

Abstract
The zebrafish embryo is now commonly used for basic and biomedical research to investigate the genetic control of developmental processes and to model congenital abnormalities. During the first day of life, the zebrafish embryo progresses through many developmental stages including fertilization, cleavage, gastrulation, segmentation, and the organogenesis of structures such as the kidney, heart, and central nervous system. The anatomy of a young zebrafish embryo presents several challenges for the visualization and analysis of the tissues involved in many of these events because the embryo develops in association with a round yolk mass. Thus, for accurate analysis and imaging of experimental phenotypes in fixed embryonic specimens between the tailbud and 20 somite stage (10 and 19 hours post fertilization (hpf), respectively), such as those stained using whole mount in situ hybridization (WISH), it is often desirable to remove the embryo from the yolk ball and to position it flat on a glass slide. However, performing a flat mount procedure can be tedious. Therefore, successful and efficient flat mount preparation is greatly facilitated through the visual demonstration of the dissection technique, and also helped by using reagents that assist in optimal tissue handling. Here, we provide our WISH protocol for one or two-color detection of gene expression in the zebrafish embryo, and demonstrate how the flat mounting procedure can be performed on this example of a stained fixed specimen. This flat mounting protocol is broadly applicable to the study of many embryonic structures that emerge during early zebrafish development, and can be implemented in conjunction with other staining methods performed on fixed embryo samples.
Keywords: Developmental Biology, Issue 89, animals, vertebrates, fishes, zebrafish, growth and development, morphogenesis, embryonic and fetal development, organogenesis, natural science disciplines, embryo, whole mount in situ hybridization, flat mount, deyolking, imaging
Introduction
The zebrafish, Danio rerio, has become a widely used organism in the study of developmental biology. Zebrafish are a tropical, freshwater vertebrate species belonging to the family Cyprinidae. Zebrafish were originally used for environmental research to gain information on the hazards of potential water pollutants, as they were easily obtainable, inexpensive, and easy to maintain1. Over the last thirty years, the zebrafish has become recognized as a genetically tractable vertebrate model system for a host of reasons. Zebrafish show a high level of anatomical and physiological conservation with higher vertebrates including mammals2,3. Recent genome sequence annotation has revealed that approximately 70% of human genes have at least one zebrafish counterpart4. For example, many orthologous genes that play an important role during kidney development have been identified between these species5-7. This dramatic degree of conservation with regards to cellular and molecular biology has enabled researchers to create models for a wide range of human diseases in the zebrafish8, and zebrafish have become a valuable tool to identify potential drug therapies using chemical genetic screens9.
Furthermore, the zebrafish model is not only able to recapitulate a variety of complex developmental processes and disease states that are seen in humans, but several additional attributes also heighten its appeal as a model organism for embryological studies. Zebrafish are oviparous and reproduce through broadcast spawning, which provides researchers with easy access to the embryos from the onset of fertilization10. Other advantages include embryo size, transparency, and rapid, external development10. Additionally, zebrafish adults possess high fecundity and typically produce relatively large clutch sizes that can range between 200-500 per week, ultimately enabling researchers to perform high throughput experiments, such as phenotype based chemical screens, with ease9.
Even so, despite the plethora of advantages that are exhibited by the zebrafish model, there are challenges that pertain to the analysis and communication of the experimental results obtained from early developmental stages. In some cases, the inherent anatomy of the zebrafish embryo could obscure the visualization of one’s data. After fertilization, the initial cell undergoes multiple cleavage events as development progresses11. Following gastrulation, the embryonic axis emerges at the tailbud stage (10 hpf) such that it is wrapped around the yolk11. As subsequent development progresses through the segmentation period, the trunk will grow in mass and also lengthen by axial elongation such that a tail along with a portion of the yolk sac itself extend out from the central yolk mass11. Between the tailbud and 20 somite stage (19 hpf), attempts to observe specific developmental features after the utilization of various staining protocols, such as WISH, can be challenging both to visualize and photograph due to the geometry of the embryo and opacity of the yolk sac. One way to eliminate these challenges is to remove the yolk and then flatten the embryo into a two-dimensional sample that can be analyzed in a more straightforward manner.
The following protocol and video resources provide a guide for how to successfully deyolk and flat mount an embryo following fixation and staining, using the example of one or two-color WISH staining. WISH is a widely used technique that enables the detection of specific nucleic acids in preserved specimens and thus allows for the site(s) of gene expression to be detected within tissues. WISH techniques have been extensively described, and can be performed with various color substrates as well as fluorescent detection systems12-20. Here we provide our modified WISH protocol used for zebrafish embryos between the tailbud and segmentation stages of development. Alternative WISH protocols, however, are compatible with the flat mount procedure described here, as noted at the outset of the protocol below. In addition, flat mounting could be utilized in conjunction with any number of existing visualization techniques, such as whole mount immunohistochemistry, or cell lineage tracking labels, which are not described in this protocol. Overall, the technique of flat mounting can better enable superior analysis and presentation of data obtained from experiments with the earliest stages of embryonic development in the zebrafish.
Protocol
The procedures for working with zebrafish embryos described in this protocol were approved by the Institutional Animal Care and Use Committee at the University of Notre Dame. Note: The procedures described in Parts 1-5 were used to generate the embryo images provided here in the representative results. The procedures described in Parts 1-5 could be substituted as desired with alternative WISH procedures12-16 or other visualization techniques such as immunohistochemical labeling for specific proteins using specific antibodies. In performing flat mounts on WISH zebrafish embryos with the protocol described in Parts 1-5, the initial embryo fixation and final staining fixation steps are the major manipulations which affect the ability to remove yolk granules from the sample for flat mounting. The best flat mounting results are obtained when the initial fixation is performed with freshly thawed ice cold 4% paraformaldehyde (PFA)/1x PBS and the fixation of the final WISH staining reaction with ice cold 4% PFA/1x PBS (which does not need to be freshly thawed), and it is advised that readers to consider this when substituting alternative staining methods in combination with Parts 6-8.
1. Embryo Collection, Fixation, and Storage
Prepare mating cages by placing adult zebrafish male(s) and female(s) in chambers of system water so that they are separated by a divider overnight.
