive aryl bromides.
Scheme 9.

Gram scale Suzuki-Miyaura cross-coupling reaction of 1e.
Substrate scope of Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic acetates with aryl bromides
We are aware of one study involving comparison of relative reactivity of sp2 C–Br bonds vs. allylic acetates with palladium catalysts. Organ demonstrated that the preference of palladium was dependent on the nucleophile (see Scheme 7F).[72]
Subjecting the 2-B(pin)-substituted allylic acetates to the Suzuki-Miyaura conditions in entry 13 of Table 4 with various aryl and heteroaryl bromides resulted in chemoselective activation of the aryl bromides over the allylic acetates. The substituents on the allylic acetate (R1 and R2) and the aryl bromide coupling partners (Ar–Br) were varied to determine the scope of the chemoselective cross-coupling (Table 6). 2-B(pin)-substituted aliphatic allylic acetate 1a underwent cross-coupling with aryl bromides bearing electron withdrawing or electron donating groups with para- and ortho-substitution to provide the arylated allylic acetates 6a–c, 6j and 6k in moderate to excellent yields (54–91%, Table 6, entries 1–5). α–Branching is tolerated as shown in the reaction of the allylic acetate 1b with bromobenzene (87% yield, entry 6). The styryl derived allylic acetate 1d gave excellent yield of the cross-coupled product 6f with 1-bromo-4-nitrobenzene (90% yield, entry 7).
Table 6.
Chemoselective Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic acetates with aryl bromides.

| |||||
|---|---|---|---|---|---|
| Entry | Acetate | Ar-Br | Product | Yield (%)[a] | |
| 1 | 1a | Ph-Br |

|
6a | 73 |
| 2 | 1a | 4-MeO-C6H4-Br |

|
6b | 54 |
| 3 | 1a | 4-O2N-C6H4-Br |

|
6c | 91 |
| 4 | 1a | 4-F3C-C6H4-Br |

|
6j | 85 |
| 5 | 1a | 2-MeO-C6H4-Br |

|
6k | 80 |
| 6 | 1b | Ph-Br |

|
6d | 87 |
| 7 | 1d | 4-O2N-C6H4-Br |

|
6f | 90 |
| 8 | 1f | 4-O2N-C6H4-Br |

|
6h | 80[b] |
| 9 | 1a |
|

|
6l | 74 |
| 10 | 1a |
|

|
6m | 82 |
Yield of isolated and purified products.
The reaction was conducted at 75 °C in THF/H2O (10:1).
With the isomeric allylic acetate 1f, the Suzuki-Miyaura cross-coupled product 6h was obtained in 80% yield (entry 8) with no observed scrambling of the acetate position. As shown in entries 9 and 10, heteroaryl bromides such as 2- or 3-bromothiophene underwent cross-coupling with 1a in 74–82% yield. Having demonstrated that aryl iodides and bromides undergo oxidative addition faster than 2-B(pin)-substituted allylic acetates undergo oxidative ionization under these conditions, we wanted to determine whether this trend would continue to hold with more reactive allylic substrates.
Substrate scope of Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic benzoates and carbonates
To further delineate how differences in substrate reactivity impact the reaction outcome, we turned our attention to increasing the reactivity of the allylic moiety by using more active leaving groups. Allylic benzoates and carbonates are known to be more reactive in allylic substitutions than acetates, with carbonates up to 200 times more reactive than acetates.[14] We, therefore, prepared several 2-B(pin)-substituted allylic benzoates and carbonates from the parent di-n-butyl-2-B(pin)-substituted allylic alcohol (see the Supporting Information). The 2-B(pin)-substituted allylic benzoates and carbonates were subjected to the optimal cross-coupling reaction conditions in entry 13 of Table 4 with aryl iodides and bromides. The 2-B(pin)-substituted allylic benzoate 7a underwent cross-coupling with iodobenzene to furnish the 2-aryl substituted allylic benzoate 9a in 96% yield (Table 7, entry 1). Bromobenzene, on the other hand, resulted in low yield of the cross-coupled product 9a (38%), with multiple byproducts (entry 2). The palladium catalyst likely activates the allylic benzoate in the presence of less reactive bromobenzene. Electron deficient aryl halides are known to undergo oxidative addition more rapidly than analogous electron rich aryl halides.[82–85] With 4-trifluoromethyl bromobenzene and allylic benzoate 7a the cross-coupled product 9b was isolated in 76% yield (entry 3). The 2-B(pin)-substituted allylic carbonates 8a and 8b underwent cross-coupling with both aryl iodides and electron deficient bromides to furnish the 2-arylated allylic carbonates 9c–f in 57–91% yield (entries 4–8). These results demonstrate that the palladium catalyst preferentially oxidatively adds aryl iodides and bromides over 1,3-dialkyl-substituted 2-B(pin) allylic benzoates and carbonates. However, more reactive 1-phenyl-2-B(pin)-substituted allylic benzoate 7b and carbonate 8c were decomposed and no cross-coupling product was observed (entries 9 and 10).
Table 7.
Chemoselective Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allyl benzoates and carbonates

