

Summary
The development of the living acute brain slice preparation for analyzing synaptic function roughly a half century ago was a pivotal achievement that greatly influenced the landscape of modern neuroscience. Indeed, many neuroscientists regard brain slices as the gold-standard model system for detailed cellular, molecular, and circuitry level analysis and perturbation of neuronal function. A critical limitation of this model system is the difficulty in preparing slices from adult and aging animals, and over the past several decades few substantial methodological improvements have emerged to facilitate patch clamp analysis in the mature adult stage. In this chapter we describe a robust and practical protocol for preparing brain slices from mature adult mice that are suitable for patch clamp analysis. This method reduces swelling and damage in superficial layers of the slices and improves the success rate for targeted patch clamp recordings, including recordings from fluorescently labeled populations in slices derived from transgenic mice. This adult brain slice method is suitable for diverse experimental applications, including both monitoring and manipulating neuronal activity with genetically encoded calcium indicators and optogenetic actuators, respectively. We describe the application of this adult brain slice platform and associated methods for screening kinetic properties of Channelrhodopsin (ChR) variants expressed in genetically-defined neuronal subtypes.
Keywords: Acute brain slice, adult animals, patch clamp recording, protective recovery method, NMDG aCSF, optogenetics, GCaMP, Channelrhodopsin
1. Introduction
For several decades the vast majority of brain slice physiologists have relied upon a ‘protective cutting’ method for preparing healthy brain slices from juvenile and adolescent animals. This method is based on the premise that passive sodium influx and subsequent water entry and cell swelling during the slice cutting step is the major insult that leads to poor survival of neurons, particularly for those neurons located in the superficial layers that are most likely to sustain direct trauma from the blade movement. Thus the implementation of a protective cutting solution having equimolar replacement of sodium chloride (NaCl) with sucrose (e.g. low sodiumaCSF, sucrose-substituted aCSF, or simply sucrose aCSF) provides a notable improvement in neuronal preservation, especially for difficult to preserve brain areas such as brainstem and other highly myelinated regions (1).
The sucrose aCSF protective cutting solution has been widely adopted for preparing acute brain slices from virtually all brain regions following the initial description over twenty years ago. Variations of the protective cutting method have subsequently been described, including modified sucrose cutting aCSF formulations with optimized osmolarity (2), mixed NaCl/sucrose (3) or alternative sodium ion substitutes such as choline (4), NMDG (5), glycerol (6) or K-Gluconate (7). While each particular study reports improved neuronal viability in juvenile or young adult brain slices using different protective cutting solutions in distinct brain regions, no clear consensus has emerged in the field to support a most effective formulation (see also (8,9), and the protective cutting method still leaves much to be desired for brain slice preparation from mature adult animals regardless of the particular aCSF formulation implemented. Through systematic investigation of the parameter space for both aCSF formulations and procedures we were able to identify and optimize several key determinants for preparing healthy acute brain slices specifically from mature adult animals:
Sodium ion replacement during the initial phases of slice recovery, but not during the slicing procedure per se, is both necessary and sufficient to drastically curb the majority of neuronal swelling and subsequent pyknosis. Thus, we call this simple but highly effective procedural modification the ‘protective recovery’ method in order to draw a clear distinction from the commonly employed methods based solely on implementation of ‘protective cutting’ solutions.
The choice of sodium ion replacement applied to protective recovery is crucial. Among those cations we have tested N-methyl-D-glucamine (NMDG) is the most versatile substitute for a wide range of adult ages and applications. In more general terms, the use of methylated organic cations (e.g. NMDG, choline, Tris) as are placement for sodium ions leads to greatly diminished permeability of sodium channel pores on neuronal membranes (10). This property may account for the enhanced neuro-protective benefits of NMDG aCSF relative to sucrose aCSF in the preparation of adult brain slices.
Adult brain slices undergo edema during the course of incubation in normal aCSF and this is ameliorated by inclusion of 20 mM HEPES.