Remove the dividers between adults and collect the fertilized eggs using a fine wire mesh tea strainer within ~15-30 min of mating to gather clutches that have equivalent embryonic stages.
Turn the strainer upside down and rinse the surface with a fine stream of E3 dispensed from a squirt bottle to transfer the embryos into incubation dishes containing approximately 25 ml of E3 solution.
After collecting the clutches, divide them into several dishes such that approximately 50-60 embryos are incubated in each dish of 25 ml of E3 solution. Note: Overcrowding of embryos can lead to dramatic developmental asynchrony within the clutch and care should be taken to avoid this to enable timely collection of the desired stage(s).
Incubate the embryos at 28.5 °C to enable developmental assessment according to the zebrafish standard staging series11.
Use a plastic transfer pipette to gently place the desired number of embryos at the selected timepoint, up to the 20 somite stage (19 hpf), into a flat bottom microcentrifuge tube. Note: Take care in handling of young embryos, as they are fragile and can be easily damaged by crushing or aggressive handling with transfer pipettes. Alternatively, glass vials can be used for fixation and processing of embryos. For all subsequent protocol washes plastic transfer pipettes can be utilized, with the exception of riboprobe addition and removal (Steps 3.11, 3.14).
Replace the E3 with ice cold 4% PFA/1x PBS, and fix the embryos in their chorions in 4% PFA/1x PBS for 4 hr at room temperature or at 4 °C overnight. Note of caution: PFA is toxic and PFA solutions should be handled at a chemical hood while wearing suitable personal protective equipment, including gloves and a lab coat. In addition, PFA powder should be handled with exceptional care when making the stock solution, as even the granulated PFA can tend to disperse through static charge. Typically PFA is prepared by heating the 1x PBS to a boil to dissolve the PFA powder, and then the solution is stored in aliquots at -20 °C. If fine sediments are present in the 4% PFA/1x PBS once the solution is cooled, filter sterilize the cooled solution before aliquoting for freeze storage by passing it through a 0.45 µm filter system. Only freshly prepared or freshly thawed PFA is used for this initial (i.e. primary) fixation of the embryo.
Remove the fix and wash the embryos twice with 1x PBST, then transfer into an incubation dish.
View the embryos under a stereomicroscope and use two pairs of fine forceps to remove the chorions surrounding the embryos, such that one pair is used to gently leverage the embryo while the second pair is used to tear open the chorion.
2. Embryo Permeablization
Transfer the dechorionated embryos back into the microcentrifuge tube or glass vial, then rinse twice with 1x PBST to remove any remaining chorion debris.
Remove the 1x PBST and wash the embryos twice with 100% methanol (MeOH).
Place the embryos at -20 °C for at least 20 min. Note: Embryos can be stored at -20 °C in MeOH for one year or more. Methanol makes the chorions sticky, therefore do not proceed to this step unless the chorions have been removed.
Rehydrate the embryos by removing the 100% MeOH and wash them at room temperature for 5 min each in 50% MeOH/1x PBST, 30% MeOH/1x PBST, then twice with 1x PBST.
Prepare a fresh proteinase K working solution (5 µg/ml) by adding 25 µl of freshly thawed proteinase K stock (10 mg/ml) to 50 ml of 1x PBST.
Remove the 1x PBST from the embryos, then replace with the proteinase K working solution and incubate based on the embryonic stage: <= 5 somites for 1 min; 10-12 somites for 1.5 min; 15 somites for 2 min, and 20 somites for 3 min.
Remove the proteinase K working solution and wash the embryos twice with 1x PBST.
Remove the 1x PBST and replace with ice cold 4% PFA/1x PBS for at least 20 min at room temperature.
3. Riboprobe Synthesis, Prehybridization, Hybridization, and Probe Removal
Assemble the antisense riboprobe in vitro transcription reaction at room temperature in a 1.5 ml microcentrifuge tube, while keeping enzymes on ice, by combining a DNA template containing sequence corresponding to the gene of interest (either 100-200 ng of PCR product or 1.5 µg of linearized DNA plasmid, prepared as described12,13), 2 µl of digoxygenin or fluorescein labeled ribonucleotides, 2 µl of the appropriate RNA polymerase (SP6, T3, T7 depending on which sequence is incorporated into the PCR product or present on the DNA plasmid), 2 µl of 10x transcription buffer, 0.5 µl of RNase inhibitor, and bring to a total volume of 20 ul with molecular grade distilled water (DNase, RNase free). Note: The 10x transcription buffer should be prewarmed by placing the tube in a 37 °C waterbath for 10-20 min, and vortexed afterward to ensure that all components are in solution. If white flakes are present, incubate the tube for another 5-10 min and vortex again. Transcription reactions can fail or have poor yields if the transcription buffer is not fully dissolved and thoroughly mixed.
Mix the components and incubate each reaction at 37 °C for 2 hr in a waterbath.
Remove the reaction tube(s) from the waterbath, and destroy the DNA template in each sample by adding 5 µl of 10x DNase I buffer, 2 µl of DNase I enzyme, and 23 µl of molecular grade distilled water to bring the total volume to 50 µl, and then incubate each tube at 37 °C for 20 min in a waterbath.
Perform an ethanol precipitation in each microcentrifuge tube by adding 5 µl of 3 M sodium acetate, pH 5.2 and 110 µl of ice cold 100% ethanol, followed by 1 µl of glycogen stock to facilitate RNA pellet visualization in subsequent steps, and then incubate the tube at -20 °C for at least 1 hr or overnight.
Centrifuge each microcentrifuge tube at maximum speed 4 °C for 20 min, then remove the 100% ethanol supernatant and replace with 100 μl of 70% ice cold ethanol.
Centrifuge each microcentrifuge tube at 4 °C for 5 min, then remove the 70% ethanol supernatant and air dry the pellet for 5 min, then finally add 100 µl of molecular grade distilled water to resuspend the pellet and quantify the riboprobe stock concentration using a nanodrop spectrophotometer. Note: Once the pellet has gone into solution, the riboprobe stock should be kept on ice and stored at -20 °C .
To prepare the fixed embryos for the prehybridization process, remove the 4% PFA/1x PBS from each sample and wash the embryos twice with 1x PBST.