| ||||||
|---|---|---|---|---|---|---|
| Entry | 2-B(pin)-substituted allyls | Ar-X | Product | Yield (%)[a] | ||
| 1 |

|
7a | Ph-I |

|
9a | 96 |
| 2 | 7a | Ph-Br | 9a | 38 | ||
| 3 | 7a | 4-CF3-C6H4-Br |

|
9b | 76 | |
| 4 |

|
8a | Ph-I |

|
9c | 91 |
| 5 | 8a | 4-NO2-C6H4-I |

|
9d | 78 | |
| 6 | 8a | 4-NO2-C6H4-Br | 9d | 75 | ||
| 7 | 8a | 4-CF3-C6H4-Br |

|
9e | 57 | |
| 8 |

|
8b | Ph-I |

|
9f | 83 |
| 9 |

|
7b | Ph-I | –[b] | ||
| 10 |

|
8c | Ph-I | –[b] | ||
Yield of isolated and purified products.
Starting materials were decomposed.
Chemoselective allene formation from 2-B(pin)-substituted allylic acetates, benzoates, and carbonates
As demonstrated in Table 7, the efficiency of Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic substrates is related to the ability of the allylic leaving group to ionize under palladium catalysis. Reactive substrates toward allylic ionization, such as benzoate, carbonates, or substrates with benzylic acetates (entries 9 and 10 in Table 7) tended to yield less cross-coupling products and more decomposition products. Analysis of 1H NMR spectroscopy of unpurified reaction mixtures suggested that decomposed products included allenes and dienes. We hypothesized that the diene products were generated from allene via palladium-catalyzed isomerization or carbopalladation.[86] If this hypothesis is correct, a third manifold of reactivity might be tapped to afford an additional family of products from 2-B(pin)-substituted allylic ester derivatives. We, therefore, set out to identify catalysts to promote chemoselective elimination of 2-B(pin)-substituted allylic substrates to provide internal allenes, which are important synthetic intermediates and common in natural products.[87–93]
Precedents for catalytic 1,2-elimination to form allenes
Tanaka and co-workers reported conversion of 2-bromo-allylic mesylate derivatives into allenes with a catalytic amount of palladium and 2 equiv of diethylzinc as reducing agent (Scheme 10A).[94] More recently, Kazmaier and co-workers demonstrated that β-stannylated allylic acetates or phenoxides undergo 1,2-elimination reactions, providing Pd-allene intermediates from which terminal allenes were isolated (Scheme 10B).[95,96] An advantage of our 2-B(pin)-substituted allylic acetates is that they do not require stoichiometric reducing agents or highly reactive mesylates and they provide 1,3-disubstituted internal allenes.
Scheme 10.
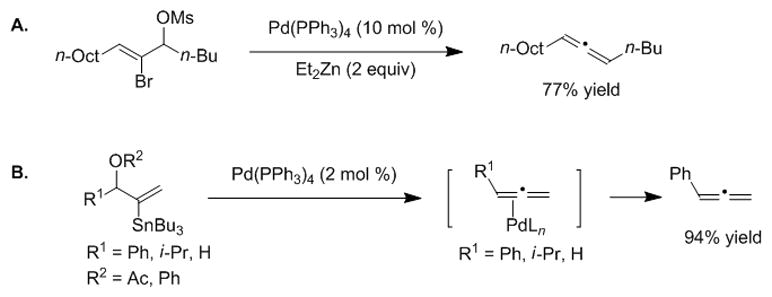
A. Tanaka’s palladium-catalyzed elimination reaction with diethylzinc. B. Kazmaier’s 2-Sn(n-Bu)3 elimination of 2-stannylated allylic acetates and phenoxides to form terminal allenes.
Optimization of allene formation
We set out to screen a variety of palladium precursors and ligands to identify catalysts that would favor allene formation from 1-phenyl-2-B(pin)-substituted allylic acetate 1f. Subjecting 1f to catalytic Pd(PPh3)4 in THF/H2O (5:1) at 60 °C resulted in 20% consumption of 1f with no allene formation (1H NMR, Table 8, entry 1). Using stoichiometric Pd(PPh3)4 resulted in only 4% allene 10a with mostly decomposition products (entry 2). We next examined bases, which dramatically changed the outcome of the reaction. Addition of 1 equiv Cs2CO3 and 1 mol % Pd(PPh3)4 led to allene formation in 87% yield, while Cs2CO3 alone exhibited no reactivity (entries 3 and 4). With the conditions in entry 3, other palladium sources and phosphines were examined (entries 5–9). Both Pd(OAc)2/2PPh3 and [Pd(allyl)Cl]2//4PPh3 formed suitable catalysts for allene formation (73 and 80% yield, respectively, entries 5 and 6). Some diene products (<5%) were also produced. With [Pd(allyl)Cl]2 and 4 equiv DPPE, the reaction did not reach completion in 1.5 h (72% yield of 10a and 12% remaining 1f, entry 7). Precatalyst [Pd(allyl)Cl]2/3 equiv PCy3 resulted in 93% yield (entry 8). Finally, [Pd(allyl)Cl]2/3 equiv CyJohnPhos and 1 equiv of Cs2CO3 at 60 °C resulted in 99% assay yield of allene product 10a (entry 9). Screening Buchwald’s phosphine ligands SPhos, XPhos, DavePhos and JohnPhos with [Pd(allyl)Cl]2 resulted in slower conversion to allenes.[97] In the absence of Cs2CO3 at 60 °C [Pd(allyl)Cl]2/3 equiv CyJohnPhos resulted in formation of only 7% of allene product 10a and 90% unreacted 1f (entry 10). Lowering the Cs2CO3 loading to 0.5 equiv did not affect the reactivity significantly (94% yield, entry 11), and 3 equiv of Cs2CO3 rendered the reaction slower (85% yield of 10a and 15% recovered 1f, entry 12). Bases such as NaOAc·3H2O, K2CO3 and NaHCO3 were less effective (entries 13–16). Ultimately, it was found that 0.5 mol % of [Pd(allyl)Cl]2, 1.5 mol % of CyJohnPhos, and 1 equiv of Cs2CO3 gave the best yield (entry 9) of the conditions examined.
Table 8.
Optimization of allene formation from 2-B(pin)-substituted allylic acetate 1f.