Each of these aspects contribute positive additive effects, and collectively the NMDG protective recovery method we describe here enables routine preparation of healthy mature adult brain slices across diverse cell types, brain regions, species, and animal ages. Earlier versions of this method have appeared in our previously published work (11–13).
2. Materials
2.1 Solutions
NMD GaCSF: 92 mM NMDG, 2.5 mM KCl, 1.25 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 2 mM thiourea, 5 mM Na-ascorbate, 3 mM Na-pyruvate, 0.5 mM CaCl2·4H2O and 10 mM MgSO4·7H2O. Titrate pH to 7.3–7.4 with concentrated hydrochloric acid (see Note 1).
HEPES holding aCSF: 92 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 2 mM thiourea, 5 mM Na-ascorbate, 3 mM Na-pyruvate, 2 mM CaCl2·4H2O and 2 mM MgSO4·7H2O.
Recording aCSF: 119 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 24 mM NaHCO3, 12.5 mM glucose, 2 mM CaCl2·4H2O and 2 mM MgSO4·7H2O.
All solutions should be made using water of high purity, such as from a MilliQ water system. Trace metals in distilled or tap water can lead to suboptimal slice quality through various pro-oxidative effects. All glassware used to contain aCSF should be thoroughly cleaned either by autoclave cycle or by rinsing with dilute (0.1M) nitric acid followed by copious amounts of MilliQ water.
All aCSF solutions must be saturated with carbogen (95% O2/5% CO2) prior to use to ensure stable pH buffering and adequate oxygenation. The pH should be adjusted to 7.3–7.4 and osmolarity measured and adjusted to 300–310 mOsm. It is recommended that solutions be made fresh on the day of the experiment (see Note 2).
Intracellular pipette solution: 145 mM K-Gluconate, 10 mM HEPES, 1 mM EGTA, 2 mM Mg-ATP, 0.3 mM Na2-GTP, and 2 mM MgCl2 (pH 7.3, 290–300 mOsm).
1.25 % Avertin stock solution (mix 2.5 g of 2,2,2-Tribromoethanol with 5 mL of 2-methyl-2-butanoland then gradually dissolve in 200 mL phosphate buffered saline, pH 7.0–7.3). Avertin stock solution should be 0.22 μm sterile filtered and warmed to body temperature before use to minimize potential adverse effects on the animal. The use of isoflurane with a vaporizer breathing system is a suitable alternative to injectable anesthetics.
2.2 Equipment and reagents
Tissue slicer machine: Various models are commercially available, many of which can provide excellent performance when optimally calibrated. We prefer the Compresstome VF-200 from Precisionary Instruments for adult brain slice applications. The machine uses agarose embedding of the tissue (see Note 3) and slight compression as the sample contacts the blade edge in order to create highly uniform slices with minimal surface chatter marks. These features produce slices with very clean surfaces, which is essential for preserving neurons in the superficial layers that are easily accessible for patch clamp recording (see Note 4).
Blades: Endurium ceramic injector style blades EF-INZ10 (Cadence/Specialty Blades). These highly durable blades outperform stainless steel and carbon steel feather blades and are best for cutting through heavily myelinated regions in adult brain. The performance of the ceramic blades is on par with the far more expensive and fragile sapphire blades.
Dissection tools: curved blunt forceps, fine dissecting ‘super cut’ scissors (for cutting through skull), large heavy duty scissors (for decapitation), fine spatula, heavy duty spatula, scalpel handle and #10 blades, plastic transfer pipette (cut off end to produce a wide mouth), super glue, filter paper, glass petri dish (for dissection surface), razor blade.
Transcardial perfusion tools: large dish filled with Sylgard for pinning anesthetized animal, dissecting pins, 30 mL syringe with 25 5/8″ needle.
pH meter: standard model—must be dedicated use with no contact with fixatives.
Vapor pressure osmometer. The osmometer should be calibrated very frequently and the thermocouple should be cleaned as needed for optimal performance.
Heater water bath with thermometer: 2.5L or other shallow bath design, large enough bath dimensions for holding the slice incubation chamber. The temperature should be maintained at 32–34°C.