Transfer between 25-40 embryos in 1x PBST into each single flat bottom microcentrifuge tube. Note: Overcrowding of embryos will lead to uneven staining later, and care should be taken to limit the sample size in each tube to approximately 40 embryos.
Replace the 1x PBST in each tube with 500-1,000 µl of HYB+.
Place the tube(s) of embryos into an oven set at 70 °C, and incubate them at this temperature for 4 hr to perform prehybridization.
Prepare the riboprobe/HYB+ solution by adding 100-500 ng of riboprobe (digoxygenin and/or fluorescein-labeled) to 500 µl of fresh HYB+ and then incubate this solution at 70 °C for at least 20 min prior to Step 3.11 to prewarm it. Note: To detect multiple gene transcripts, using single and/or double colorimetric labeling, each of the riboprobes corresponding to these genes need to be combined into one riboprobe/HYB+ cocktail.
Remove the prehybridization HYB+ from each tube of embryos and replace with sufficient riboprobe/HYB+ to cover the embryos. Note: Typically, 200-250 µl of riboprobe/HYB+ is necessary to cover embryos in one flat bottom microcentrifuge tube. Riboprobe/HYB+ addition and removal is typically performed using a pipette and barrier tips to prevent cross-contamination between samples.
Incubate the embryos at 70 °C overnight with ribroprobe/HYB+.
Prepare the following set of wash solutions and pre-warm them overnight at 70 °C: 50% formamide/2x SSCT, 2x SSCT, and 0.2x SSCT. Note: It is convenient to prepare 50 ml aliquots of each wash solution, because the excess can be stored at room temperature and subsequently reheated for future use.
Remove the riboprobe/HYB+ using a pipette and barrier tips and store at -20 °C. Note: Typically each riboprobe/HYB+ mixture can be used several times (e.g., 3 or more) without diminution of signal. While reused riboprobe/HYB+ is associated with lower background, however, keep in mind that reused riboprobe mixtures can be subject to degradation.
Wash the embryos twice with 50% formamide/2x SSCT for 30 min at 70 °C.
Wash the embryos once with 2x SSCT for 15 min at 70 °C.
Wash the embryos twice with 0.2x SSCT for 30 min at 70 °C.
Remove the 0.2x SSCT and wash the embryos twice with MABT at room temperature.
4. Anti-digoxygenin Antibody Incubation and Detection
Prepare fresh block solution.
Remove the MABT and replace with block solution.
Incubate the embryos in block solution for 2-4 hr at room temperature. Note: Longer block incubations provide optimal results with young zebrafish embryos stages (<15 somites). To do the lengthy block procedure, perform the block incubation for 8-12 hr at room temperature or a combination of room temperature plus (up to ~8 hr) a subsequent 4 °C incubation overnight.
Prepare the anti-digoxygenin antibody solution by making a 5,000 fold dilution in fresh block solution (e.g., 1 µl per 5 ml of block).
Remove the block and add the antibody/block solution.
Cover the sample in foil and incubate overnight at 4 °C or for 4 hr at room temperature.
Remove the anti-digoxygenin/block and wash the embryos at least 10 times with MABT. Note: Transfer the embryos to 12-well dishes for the MABT washes. This increases the speed at which the embryos can be washed, which is useful when processing large sample quantities, and makes it easy to monitor the progress of the staining reaction (Step 4.11) under a stereomicroscope. Note: At this step embryos can be ‘super-washed’ by incubating them overnight in MABT at 4 °C before proceeding to Step 4.8, which is a useful treatment for probes that tend to give high background or stains that require lengthy monitoring during the work day.
Prepare the pre-staining buffer and the desired staining solution (purple or red substrate).
Remove the MABT and wash the embryos in pre-staining buffer for 5 min at room temperature.
Remove the pre-staining buffer and add the staining solution.
Monitor the progress of the room temperature staining reaction every 15-30 min by checking the appearance of the embryos under a stereomicroscope. Note: The rate of the staining reaction can be slowed down by transferring the samples to 4 °C, if it is necessary to give more time for completion of the stain. Once chilled to 4 °C, however, the reaction cannot be accelerated by warming to room temperature.
When the staining reaction is complete, remove the staining solution and replace with 1x PBST.
Wash twice with 1x PBST for 5 min. Note: If not detecting other genes with another substrate, then fix the samples in ice cold 4% PFA/1x PBS for at least 20 min, and proceed to step 6.1. Otherwise proceed to step 5.1.
5. Anti-fluorescein Antibody Incubation and Detection
Remove the 1x PBST and wash the embryos twice with 0.1 M glycine, pH 2.2 for 15 min each at room temperature. Note: The time for the glycine washes is essential to completely deactivate the first staining reaction.
Remove the 0.1 M glycine and wash the embryos twice with MABT for 5 min at room temperature.
Prepare the anti-fluorescein antibody solution by making a 5,000 fold dilution in fresh block solution (e.g., 1 µl per 5 ml of block).
Remove the MABT and add the antibody/block solution. Follow steps 4.6-4.13 to perform the detection of the fluorescein-labeled ribroprobe. Note: When the final staining reaction is completed, it is essential that the last fixation of the sample is done in ice cold 4% PFA/1X PBS. Fixation with warmer PFA solutions tends to be associated with sticky yolk that is difficult to remove from the embryo during dissection and deyolking for the flat mount procedure.
6. Embryo Dissection and Initial Deyolking of the Embryo
Select a fixed, stained embryo for flat mounting.
Use a transfer pipette to place the selected embryo sample into a plastic or glass Petri dish containing 1x PBST.
Place the dish under a stereomicroscope, and roll the embryo using a pair of fine forceps or lash tool so that a lateral view is obtained and the head and tail ends are visualized.
Use one pair of fine forceps to make an incision in the yolk at a position located centrally between the head and the tail of the embryo, and meanwhile use a second pair of fine forceps to hold the embryo so as to provide leverage for the yolk incision. Note: Alternatively, make two major incisions in the yolk, one close to the head and the other close to the tail of the embryo.
Use fine forceps and/or a lash tool to scoop out the yolk from the yolk cell cavity.
Rinse the embryo gently with 1x PBST to remove stray yolk.
7. Fine Deyolking of the Embryo
Transfer the embryo to a flat glass slide with 1-2 drops of 1x PBST.