| ||||||
|---|---|---|---|---|---|---|
| Entry | [Pd] | Ligand (mol %) | Base (equiv) | Time (h) | Yield (%)[b] | |
| recovered 1f | 10a | |||||
| 1 | Pd(PPh3)4 | – | – | 1.5 | 80 | 0 |
| 2 | Pd(PPh3)4[c] | – | – | 1.5 | 0 | 4 |
| 3 | Pd(PPh3)4 | – | Cs2CO3 (1) | 1.5 | 0 | 87 |
| 4 | – | – | Cs2CO3 (3) | 1.5 | 95 | <2 |
| 5 | Pd(OAc)2 | PPh3 (2) | Cs2CO3 (1) | 1.5 | 0 | 73 |
| 6 | [Pd(allyl)Cl]2 | PPh3 (2) | Cs2CO3 (1) | 1.5 | 0 | 80 |
| 7 | [Pd(allyl)Cl]2 | DPPE (2) | Cs2CO3 (1) | 1.5 | 12 | 72 |
| 8 | [Pd(allyl)Cl]2 | PCy3 (1.5) | Cs2CO3 (1) | 1.0 | 0 | 93 |
| 9 | [Pd(allyl)Cl]2 | CyJohnPhos (1.5) | Cs2CO3 (1) | 1.0 | 0 | 99 |
| 10 | [Pd(allyl)Cl]2 | CyJohnPhos (1.5) | – | 1.0 | 90 | 7 |
| 11 | [Pd(allyl)Cl]2 | CyJohnPhos (1.5) | Cs2CO3 (0.5) | 1.0 | 0 | 94 |
| 12 | [Pd(allyl)Cl]2 | CyJohnPhos (1.5) | Cs2CO3 (3) | 1.0 | 15 | 85 |
| 13 | [Pd(allyl)Cl]2 | CyJohnPhos (1.5) | NaOAc·3H2O (3) | 1.0 | 78 | 19 |
| 14 | [Pd(allyl)Cl]2 | CyJohnPhos (1.5) | K2CO3 (3) | 1.0 | 59 | 41 |
| 15 | [Pd(allyl)Cl]2 | CyJohnPhos (1.5) | NaHCO3 (3) | 1.0 | 0 | 76 |
| 16 | [Pd(allyl)Cl]2 | CyJohnPhos (1.5) | NaHCO3 (1) | 1.0 | 0 | 90 |
Reactions conducted at 0.17 M concentration on 0.04 mmol scale.
Yield determined by 1H NMR integration of the crude reaction mixture using 1,4-dimethoxybenzene as the internal standard.
1 equiv of Pd(PPh3)4.
Substrate scope for allene formation from 2-B(pin)-substituted allylic acetates, benzoates and carbonates
Subjecting various 1-aryl-2-B(pin)-3-butyl allylic acetates to the condition in entry 9 of Table 8 resulted in formation of allene products. For example, aryl substrates bearing electron withdrawing or electron donating groups at the para-position provided excellent yields of allene products 10a–d (87–92%, Table 9, entries 1–4). The styryl derived allylic acetate 1l gave 55% yield of 10e without noticeable isomerization of the vinyl group (entry 5). Although 1,3-di-n-butyl-2-B(pin) allylic acetate 1a was not suitable for this transformation (entry 6), benzoate substrates 7a and 7c were converted to allene products 10f and 10g in 53% and 79% isolated yields (entries 7 and 8). With the dialkyl 2-B(pin)-substituted derivatives, the greater leaving group ability of the carbonates 8a and 8d resulted in higher yields of allene products 10f and 10g (85 and 83% yield, entries 9 and 10). The results in Tables 8 and 9 indicate that proper choice of catalyst, substrates and conditions enable the facile generation of allenes from 2-B(pin)-substituted allylic esters and carbonates.
Table 9.
Allene formation from 2-B(pin)-substituted allylic acetates, benzoates and carbonates.

| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Yield (%)[a] | ||
| 1 |

|
1f |

|
10a | 90 |
| 2 |

|
1i |

|
10b | 92 |
| 3 |

|
1h |

|
10c | 87 |
| 4 |

|
1k |
|
10d | 92 |
| 5 |

|
1m |
|
10e | 55 |
| 6 |

|
1a |
|
10f | –[b] |
| 7 |

|
7a |
|
10f | 53 |
| 8 |

|
7c |
|
10g | 79 |
| 9 |

|
8a |
|
10f | 85 |
| 10 |

|
8d |
|
10g | 83 |
Yield of isolated and purified products.
Starting material decomposed during course of the reaction.
Plausible catalytic cycle of allene formation reaction and racemization of allene
To rationalize the formation of allylic substitution vs. allene products, a plausible catalytic cycle for allene formation is illustrated in Figure 3. Electron donating ligands, such as PCy3 and CyJohnPhos, easily lead to π-allyl palladium intermediates. In the presence of good nucleophiles, such as malonates or amines, allylic substitution occurs by addition of the nucleophile to the π-allyl palladium. In contrast, in the absence of a suitable nucleophile, the base is proposed to attack the empty coordination site on boron or hydrolyze the boronate ester. Several possible explanations for the observed dependency on base strength can be considered. The stronger bases are more likely to promote hydrolysis of the B(pin) group to the boronic acid. Also, more reactive carbonate bases are harder and will be better nucleophiles toward boron than either bicarbonate or acetate. Finally, elimination of 3-coordinate boron can be envisioned to lead to a palladium-allene π-complex before allene dissociation.
Figure 3.
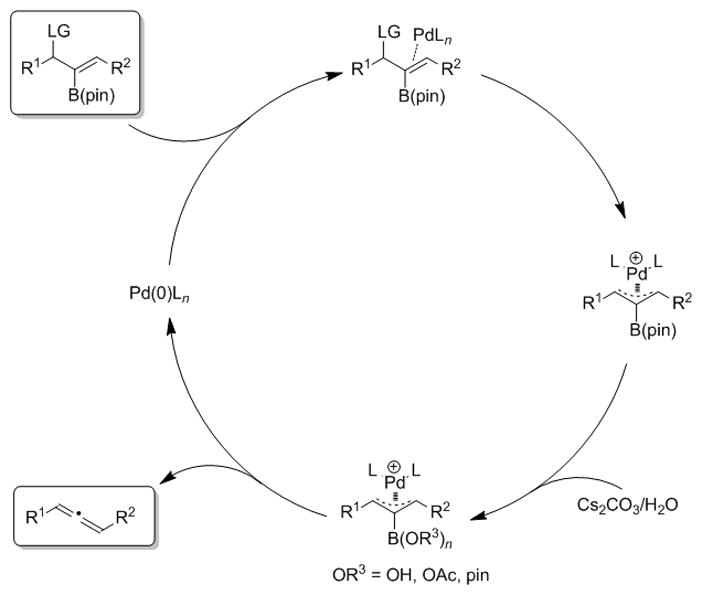
Plausible catalytic cycle for allene formation.
Optically active allenes are ubiquitous intermediates and targets for the synthesis of natural products and biologically active compounds.[98,99] To investigate the possibility of chirality transfer in catalytic cycle of Figure 3, we performed the allene formation reaction using enantioenriched 2-B(pin)-allylic acetate 1f to generate allene product 10a. The reaction of 1f (60% ee) in the presence of the Pd/CyJohnPhos catalyst afforded enantioenriched allene 10a with 36% ee in 18% yield in 10 min (Table 10, entry 2). When the reaction reached completion in 35 min, however, a significant loss of ee was observed (Table 10, entries 3 and 4). Finally, allene product 10a was racemized completely in 1 h (Table 10, entry 6). It is noteworthy that B(pin)-substituted 1f (60% ee) was not racemized during the course of the reaction. These results suggest that 2-B(pin)-allylic acetate undergoes stereoselective allene formation, however, a competing racemization of the allene product in the presence of the palladium catalyst, probably via hydropalladation, is responsible for the observed loss of ee over time.[100,101]
Table 10.
Palladium-catalyzed allene formation reaction of enantioenriched 1f.