Thermomixer with 24 × 1.5 mL thermoblock. The temperature should be set to 42°C and mixing speed at 600 rpm to maintain the 2% low melting point agarose in the molten state prior to use.
Multi-slice incubation chambers: Brain Slice Keeper-4 (BSK-4, AutoMate Scientific), or similar custom design. Slice chamber must have a minimally submerged netting for the slices to rest on, a fine gas diffuser stone for infusion of carbogen into the aCSF, and ideally some gentle constant flow to circulate solution around and/or through the slices.
Carbogen supply: either provided by refillable compressed gas cylinders or an in-wall line served by the building facilities, depending on existing infrastructure.
Electrophysiology rig suitable for patch clamp recording in acute brain slices. This will generally be composed of an upright microscope equipped with infrared differential interference contrast (IR-DIC) optics and a fluorescence illumination system, a patch clamp amplifier and data digitizer, motorized micromanipulators and microscope platform, vibration isolation table, Faraday cage, and solution heating and perfusion system. The sample chamber and platform should be designed for submerged slice recording.
Data acquisition software package for data analysis.
Laser scanning confocal microscope equipped with a 488 nm laser line for live calcium imaging experiments with GCaMP. The sample chamber must be adapted for continuous solution flow.
Blue laser for ChR2 photostimulation experiments with 200 μm core diameter optical fiber patch cord. Green laser for VChR1 photostimulation experiments. The lasers should have a digital or analog power controller box and option for TTL trigger. High power LED systems may alsobe suitable.
Boroscillicate glass capillaries type 8250 or KG-33 and pipette puller for fabricating microelectrodes.
2.3 Mice
-
Many unique lines of transgenic mice for fluorescently labeling diverse genetically-defined neuronal subtypes are commercially available. Such lines exhibit stable and heritable expression of fluorophoresin the nervous system and thereby greatly facilitate patch clamp recording from rare or broadly distributed cell populations. We have developed several transgenic mouse lines for optogenetics-based investigations (both manipulation and monitoring of neuronal activity), and these lines have been previously deposited to the Jackson Laboratory (JAX). Please see (14) for a comprehensive review of these lines and their demonstrated utility in diverse experimental applications. The stock numbers are provided below for many of the lines we used in developing our brain slice methods (see also (13,12,15–20)):
Thy1-GCaMP3 (JAX stock #017893)
VGAT-ChR2 (H134R)-EYFP (JAX stock #014548)
ChAT-ChR2 (H134R)-EYFP line 6 (JAX stock #014546)
TPH2-ChR2 (H134R)-EYFP (JAX stock #014555)
Pvalb-ChR2 (H134R)-EYFP (JAX stock # 012355)
Thy1-ChR2 (H134R)-EYFP line 18 (JAX stock #007612)
Thy1-vChR1-EYFP line 8 (JAX stock #012348)
Thy1-eNpHR2.0-EYFP line 2 (JAX stock #012332)
R26-2xChETA (JAX stock #017455)
Ai32 (JAX stock #012569)
3. Methods
3.1 Brain slice procedure
Prepare for the slicing procedure by melting the required 2% agarose block in the microwave (in appropriate container) and then aliquot the molten agarose into 1.5 mL tubes. Maintain the agarose in the molten state using a thermomixer set to 42°C at 600 rmp. Care must be taken to ensure that the molten agarose does not solidify prematurely. Place the chilling block accessory device on ice to pre-cool.
Glue a ceramic injector blade onto the blade holder and optimally align the blade for slicing. If the blade edge is not physically damaged it can be reused for many weeks or even months without replacement.
Deeply anesthetize mature adult mice ages 2–8 months by intraperitoneal injection of Avertin (250 mg/kg: 0.2 mL of 1.25% Avertin working stock solution per 10 g body weight) and then perform transcardial perfusion with 25–30 mL of room temperature carbogenated NMDG aCSF (see Note 5). If the perfusion is successful the liver will change in color from deep red to pale yellow, and in some cases clear fluids can be observed exiting the nostrils towards the end of the procedure. The transcardial perfusion is an important step when working with adult animals. For best results it is not advisable to omit transcardial perfusion.