Use a lash tool and fine forceps to gently position the embryo with the ventral (yolk) side facing up on the glass slide.
Drag the embryo to the edge of the 1x PBST solution using the lash tool so that the embryo remains in a flat position against the glass slide.
Scrape the ventral surface of the embryo gently using the lash tool in order to remove the yolk granules without ripping the embryo. Simultaneously use a pair of fine forceps to anchor the embryo while scraping with the lash tool. Note: Removal of the yolk granules can require repetitive scraping for 5-10 min.
If the PBST solution becomes cluttered with excess yolk or begins to evaporate, add 1-2 fresh drops of 1x PBST to the slide using a plastic or glass transfer pipette, and then drag the embryo to the area with the clean solution for further yolk granule removal.
When the yolk has been satisfactorily removed, use a transfer pipette to gently move the embryo back into a plastic or glass Petri dish containing 1x PBST for a final rinse in order to remove stray yolk granules.
Transfer the deyolked embryo using a plastic or glass transfer pipette into a plastic or glass Petri dish containing 100% glycerol.
Incubate the embryo in 100% glycerol for ~5 min to acclimate the embryo.
8. Mounting on a Glass Slide
Transfer the deyolked embryo to a clean glass slide in 1-2 drops of 100% glycerol.
Position the embryo with the ventral (yolk) side facing the glass slide.
Using a lash tool, drag the embryo to the edge of the glycerol drop so that the embryo remains in a flat position against the glass slide. If the embryonic axis is curved, use the fine forceps to gently make very small incisions in the remaining yolk cell membrane to relieve tension so that the embryo can be positioned flat on the slide with a straight axis.
Place small divots of modeling clay on the four corners of an 18 x 18 mm glass coverslip. Note: As an alternative, small drops of vacuum grease15 can be substituted for modeling clay.
Place the glass coverslip slowly on the embryo, angling the coverslip in order to minimize the generation of air bubbles in the glycerol around the sample.
Add an additional drop of 100% glycerol to the side of the glass coverslip to fill the space between the coverslip and the glass slide.
Press down slowly and carefully on the four corners of the coverslip to firmly position the embryo between the glass and eliminate drifting. Note: Be careful not to crush the embryo.
If the embryo is not positioned as desired, use fine forceps to remove the coverslip. Use the lash tool to reposition the embryo and/or fine forceps to make additional incisions so that the embryo can be repositioned. Repeat steps 8.4-8.7.
Observe and/or image the specimen using either a stereo- or compound microscope.
Store the flat mount preparation flat with a cardboard slide holder in the dark at 4 °C for several weeks or temporarily at room temperature. Note: The WISH stain will fade progressively over time, particularly red substrate reactions, and cannot be preserved in glycerol indefinitely. Purple stains also fade more readily if kept in glycerol compared to PFA (See Step 8.11). Interestingly, it has been published that mounting embryos in permount rather than glycerol can enable indefinite storage at room temperature15.
For long term storage of purple substrate stained samples, remove the glass coverslip and rinse the embryo rinsed twice in 1x PBST, then transfer to a microcentrifuge tube with 4% PFA/1x PBS for long-term storage. Note: Alternatively, embryos in 1x PBST can be processed for additional analyses, such as DNA isolation for genotyping.
Representative Results
The flat mounting protocol procedure involves transformation of the zebrafish embryo from its normal anatomy, in which the body axis is wrapped around a centrally located yolk ball, into a two-dimensional preparation (Figure 1A). Whole mount imaging of the young zebrafish embryo is limited by the nature of how the embryonic axis is wrapped around the yolk. As a result, lateral or dorsal views of whole mount stained embryo specimens can only capture a portion of the axis or obscure particular tissues (Figure 1B). By comparison, deyolking and flattening of the embryo enables the entire embryonic axis to be visualized at one time (Figure 1B). These limitations are demonstrated by examining a WISH stained embryo that was labeled with antisense riboprobes to detect irx3b (purple), which marks a subset of the renal progenitors located adjacent to somites 7 through 13, and myod1 (red) which labels the somites (Figure 1B). The field of irx3b+ renal progenitors is obscured in the whole mount lateral view because irx3b transcripts are abundant in the central nervous system, making the renal field practically impossible to visualize with the lateral camera angle; further, the field of irx3b+ renal progenitors is only partly visible when the embryo is rotated into a dorsal camera view because the field is wrapped around the yolk (Figure 1B). However, a dorsal view of the same embryo following flat mounting enables the entire field of irx3b+ renal progenitors to be analyzed in a straightforward fashion in relation to the somites along the trunk and other embryonic structures (Figure 1B). Thus, elaborate spatial domains can be ideally documented with this method.
Several major steps are involved in the successful execution of the flat mount procedure. To perform this technique, the yolk ball must be first detached from the embryo proper (Figure 2A), which can be done with fine instruments such as a pair of fine forceps while the embryo is visualized using a stereomicroscope. Following the crude removal of the yolk ball, a fine lash tool is utilized to scrape away yolk granules that have remained attached to the embryo (Figure 2B). The removal of remaining yolk granules helps to facilitate visualization of the embryonic tissues. Finally, the deyolked embryo is manipulated to position it stably on a glass slide (Figure 2C), which enables observation and aids documentation of the sample using imaging software and a camera attached to the microscope. The lash tools are homemade devices that can be constructed by affixing a suitable lash to a pipette tip using superglue, which can be mounted onto a handle if desired (Figure 3A, Materials table). Different lash samples can be procured to produce lash tools with slightly different characteristics in terms of length and taper (Figure 3B), which each user may have different predilections for once they begin to experiment with the flat mount procedure. Examples of lash samples include naturally shed human eyelashes, naturally shed animal wiry hair or whiskers, and finally synthetic varieties of commercially available lashes found in retail cosmetic departments.