| ||||
|---|---|---|---|---|
| Entry | Time (min) | Yield [%][a] | ee[b] of 10a | |
| 1f | 10a | |||
| 1 | 0 | 100 | – | – |
| 2 | 10 | 82 | 18 | 36 |
| 3 | 20 | 50 | 50 | 29 |
| 4 | 35 | 0 | 100 | 13 |
| 5 | 45 | 0 | 100 | 3 |
| 6 | 60 | 0 | 100 | 0 |
Conversion yield determined by 1H NMR of aliquots of unpurified reaction mixture.
Enantiomeric excess of allene 10a was determined by HPLC analysis (see Supporting Information).
Conclusion
Application of multicomponent tandem reactions to molecules bearing more than one reaction site is a powerful synthetic strategy for rapid generation of molecular complexity and diversity. Key to development of such methodology is to tailor the catalyst, reagents and conditions to afford chemoselective activation of the substrate. Toward this goal, we introduced a new class of bifunctional reagents that contain vinyl boronate esters embedded within allylic acetates. The requisite 2-B(pin)-substituted allylic acetates are synthesized on gram scale in one-pot procedures from readily available materials using the heterobimetallic chemistry developed in our laboratory.[15–18]
To facilitate applications of our bifunctional substrates, methods to control the chemoselectivity of their activation is crucial. The challenge is to employ common palladium-phosphine-based catalysts and modulate their activation of 2-B(pin)-substituted allylic acetates through manipulation of reagents, substrates, and conditions. In the case of allylic substitutions, reactions were conducted under conditions where the B(pin) group was stable to hydrolysis (and palladium hydroxide intermediates are not likely formed), [13,102]inhibiting B to Pd transmetallation. Under such conditions, oxidative ionization of the acetate and nucleophilic addition to the π-allyl palladium intermediate were much faster than transmetallation. In the Suzuki-Miyaura cross-coupling of 2-B(pin)-substituted allylic acetates, we found that the chemoselectivity of the Pd(0) is Ar–I > Ar–Br > allylic acetate. Once the Pd(0) is converted to Pd(II) by the aryl halide, ionization of the allylic acetate does not interfere with cross-coupling. Under conditions where the B(pin) group hydrolyzes to the boronic acid (or palladium hydroxides can form), transmetallation is also fast. Detailed study of this system indicates the relative order of reactivity toward Pd(0) is Ar–I > Ar–Br > allylic carbonate > allylic benzoate > allylic acetate, with the reactivity of allylic carbonates approaching that of aryl bromides. For allylic acetates with the acetate in benzylic position, the allylic acetate is more reactive then aryl bromides in the presence of L2Pd. The observations outlined herein enabled a complete reversal of chemoselectivity from allylic substitution reactions to Suzuki-Miyaura cross-coupling of allylic acetates, benzoates, and carbonates.
In the case of the third reaction manifold, allene formation, oxidative ionization of the allylic acetate occurs to generate the π-allyl palladium intermediate. In the absence of nucleophile, we hypothesize that hard bases (CO32−, HCO3−, AcO−) or hydroxide generated from them, attack the unsaturated boron to make ate complexes that can undergo elimination of 3-coordinate boron and to generate an intermediate Pd(η2-allene). The rate of allene formation is base dependent, with the more basic carbonate more reactive than the less basic acetate. Our method provides high yields of 1,3-disubstituted allenes under mild conditions. While the allene formation reaction of enantioenriched 2-B(pin) allylic acetate provided the enantioenriched allene, palladium-catalyzed racemization of the allene resulted in the racemic product over time.
With the ability to control chemoselectivity, various tandem reactions were investigated. The allylic substitution of 2-B(pin)-substituted allylic acetates can be followed with a Suzuki-Miyaura cross-coupling to afford (E)-trisubstituted allylic amines and malonates with four points of diversity. Enantioenriched 2-B(pin)-substituted allylic acetates undergo allylic substitution with net stereochemical retention, allowing isolation of B(pin)-containing substitution products or α-amino ketones with little or no loss of ee. Alternatively, the allylic substitution of 2-B(pin)-substituted allylic acetates can be paired with oxidation of vinyl boronates to provide valuable α-amino ketones and 1,4-dicarbonyl compounds.