Following perfusion the mice are decapitated, and the brainsgently extracted from the skull within 1 minute and placed into the cutting solution for an additional 30 seconds. Block the brain for the desired brain region and slicing angle (coronal for cortico-striatal slices or transverse for hippocampal slices) and glue the tissue onto the mounting cylinder, which looks similar to a lipstick tube.
The tissue block is withdrawn into the lipstick tube and rapidly embedded by pouring a 1.5 mL tube of molten agarose directly over the tissue until completely submerged, taking care to avoid air bubbles. The agarose is rapidly solidified by clamping the tube with the cold chilling block for 5–10 seconds, and then the lipstick tube with embedded brain block is inserted into the receiver of the slicing platform of the Compresstome VF-200 (see Figure 1A and 1B).
Fill the slicing chamber with NMDG aCSF solution and ensure continued carbogenation throughout the procedure. The micrometer should now be adjusted and locked in place. Manually advance the tissue for sectioning at 300 μm thickness. The optimal settings on each machine should be determined empirically. Set the advance to the slowest possible speed (~20 s per pass). Adjust the oscillation to a low-moderate setting where the blade is moving rapidly. The optimal blade movement produces a gentle humming sound but without any harsh buzzing noise. The total time for the slicing procedure should be less than 15 minutes.
Critical protective recovery step: transfer slices using a cut-off plastic Pasteur pipet into a pre-warmed BSK-4 (see Figure 1C and 1D) containing carbogenated NMDG aCSF. Allow the initial protective recovery to proceed for ≤12 min at 32–34 °C. Care must be taken to ensure that this step is precisely timed and that the temperature is properly maintained (see Notes 6 and 7).
After the initial recovery period, transfer the slices into a new holding chamber (additional BSK-4 or similar design) containing room-temperature HEPES holding aCSF under constant carbogenation. Care should be taken to ensure minimal carry-over of the NMDG aCSF into the new holding chamber. At this point the slices can be stored for 1–5 hours before transfer to the recording chamber for use. The presence of the 20 mM HEPES and ascorbate/thiourea combination reduces slice edema and slows deterioration.
Figure 1.

Equipment for preparation of brain slices.(A) Compresstome VF-200 slicing machine with major components labeled as follows: 1-slicing chamber, 2-blade arm, 3-micrometer, 4-controller box.(B) Alternate view of the blade arm with lipstick inserted into the receiver of the slicing platform.(C) Brain Slice Keeper-4 apparatus.(D) Disassembled BSK-4 with major components labeled as follows: 1-lid, 2-four chambers with netting, 3-gas diffuser stone, 4-outer container.
3.2 Evaluation of slice quality: morphological integrity
The slices were transferred one at a time to the recording chamber of the microscope equipped with epifluorescence and IR-DIC optics (900 nm IR filter) aligned for Kohler illumination (see Note 8). The slices were constantly perfused with room-temperature (22–25°C) carbogenated recording aCSF at a rate of 4 mL per minute.
A direct comparison of the traditional sucrose protective cutting method and the NMDG protective recovery method was performed using age-matched littermate mice at 5 months old. Acute brain slices are prepared and IR-DIC images acquired at defined early time windows (0–5 min and 25–30 min) following the slicing procedure to evaluate the extent of morphological preservation. This approach revealed a dramatic improvement with the NMDG protective recovery method across brain regions, with much less initial neuronal swelling evident at 0–5 minutes and much reduced subsequent neuronal shrinkage and pyknosis at 25–30 minutes post slicing (see Figure 2). Thus, the number of neurons that appear accessible for patch clamp recordings at later time points throughout the recording session is greatly increased, particularly for superficial layers of the slices (see Note 9).
Figure 2.