The local area surrounding and containing the sample(s) positioned on a glass slide (Figure 4A) can be imaged for high-resolution analysis. A combination of probes were used to to label the developing renal progenitors that give rise to the kidney in the wild type embryo at early developmental stages with two-color WISH, and then flat mount preparations were performed to view the samples (Figure 4B, 4C). During the early somite stages, renal progenitors are demarcated in a U-shaped pattern by their expression of pax2a transcripts (purple) surrounding the paraxial mesoderm that concomitantly expresses delta C (dlc) (red) (left column, Figure 4B)6,7. In comparison, when dlc transcripts were detected with a purple substrate, and were labeled in combination with the hindbrain marker krox20 (red), a rostral subdomain of the renal progenitors was visualized that expresses dlc (right column, Figure 4B), as noted previously6,7. Flat mount preparations can be used similarly to study later somitogenesis stages. At the 14 somite stage, we analyzed the expression of renal markers pax2a, slc4a4, and slc12a3 (purple) along with smyhc1 (red), which marks the somites (Figure 4C). These combinations enable the mapping of the entire renal progenitor domain with pax2a, while revealing that a subset of cells in a rostral subdomain expressed slc4a4a and were distinguished by a non-overlapping caudal subdomain of renal progenitors that expressed slc12a3.
Two-color WISH and the flat mount preparation are also valuable for the study of cellular domains during organogenesis in zebrafish with genetic mutations or other perturbations from the environmental exposure to small molecules6,7 (Figure 5). The zebrafish nls and lib mutants harbor mutations in aldehyde dehydrogenase 1a2 (aldh1a2, formerly known as raldh2), which encodes an enzyme required for the biosynthesis of retinoic acid (RA). RA is essential for the proper development of many renal cell types including the formation of podocyte cells that contribute to make the blood filter of the embryonic kidney, known as the glomerulus6,7. WISH was performed to label the podocyte progenitors with wt1a concomitantly with demarcation of the developing somites with smyhc1 and hindbrain with krox20 in wildtype embryos, nls mutant embryos, lib mutant embryos, and embryos treated with an aldh enzyme chemical inhibitor, DEAB (Figure 5). Embryos with deficient aldh1a2 expression showed reduced wt1a expression compared to wild type embryos, while DEAB-treated embryos showed an abrogation of wt1a transcripts (Figure 5, right column). Taken together, these representative results demonstrate how the flat mount technique can be used to analyze and document anatomical differences with precision in the early embryo, and thus be implemented for valuable developmental studies.
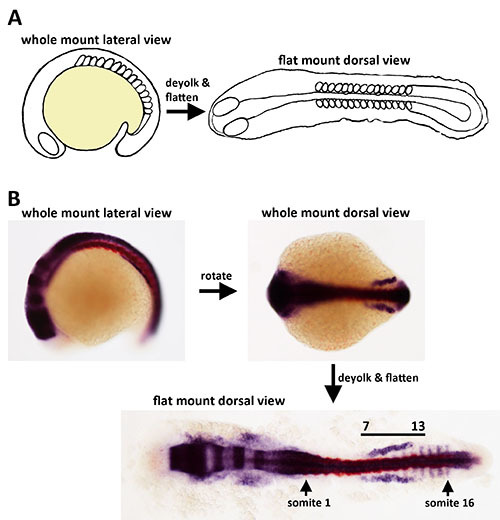 Figure 1. Overview of the flat mount preparation for zebrafish embryos. A) The procedure of flat mounting enables the simultaneous view of tissues along the embryonic axis because the central yolk mass is removed and the embryo stably positioned on a flat surface. B) Images of a whole mount WISH stained embryo in lateral and dorsal views, and then after a flat mount preparation was conducted. The embryo was stained with antisense probes to detect gene transcripts encoding irx3b (purple) and myod1 (red).
Figure 1. Overview of the flat mount preparation for zebrafish embryos. A) The procedure of flat mounting enables the simultaneous view of tissues along the embryonic axis because the central yolk mass is removed and the embryo stably positioned on a flat surface. B) Images of a whole mount WISH stained embryo in lateral and dorsal views, and then after a flat mount preparation was conducted. The embryo was stained with antisense probes to detect gene transcripts encoding irx3b (purple) and myod1 (red).
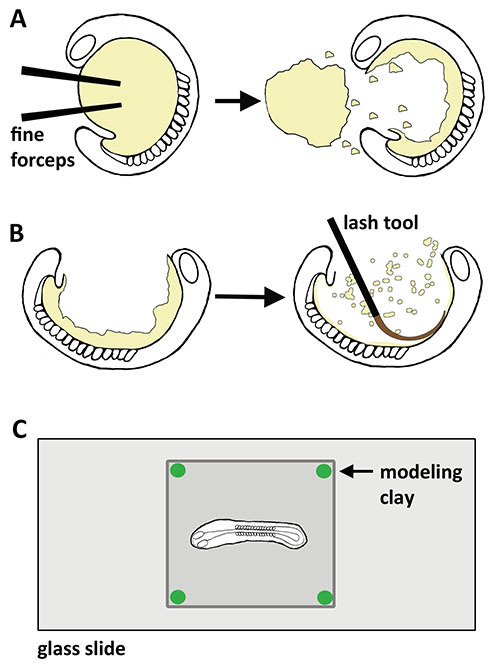 Figure 2. Schematic of the flat mount procedure for zebrafish embryos. A) The embryo is first grossly deyolked to remove the majority of the yolk mass, then (B) the ventral surface is finely deyolked to remove remaining yolk granules, and after washing the embryo is (C) mounted dorsal side up on a glass slide for visual analysis and/or photographic imaging.
Figure 2. Schematic of the flat mount procedure for zebrafish embryos. A) The embryo is first grossly deyolked to remove the majority of the yolk mass, then (B) the ventral surface is finely deyolked to remove remaining yolk granules, and after washing the embryo is (C) mounted dorsal side up on a glass slide for visual analysis and/or photographic imaging.
 Figure 3. Photographs of example lash tools compared to fine forceps. A) Homemade lash tools were imaged alongside a standard pair of fine forceps, positioned adjacent to a metric ruler to provide a reference. B) Magnified view of lash tools next to the fine forceps, with the millimeter reference provided along the top of the view. Two different lash tools are shown, with the asterisk (*) used to mark the lash obtained from a naturally shed human eyelash, and double asterisk (**) used to mark a lash that was obtained from a naturally shed feline whisker and trimmed to make a somewhat blunt end. In each case, the lash was threaded through the pipette tip and affixed with several coats of superglue to progressively create a strong seal to stabilize/anchor the lash to the pipette attached to a handling device.