Given the high levels of control over chemoselectivity in our optimized palladium-catalyzed reactions of 2-B(pin)-substituted allylic acetate derivatives, we are optimistic that these bifunctional substrates will serve as useful intermediates in synthesis.
Supplementary Material
Acknowledgments
We thank the NSF (CHE-0848467 and 1152488) and NIH (NIGMS 104349) for support of this work. We also thank Dr. Alice E. Lurain for helpful suggestions in the preparation of this manuscript.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.Trost BM. Science. 1983;219:245. doi: 10.1126/science.219.4582.245. [DOI] [PubMed] [Google Scholar]
- 2.Shenvi RA, O’Malley DP, Baran PS. Acc Chem Res. 2009;42:530. doi: 10.1021/ar800182r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wuts PGM, Greene TW. Greene’s Protective Groups in Organic Synthesis. 4. Wiley; Hoboken, NJ: 2007. [Google Scholar]
- 4.Hegedus LS, Söderberg BCG. Transition Metals in the Synthesis of Complex Organic Molecules. University Science Books; Sausalito: 2009. [Google Scholar]
- 5.Trost BM. Angew Chem Int Ed. 1989;28:1173. [Google Scholar]
- 6.Tsuji J. Palladium Reagents and Catalysts : Innovations in Organic Synthesis. Chichester; New York: 1995. [Google Scholar]
- 7.Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- 8.Graening T, Schmalz HG. Angew Chem Int Ed. 2003;42:2580. doi: 10.1002/anie.200301644. [DOI] [PubMed] [Google Scholar]
- 9.Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- 10.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]
- 11.Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
- 12.Barrios-Landeros F, Carrow BP, Hartwig JF. J Am Chem Soc. 2009;131:8141. doi: 10.1021/ja900798s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carrow BP, Hartwig JF. J Am Chem Soc. 2011;133:2116. doi: 10.1021/ja1108326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans LA, Fey N, Harvey JN, Hose D, Lloyd-Jones GC, Murray P, Orpen AG, Osborne R, Owen-Smith GJJ, Purdie M. J Am Chem Soc. 2008;130:14471. doi: 10.1021/ja806278e. [DOI] [PubMed] [Google Scholar]
- 15.Hussain MM, Walsh PJ. Angew Chem Int Ed. 2010;49:1834. doi: 10.1002/anie.200905399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Carroll PJ, Walsh PJ. J Am Chem Soc. 2008;130:3521. doi: 10.1021/ja077664u. [DOI] [PubMed] [Google Scholar]
- 17.Hussain MM, Li H, Hussain N, Ureña M, Carroll PJ, Walsh PJ. J Am Chem Soc. 2009;131:6516. doi: 10.1021/ja900147s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hussain N, Hussain MM, Carroll PJ, Walsh PJ. Chem Sci. 2013;4:3946. [Google Scholar]
- 19.Kim BS, Hussain MM, Norrby PO, Walsh PJ. Chem Sci. 2014;5:1241. doi: 10.1039/C3SC53035C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown HC, Sinclair JA. J Organomet Chem. 1977;131:163. [Google Scholar]
- 21.Brown HC, Bhat NG, Srebnik M. Tetrahedron Lett. 1988;29:2631. [Google Scholar]
- 22.Blanchard C, Framery E, Vaultier M. Synthesis. 1996:45. [Google Scholar]
- 23.Brown HC, Roy CD, Soundararajan R. Tetrahedron Lett. 1997;38:765. [Google Scholar]
- 24.Renaud J, Graf C-D, Oberer L. Angew Chem Int Ed. 2000;39:3101. doi: 10.1002/1521-3773(20000901)39:17<3101::aid-anie3101>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 25.Chong JM, Shen L, Taylor NJ. J Am Chem Soc. 2000;122:1822. [Google Scholar]
- 26.Molander GA, Ellis NM. J Org Chem. 2008;73:6841. doi: 10.1021/jo801191v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawatsura M, Ikeda D, Komatsu Y, Mitani K, Tanaka T, Uenishi Ji. Tetrahedron. 2007;63:8815. [Google Scholar]
- 28.Nemoto T, Fukuyama T, Yamamoto E, Tamura S, Fukuda T, Matsumoto T, Akimoto Y, Hamada Y. Org Lett. 2007;9:927. doi: 10.1021/ol0700207. [DOI] [PubMed] [Google Scholar]