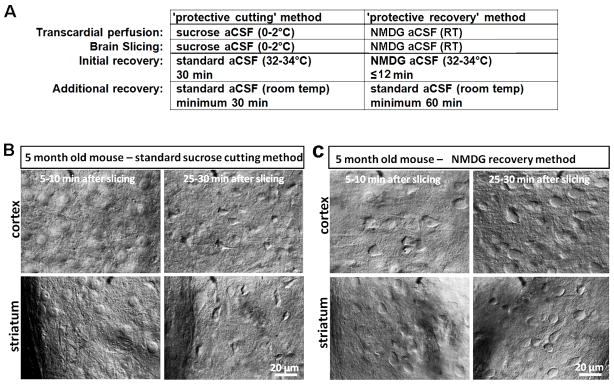
The protective recovery method yields superior neuronal preservation for acute brain slice preparation from mature adult animals. (A) Comparison of procedural steps in the protective cutting versus protective recovery methods. (B) Rapid neuronal swelling and subsequent shriveling in acute brain slices prepared from 5 month old adult mice with the sucrose aCSF protective cutting method.(C) Reduced swelling and improved neuronal preservation in acute brain slices prepared from 5 month old adult littermate mice with the protective recovery method.
3.3 Evaluation of slice quality: Functional integrity
Additional testing was conducted to broadly assess functional responding of large populations of neurons in adult brain slices prepared with the protective recovery method. This was carried out using population calcium imaging and high-potassium depolarization in brain slices prepared from 5 month old Thy1-GCaMP3 transgenic mice.
The slices were transferred one at a time to the sample chamber of the laser scanning confocal system. The slices were constantly perfused with room-temperature (22–25 °C) carbogenated recording aCSF at a rate of 4 mL per minute.
GCaMP3 was excited with a 488 nm laser line at 5–10% power and the green fluorescent signal was detected using a band-pass filter (505–525 nm). The selected region was imaged at 3 frames per second. We primarily focused on imaging calcium transients in hippocampal dentate gyrus granule cells because this population is uniformly labeled with high level GCaMP3 expression in the selected transgenic mouse line. We imaged a focal plane below the superficial layers (~20–50 μm deep) to avoid damaged neurons and auto-fluorescence from cellular debris.
To induce synchronous depolarization and neuronal firing we chose transient bath perfusion of high K+aCSF using a peristaltic pump for solution delivery (see Note 10). The time of stimulation was marked from the time that the high K+ solution first entered the bath and was terminated after 60 sec, followed by return to perfusion with normal aCSF.
Images were analyzed as previously reported (13). For each image series we selected at least 20 regions of interest (ROI) for analysis. Each ROI corresponded to single granule cell bodies. The fluorescence intensity increase upon high K+ stimulation was calculated as peak fluorescence intensity divided by baseline fluorescence intensity (ΔF/F), corrected for background fluorescence.
The imaged neuronal populations in brain slices from the protective recovery group exhibited larger calcium transients at moderate stimulation levels (5 mM and 10 mM K+ stimulation) and there were far more total numbers of functionally responding neurons with nearly every granule cell showing robust fluorescence increases (see Figure 3). In addition, in several brain regions there were fewer neurons exhibiting elevated fluorescence in the basal condition, indicating fewer dead or damaged neurons as compared to slices prepared with the sucrose protective cutting method.
Figure 3.
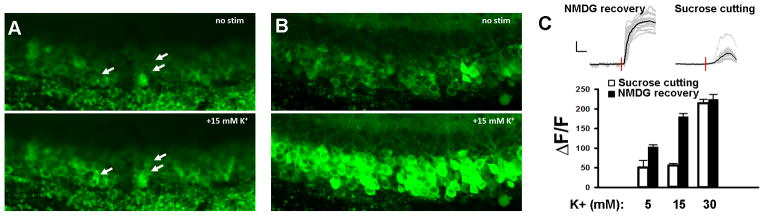
GCaMP3 calcium imaging to assess functional integrity of mature adult brain slices. (A) Transient bath application of high K+ solution (15 mM) evokes weak fluorescence increase in hippocampal dentate granule cells of brain slices prepared from a 5 month old Thy1-GCaMP3 transgenic mouse using the standard sucrose aCSF protective cutting method. Arrows mark examples of weakly responding neurons.(B) The same high K+ (15 mM) perfusion evokes a robust increase in fluorescence throughout the entire granule cell layer in brain slices prepared from an age matched littermate animal using the NMDG protective recovery method.(C) Example raw traces of fluorescence intensity measured over time for selected regions of interest (ROIs) demonstrating the effect of high K+ bath perfusion. Scale bars: 50%, 50 s. Summary data comparing ΔF/F for the two slice preparation methods over a range of extracellular K+ concentrations.