Figure 3. Photographs of example lash tools compared to fine forceps. A) Homemade lash tools were imaged alongside a standard pair of fine forceps, positioned adjacent to a metric ruler to provide a reference. B) Magnified view of lash tools next to the fine forceps, with the millimeter reference provided along the top of the view. Two different lash tools are shown, with the asterisk (*) used to mark the lash obtained from a naturally shed human eyelash, and double asterisk (**) used to mark a lash that was obtained from a naturally shed feline whisker and trimmed to make a somewhat blunt end. In each case, the lash was threaded through the pipette tip and affixed with several coats of superglue to progressively create a strong seal to stabilize/anchor the lash to the pipette attached to a handling device.
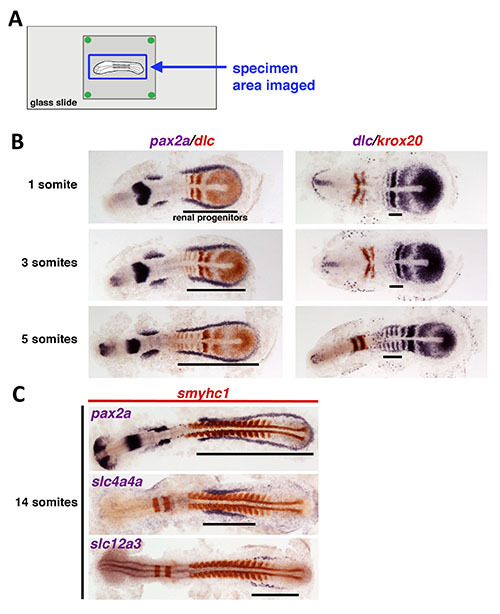 Figure 4. Characterization of the developmental changes in the renal progenitor field in the wild type zebrafish embryo. A) Slide schematic indicating the area imaged for analysis in (B, C). B) Wild type embryos at the 1, 3, and 5 somite stage were stained by two-color WISH to assess the composition of the renal progenitor field that gives rise to the embryonic kidney, or pronephros. (Left column) Transcripts encoding the transcription factor pax2a (stained in purple) mark several populations in the embryo, including the renal progenitor field that emerges from the intermediate mesoderm. Transcripts encoding the Notch ligand dlc mark the somites that form from the paraxial mesoderm (stained in red). (Right column) When the expression of dlc transcripts (stained in purple) and the hindbrain rhombomere marker krox20 (stained in red) are examined in the same embryos, dlc expression can be observed in a rostral subdomain of the renal progenitors located adjacent to somites 1-5, which was obscured during co-staining of dlc with pax2a. C) Wild type embryos at the 14 somite stage were double or triple-stained with a combination of a renal marker (purple), the somite marker smyhc1 (red), and the hindbrain rhombomere marker krox20 (also red). (Top panel) At this stage, pax2a transcripts continue to demarcate the renal progenitors. (Middle, Lower panels) Within the renal progenitor territory, a rostral subdomain is marked by slc4a4 and transcripts that encode slc12a3 mark a caudal subdomain.
Figure 4. Characterization of the developmental changes in the renal progenitor field in the wild type zebrafish embryo. A) Slide schematic indicating the area imaged for analysis in (B, C). B) Wild type embryos at the 1, 3, and 5 somite stage were stained by two-color WISH to assess the composition of the renal progenitor field that gives rise to the embryonic kidney, or pronephros. (Left column) Transcripts encoding the transcription factor pax2a (stained in purple) mark several populations in the embryo, including the renal progenitor field that emerges from the intermediate mesoderm. Transcripts encoding the Notch ligand dlc mark the somites that form from the paraxial mesoderm (stained in red). (Right column) When the expression of dlc transcripts (stained in purple) and the hindbrain rhombomere marker krox20 (stained in red) are examined in the same embryos, dlc expression can be observed in a rostral subdomain of the renal progenitors located adjacent to somites 1-5, which was obscured during co-staining of dlc with pax2a. C) Wild type embryos at the 14 somite stage were double or triple-stained with a combination of a renal marker (purple), the somite marker smyhc1 (red), and the hindbrain rhombomere marker krox20 (also red). (Top panel) At this stage, pax2a transcripts continue to demarcate the renal progenitors. (Middle, Lower panels) Within the renal progenitor territory, a rostral subdomain is marked by slc4a4 and transcripts that encode slc12a3 mark a caudal subdomain.
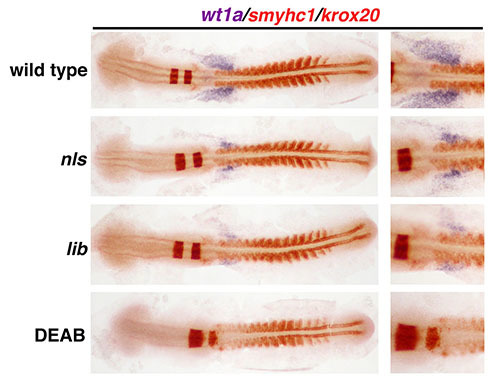 Figure 5. The use of flat mounted preparations to characterize the phenotypes caused by genetic mutations or chemical genetic perturbations that modulate retinoic acid biosynthesis. The WISH expression pattern at the 15 somite stage of wt1a (purple) and smyhc1/krox20 (red) was compared between wild types and embryos with deficiencies in retinoic acid (RA) production due to defects in aldehyde dehydrogenase 1a2 (nls and lib mutations) or chemical inhibition of retinaldehyde dehydrogenase activity with diethylaminobenzaldehyde (DEAB). The reduction of RA production in lib is slightly more severe than nls, such that lib embryos express slightly reduced staining of wt1a transcripts than similarly staged nls mutant embryos, while DEAB treatment of wild types is associated with complete abrogation of wt1a expression.
Figure 5. The use of flat mounted preparations to characterize the phenotypes caused by genetic mutations or chemical genetic perturbations that modulate retinoic acid biosynthesis. The WISH expression pattern at the 15 somite stage of wt1a (purple) and smyhc1/krox20 (red) was compared between wild types and embryos with deficiencies in retinoic acid (RA) production due to defects in aldehyde dehydrogenase 1a2 (nls and lib mutations) or chemical inhibition of retinaldehyde dehydrogenase activity with diethylaminobenzaldehyde (DEAB). The reduction of RA production in lib is slightly more severe than nls, such that lib embryos express slightly reduced staining of wt1a transcripts than similarly staged nls mutant embryos, while DEAB treatment of wild types is associated with complete abrogation of wt1a expression.