- 29.Benfatti F, Cardillo G, Gentilucci L, Mosconi E, Tolomelli A. Org Lett. 2008;10:2425. doi: 10.1021/ol8006919. [DOI] [PubMed] [Google Scholar]
- 30.Park JK, Ondrusek BA, McQuade DT. Org Lett. 2012;14:4790. doi: 10.1021/ol302086v. [DOI] [PubMed] [Google Scholar]
- 31.Moure AL, Mauleón P, Arrayás RG, Carretero JC. Org Lett. 2013;15:2054. doi: 10.1021/ol4007663. [DOI] [PubMed] [Google Scholar]
- 32.Pelz NF, Woodward AR, Burks HE, Sieber JD, Morken JP. J Am Chem Soc. 2004;126:16328. doi: 10.1021/ja044167u. [DOI] [PubMed] [Google Scholar]
- 33.Sieber JD, Morken JP. J Am Chem Soc. 2006;128:74. doi: 10.1021/ja057020r. [DOI] [PubMed] [Google Scholar]
- 34.Tonogaki K, Itami K, Yoshida J-i. J Am Chem Soc. 2006;128:1464. doi: 10.1021/ja057778a. [DOI] [PubMed] [Google Scholar]
- 35.Tonogaki K, Itami K, Yoshida J-i. Org Lett. 2006;8:1419. doi: 10.1021/ol060197l. [DOI] [PubMed] [Google Scholar]
- 36.Burks HE, Liu S, Morken JP. J Am Chem Soc. 2007;129:8766. doi: 10.1021/ja070572k. [DOI] [PubMed] [Google Scholar]
- 37.Carosi L, Hall DG. Angew Chem Int Ed. 2007;46:5913. doi: 10.1002/anie.200700975. [DOI] [PubMed] [Google Scholar]
- 38.Peng F, Hall DG. Tetrahedron Lett. 2007;48:3305. [Google Scholar]
- 39.Štambaský J, Malkov AV, Kočovský P. Collect Czech Chem Commun. 2008;73:705. [Google Scholar]
- 40.Fernández E, Pietruszka J. Synlett. 2009:1474. [Google Scholar]
- 41.Fernández E, Pietruszka J, Frey W. J Org Chem. 2010;75:5580. doi: 10.1021/jo1008959. [DOI] [PubMed] [Google Scholar]
- 42.Kukkadapu KK, Ouach A, Lozano P, Vaultier M, Pucheault M. Org Lett. 2011;13:4132. doi: 10.1021/ol201661s. [DOI] [PubMed] [Google Scholar]
- 43.Touchet S, Carreaux F, Molander GA, Carboni B, Bouillon A. Adv Synth Cat. 2011;353:3391. doi: 10.1002/adsc.201100407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meng F, Jang H, Jung B, Hoveyda AH. Angew Chem Int Ed. 2013;52:5046. doi: 10.1002/anie.201301018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibson JR, Manna VK, Salisbury J. J Int Med Res. 1989;17:28B. [PubMed] [Google Scholar]
- 46.Posner GH. Chem Rev. 1986;86:831. [Google Scholar]
- 47.Ho T-L. Tandem Organic Reactions. Wiley and Sons; New York: 1992. [Google Scholar]
- 48.Tietze LF, Beifuss U. Angew Chem Int Ed. 1993;32:131. [Google Scholar]
- 49.Wender PA, Miller BL. In Organic Synthesis: Theory and Applications. Vol. 2. JAI Press; Greenwich, CT: 1993. [Google Scholar]
- 50.Parsons PJ, Penkett CS, Shell AJ. Chem Rev. 1996;96:195. doi: 10.1021/cr950023+. [DOI] [PubMed] [Google Scholar]
- 51.Hussain MM, Walsh PJ. Acc Chem Res. 2008;41:883. doi: 10.1021/ar800006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown HCZM. Organic Synthesis Via Boranes. Vol. 2. Aldrich Chemical Company, Inc; Milwaukee: 2001. [Google Scholar]
- 53.Kabalka GW, Shoup TM, Goudgaon NM. J Org Chem. 1989;54:5930. [Google Scholar]
- 54.Kabalka GW, Shoup TM, Goudgaon NM. Tetrahedron Lett. 1989;30:1483. [Google Scholar]
- 55.Trost BM, Gowland FW. J Org Chem. 1979;44:3448. [Google Scholar]
- 56.Trost BM, Curran DP. J Am Chem Soc. 1980;102:5699. [Google Scholar]
- 57.Negishi E, Luo FT, Pecora AJ, Silveira A., Jr J Org Chem. 1983;48:2427. [Google Scholar]
- 58.Schuda PF, Bernstein B. Synth Commun. 1984;14:293. [Google Scholar]
- 59.Organ MG, Arvanitis EA, Dixon CE, Cooper JT. J Am Chem Soc. 2002;124:1288. doi: 10.1021/ja011508k. [DOI] [PubMed] [Google Scholar]
- 60.Johannsen M, Jørgensen KA. Chem Rev. 1998;98:1689. doi: 10.1021/cr970343o. [DOI] [PubMed] [Google Scholar]
- 61.Martín VS, Woodward SS, Katsuki T, Yamada Y, Ikeda M, Sharpless KB. J Am Chem Soc. 1981;103:6237. [Google Scholar]
- 62.Slater JW, Zechnich AD, Haxby DG. Drugs. 1999;57:31. doi: 10.2165/00003495-199957010-00004. [DOI] [PubMed] [Google Scholar]
- 63.Ashton D, Reid K, Willems R, Marrannes R, Wauguier A. Drug Dev Res. 1986;8:397. [Google Scholar]
- 64.Straub H, Kohling R, Speckmann EJ. Brain Res. 1994;658:119. doi: 10.1016/s0006-8993(09)90017-8. [DOI] [PubMed] [Google Scholar]
- 65.Andersen KE, Sorensen JL, Huusfeldt PO, Knutsen LJ, Lau J, Lundt BF, Petersen H, Suzdak PD, Swedberg MD. J Med Chem. 1999;42:4281. doi: 10.1021/jm980492e. [DOI] [PubMed] [Google Scholar]
- 66.Bukovec C, Kazmaier U. Org Lett. 2009;11:3518. doi: 10.1021/ol901415p. [DOI] [PubMed] [Google Scholar]
- 67.Bukovec C, Wesquet AO, Kazmaier U. Eur J Org Chem. 2011:1047. [Google Scholar]
- 68.Nwokogu GC. Tetrahedron Lett. 1984;25:3263. [Google Scholar]
- 69.Nwokogu GC. J Org Chem. 1985;50:3900. [Google Scholar]
- 70.Trost BM, Oslob JD. J Am Chem Soc. 1999;121:3057. [Google Scholar]
- 71.Yamamoto K, Heathcock CH. Org Lett. 2000;2:1709. doi: 10.1021/ol000064e. [DOI] [PubMed] [Google Scholar]
- 72.Comer E, Organ MG, Hynes SJ. J Am Chem Soc. 2004;126:16087. doi: 10.1021/ja045416h. [DOI] [PubMed] [Google Scholar]
- 73.Delcamp JH, White MC. J Am Chem Soc. 2006;128:15076. doi: 10.1021/ja066563d. [DOI] [PubMed] [Google Scholar]
- 74.Lin S, Song CX, Cai GX, Wang WH, Shi ZJ. J Am Chem Soc. 2008;130:12901. doi: 10.1021/ja803452p. [DOI] [PubMed] [Google Scholar]
- 75.Ohmiya H, Makida Y, Tanaka T, Sawamura M. J Am Chem Soc. 2008;130:17276. doi: 10.1021/ja808673n. [DOI] [PubMed] [Google Scholar]
- 76.Su Y, Jiao N. Org Lett. 2009;11:2980. doi: 10.1021/ol9009865. [DOI] [PubMed] [Google Scholar]
- 77.Lautens M, Tayama E, Herse C. J Am Chem Soc. 2004;127:72. doi: 10.1021/ja043898r. [DOI] [PubMed] [Google Scholar]
- 78.Mariampillai B, Herse C, Lautens M. Org Lett. 2005;7:4745. doi: 10.1021/ol051947e. [DOI] [PubMed] [Google Scholar]
- 79.Delin P, Anjun C, Yijin S, Wang Z, Si L, Wei J, Juan X, Qingjian L, Liangren Z, Jiao N. Angew Chem Int Ed. 2008;47:4729. [Google Scholar]
- 80.Genet JP, Linquist A, Blart E, Mouries V, Savignac M, Vaultier M. Tetrahedron Lett. 1995;36:1443. [Google Scholar]
- 81.Trost BM, Malhotra S, Chan WH. J Am Chem Soc. 2011;133:7328. doi: 10.1021/ja2020873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Amatore C, Pfluger F. Organometallics. 1990;9:2276. [Google Scholar]
- 83.Bedford RB, Welch SL. Chem Commun. 2001:129. [Google Scholar]
- 84.Yin J, Rainka MP, Zhang XX, Buchwald SL. J Am Chem Soc. 2002;124:1162. doi: 10.1021/ja017082r. [DOI] [PubMed] [Google Scholar]
- 85.Walker SD, Barder TE, Martinelli JR, Buchwald SL. Angew Chem Int Ed. 2004;43:1871. doi: 10.1002/anie.200353615. [DOI] [PubMed] [Google Scholar]
- 86.Zimmer R, Dinesh CU, Nandanan E, Khan FA. Chem Rev. 2000;100:3067. doi: 10.1021/cr9902796. [DOI] [PubMed] [Google Scholar]
- 87.Hashmi ASK. Angew Chem Int Ed. 2000;39:3590. [PubMed] [Google Scholar]
- 88.Bates RW, Satcharoen V. Chem Soc Rev. 2002;31:12. doi: 10.1039/b103904k. [DOI] [PubMed] [Google Scholar]
- 89.Krause N, Hashmi ASK. Modern Allene Chemistry. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 90.Krause N, Hoffmann-Röder A. Tetrahedron. 2004;60:11671. [Google Scholar]
- 91.Ma S. Chem Rev. 2005;105:2829. doi: 10.1021/cr020024j. [DOI] [PubMed] [Google Scholar]
- 92.Brummond KM, DeForrest JE. Synthesis. 2007. p. 795. [Google Scholar]
- 93.Ogasawara M. Tetrahedron: Asymmetry. 2009;20:259. [Google Scholar]
- 94.Ohno H, Miyamura K, Tanaka T, Oishi S, Toda A, Takemoto Y, Fujii N, Ibuka T. J Org Chem. 2002;67:1359. doi: 10.1021/jo016320y. [DOI] [PubMed] [Google Scholar]
- 95.Wesquet AO, Kazmaier U. Angew Chem Int Ed. 2008;47:3050. doi: 10.1002/anie.200705976. [DOI] [PubMed] [Google Scholar]
- 96.Bukovec C, Kazmaier U. Org Biomol Chem. 2011;9:2743. doi: 10.1039/c0ob00945h. [DOI] [PubMed] [Google Scholar]
- 97.Martin R, Buchwald SL. Acc Chem Res. 2008;41:1461. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ogasawara M, Watanabe S. Synthesis. 2009:1761. [Google Scholar]
- 99.Yu S, Ma S. Angew Chem Int Ed. 2012;51:3074. doi: 10.1002/anie.201101460. [DOI] [PubMed] [Google Scholar]
- 100.Molander GA, Sommers EM, Baker SR. J Org Chem. 2011;71:1563. doi: 10.1021/jo052201x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Crouch IT, Neff RK, Frantz DE. J Am Chem Soc. 2013;135:4970. doi: 10.1021/ja401606e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Christian AH, Müller P, Monfette S. Organometallics. ASAP; 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.