Discussion
Zebrafish have proven to be an extremely valuable model organism throughout the research community during recent years. Zebrafish exhibit a substantial degree of genetic conservation with that of higher vertebrates, and advantages pertaining to their anatomy, breeding, and life cycle make this species very amenable to experimental analyses. Moreover, the advancement of molecular techniques applicable to the zebrafish has further promoted the use of this model organism in scientific research. For instance, a large variety of transgenic zebrafish lines have been generated to study disease progression and to answer many fundamental questions that underlie the key mechanisms of development including organogenesis.
Accordingly, based on the dynamic use of zebrafish in both disease and developmental research, cell labeling methodologies and signal quantification are a crucial aspect of data analyses. While methods that employ fluorescent antibodies can be used to detect protein expression in transgenic zebrafish, researchers typically rely on standard procedures like WISH12-16 to examine the spatiotemporal localization of gene transcripts in a fixed, non-transgenic zebrafish sample. In fact, WISH is one of the most widely used techniques in biology16, and is a vital tool used to characterize the phenotype of genetic mutations and chemical genetic perturbations. Nevertheless, WISH is associated with a number of potential pitfalls and challenges. To confirm specificity of the antisense ribroprobe, sense riboprobes can be used as a control to assess the specificity of antisense riboprobe staining patterns. While sections 4 and 5 are presented in the order of labeling and detection of a digoxygenin-labeled probe followed by fluorescein-labeled probe, this order can be reversed. Typically, weaker signals (genes with lower expression levels) are best detected with digoxygenin-labeled probes and this stain developed first with a purple substrate, followed by detection and staining of the stronger signal (more abundantly expressed gene) using the red substrate. However, if two-color reactions are going to be performed, it is recommended that various combinations of labels and substrates are tested to find the procedure that is most suited for data collection and analysis. In addition, the times provided for blocking, antibody incubation and washing provided in sections 4-5 are tailored for embryos younger than 24 hr post fertilization; lengthy intervals of these same steps with older embryos can lead to salt accumulation on the sample. Extensive tips for troubleshooting standard zebrafish embryo WISH have already been documented in the literature12-16. Arguably the most common dilemma is high background. We recommend increasing the number of MABT washes in Step 4.7 to 15-20 washes if needed, though, in our experience the addition of the overnight MABT ‘super-wash’ gives outstanding results when used in conjunction with 10 or more short MABT washes before proceeding to Step 4.8. Another example dilemma is poor probe infiltration, which can occur with lengthy riboprobes. Shearing the riboprobe following Step 3.6 through alkaline hydrolysis can counteract this. In our experience with young samples, probe shearing is not typically required. However, one should keep in mind that parameters like this can be contributing factors in the outcome of a WISH experiment, and we suggest examination of these useful troubleshooting instructions already publicly available in the community as needed to refine the experimental parameters for WISH in your own research12.
In addition to traditional WISH techniques12-16, there has been a continual emergence of advances in WISH methodologies and alternate protocols that have been formulated. These include new ways of signal detection, such as through the use of fluorescence17-19, to methods that enable the localization of microRNAs20. Even so, despite the many improvements that have been made to current cell labeling methods, the effectiveness of these procedures are negated if the stain(s) cannot be easily visualized within the sample of interest at a desired time point. This limitation is problematic for researchers especially when it pertains to the early embryonic stages of zebrafish ontogeny, which could potentially lead to gaps in our understanding of various developmental processes that arise at these times. During normal zebrafish development, the yolk sac will progressively decrease in size as the embryo ages11. Therefore, at these later developmental stages (>24 hours post fertilization), WISH staining is fairly straightforward to analyze because the embryo is larger in comparison to its adjoining yolk sac. However, this is not the case from the tail bud stage to early somitogenesis (when the embryonic mass is positioned around the yolk) where the opaque yolk sac can sometimes obscure the visualization of the stain. Consequently, the method of flat mounting was devised to help alleviate the imaging and data analysis problems associated with the characterization of early zebrafish embryo samples (schematized in Figure 1 and Figure 2), and has been broadly used since the systematic phenotyping of genetic mutations isolated from the first large scale genetic screens21.
Since the advent of the flat mounting technique, the analysis of early developmental stages has been significantly enhanced. Once the yolk is removed from the embryo, it is possible to lay the embryo flat on a glass slide in the desired orientation for imaging. This two-dimensional position enables viewing of the entire embryo at once, which eliminates the need to rotate the sample. Numerous tissues are located in broad domains of the embryo, such as the precursors that form the blood and kidney. Further, one may need to compare several different developing tissues at one time. Flat mounting enables the researcher to simultaneously view the entire domain of such cell groups, and thus can greatly aid quantifications such as an area measurement for a tissue or cell counts. This technique has ultimately helped lead to many discoveries concerning a variety of important developmental mechanisms underlying hematopoiesis, neurogenesis, and organogenesis. Thus, once mastered, the applications for this simple technique for researchers are quite substantial.
For example, in a study examining lineage choice during hematopoiesis, flat mount preparations of zebrafish embryos at the 14 and 18 somite stages (ss) were used to illustrate the changes that occurred in the expression pattern of the pu.1 zinc finger transcription factor between wild-type and gata1 morpholino injected embryos22. This method enabled the detection of an expanded pu.1 domain at 18 ss, indicating that gata1 is most likely regulating the expression of pu.1 in the intermediate cell mass (ICM) around this time point. Additional flat mounted embryos stained for different erythroid genes including gtpbp1 and epsin demonstrated a loss in the expression of these genes in vlt mutants that were gata1-/-. Further analyses showed no change in the expression of erythoid genes like biklf and testhymin that were known to be independent of gata activity. Therefore, in conjunction with their other experimental results, it was revealed that gata1 is essential in regulating the differentiation of cells within the myelo-erythroid lineage. In a study of neural crest development, flat mounts were used to examine crestin expression in mont blanc (mobm610) mutants in a study examining the roles of mont blanc and tfap2a gene function23. At 10 and 20 ss, crestin expression was entirely abrogated in the head and was decreased in the trunk region of mobm610 mutants in comparison to the normal wild-type expression, ultimately aiding this group to conclude on the importance of mont blanc and tfap2a regulation during neural crest formation. Additionally, in terms of organogenesis, flat mounts have enabled the discovery of the presence of distinct domains in the early renal progenitor field during development of the zebrafish embryonic kidney, known as the pronephros. The zebrafish embryo provides a conserved and yet anatomically straightforward system to study how renal progenitors give rise to the nephron functional units that comprise the pronephros, a process known as nephrogenesis6,7 (Figure 4). Flat mount analysis has been useful to document domains of renal progenitors and to dissect the outcome of changes in RA signaling during pronephros patterning6,7 (Figure 5), which was previously unknown. Taken together, these examples suggest that there are indeed many broad applications for this flat mount protocol in the study of diverse processes that occur during normal development and diseased states.
Conceptually, this flat mount procedure is fairly simple overall. However, the manipulations and degree of finesse required to master this method can be quite challenging to master in the absence of visual demonstration. Therefore, this protocol was compiled to enable researchers, especially those new to the zebrafish model, with the opportunity to better understand how to perform this technique and share our advice on the reagents that can optimize tissue handling. We hope that this protocol will ultimately allow others in the community to refine the most ideal sample conditions to be used for premium imaging, data analysis, and communication of their results in publications. Ultimately, this should enable researchers to overcome the anatomical hindrances of the zebrafish during early embryogenesis, which can obscure the presentation of experimental results, and resolve these through the use of this simple but significant technique.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was partly supported by funding to RAW from each of the following: NIH grants K01 DK083512, DP2 OD008470, and R01 DK100237; March of Dimes Basil O’Connor Starter Scholar grant award #5-FY12-75; start up funds from the University of Notre Dame College of Science and Department of Biological Sciences. We would particularly like to thank Elizabeth and Michael Gallagher, along with the entire Gallagher Family, who imparted a generous gift to the University of Notre Dame to foster stem cell research. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank the staffs of the Department of Biological Sciences for their support, and the Center for Zebrafish Research at Notre Dame for their outstanding dedication in the care and welfare of our zebrafish colony. Finally, we thank the members of our research lab for their comments, discussions and insights about this work, as well as Marigold (Maripooka) and Zinnia Wingert for providing naturally shed feline whiskers to make most excellent lash tools for our research.
References
- Laale HW. The biology and use of zebrafish, Brachydanio rerio in fisheries research. A literature review. J Fish Biol. 1977;10:121–173. [Google Scholar]
- Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet. 2007;8:353–367. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- Goldsmith JR, Jobin C. Think small: zebrafish as a model system of human pathology. J Biomed Biotechnol. 2012;2012 doi: 10.1155/2012/817341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond IA. Kidney development and disease in the zebrafish. J Am Soc Nephrol. 2005;16:299–304. doi: 10.1681/ASN.2004090754. [DOI] [PubMed] [Google Scholar]
- Wingert RA, et al. The cdx genes and retinoic acid control the positioning and segmentation of the zebrafish pronephros. PLoS Genet 3. 1922. [DOI] [PMC free article] [PubMed]
- Wingert RA, Davidson AJ. Zebrafish nephrogenesis involves dynamic spatiotemporal expression changes in renal progenitors and essential signals form retinoic acid and irx3b. Dev Dyn. 2011;240:2011–2027. doi: 10.1002/dvdy.22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoriello C, Zon LI. Hooked! Modeling human disease in zebrafish. J Clin Invest. 2012;122:2337–2343. doi: 10.1172/JCI60434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamplin OJ, et al. Small molecule screening in zebrafish: swimming in potential drug therapies. WIREs Dev Biol. 2012;1:459–468. doi: 10.1002/wdev.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book. 3rd ed. Eugene: University of Oregon Press; 2000. [Google Scholar]
- Kimmel CB, et al. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- Moens C. Whole mount RNA in situ hybridization on zebrafish embryos: probe synthesis. CSH Protoc. 3, DOI: 10.1101/pdb.prot5036. 2008. [DOI] [PubMed]
- Moens C. Whole mount RNA in situ hybridization on zebrafish embryos: hybridization. CSH Protoc. 3, DOI: 10.1101/pdb.prot5037. 2008. [DOI] [PubMed]
- Moens C. Whole mount RNA in situ hybridization on zebrafish embryos: mounting. CSH Protoc. 3, DOI: 10.1101/pdb.prot5038. 2008. [DOI] [PubMed]
- Machluf Y, Levkowitz G. Visualization of mRNA expression in the zebrafish embryo. Methods Mol Biol. 2011;714:83–102. doi: 10.1007/978-1-61779-005-8_6. [DOI] [PubMed] [Google Scholar]
- Brend T, Holley SA. Zebrafish whole mount high-resolution double fluorescent in situ hybridization. J Vis Exp. 2009;25(25) doi: 10.3791/1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauter G, et al. Two-color fluorescent in situ hybridization in the embryonic zebrafish brain using differential detection systems. BMC Dev Biol. 2011;11(43):1–11. doi: 10.1186/1471-213X-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauter G, et al. Multicolor fluorescent in situ hybridization to define abutting and overlapping gene expression in the embryonic zebrafish brain. Neural Dev. 2011;6(10):1–13. doi: 10.1186/1749-8104-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagendijk AK, et al. Revealing details: whole mount microRNA in situ hybridization protocol for zebrafish embryos and adult tissues. Biol Open. 2012;1:566–569. doi: 10.1242/bio.2012810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffter P, Nüsslein-Volhard C. Large scale genetics in a small vertebrate, the zebrafish. Int J Dev Biol. 1996;40:221–227. [PubMed] [Google Scholar]
- Galloway JL, et al. Loss of Gata1 but not Gata2 converts erythropoiesis to myelopoiesis in zebrafish embryos. Dev Cell. 2005;8:109–116. doi: 10.1016/j.devcel.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Barrallo-Gimeno A, et al. Neural crest survival and differentiation in zebrafish depends on mont blanc/tfap2a gene function. Development. 2003;131:1463–1477. doi: 10.1242/dev.01033. [DOI] [PubMed] [Google Scholar]