3.4 Application: Measuring kinetic properties of Channelrhodopsin variants
The patch clamp electrophysiology rig was modified to enable focal delivery of laser light for precisely timed photostimulation of ChR-expressing neurons in adult brain slices. This was accomplished by adding a micromanipulator for positioning of a 200 μm core diameter optic fiber directly above patch clamped neurons in submerged slices (see Figure 4). A glass capillary was mounted on the probe holder and the optic fiber was inserted through the glass capillary and secured in place with the flat cleaved end extending approximately 1 cm beyond the glass. The other end of the optic fiber was connected to a blue or green laser source under TTL control in the pClamp10 data acquisition software (see Note 11).
A simple stimulation protocol was designed for triggering a brief 2-ms light pulse using the pClamp10 software. Each sweep was 1-s long with the stimulus was triggered at the 100 ms time point.
Adult brain slices were prepared using the NMDG recovery method and transgenic mouse lines with stable expression of ChR variants in cortical pyramidal neurons (Thy1-ChR2R-EYFP line 18 and Thy1-VChR1-EYFP line 8).
Slices were transferred one at a time to the recording chamber of the patch clamp rig. Transgene-expressing neurons were targeted for whole-cell patch clamp recordings by combined IR-DIC imaging and fluorescence.
Recordings were obtained under identical conditions in order to standardize the light-evoked photocurrents mediated by transient activation of the ChR variants. The rate of channel closure at the termination of the light stimulus (Tauoff) was quantified using a mono-exponential fit of the decay of the photocurrent after light offset (see Note 12). To perform the decay fitting data files were opened in Clampfit and cursor 1 aligned to the peak of the response. A second cursor was set to +300 ms after cursor 1 (to ensure complete decay of the response). Select analyze>fit>standard exponential (n=1), then click OK. The tau value is now exported into the results sheet for the selected traces.
Tauoff was measured for cortical pyramidal neurons expressing ChR2R (mean = 26.3 ms) or VChR1 (mean = 93.6 ms) (see Figure 5A).
A screen was conducted to determine Tauoff for cortical pyramidal neurons expressing the novel ChR variants ChETAARC and oChIEFAC. ChETAARC denotes the ChR2 variant with E123A/H134R/T159C mutations, a novel fast kinetic variant designed for higher photocurrent amplitude than the previously reported ChETA variants. oChIEFAC denotes the oChIEF variant with the dual E162A/T198C mutations, a second novel fast kinetic variant designed for large photocurrents and fast kinetics. By comparing data sets across experiments but obtained in the identical cells type we found that Tauoff was reduced for both ChETAARC (19.3 ms) and oChIEFAC (9.1 ms) relative to the ChR2R variant (26.3 ms) (see Figure 5B). With a Tauoff of nearly half that of ChETAARC, we propose that oChIEFAC is the most promising new fast kinetic variant for applications requiring ultrafast optogenetic stimulation. We also found that photocurrents measured from oChIEFAC expressing neurons were approximately 5 times larger than those measured from ChETAARC expressing neurons.
Additional experiments were conducted to evaluate the effect of the ChETATR fast kinetic variant compared to ChR2R in cortical fast-spiking interneurons. For these experiments the NMDG recovery method was used to prepare adult brain slices from transgenic mouse lines with stable expression of ChR variants in cortical interneurons (VGAT-ChR2R-EYFP line 8 or R26-2xChETATR/Pvalb-IRES-Cre double transgenic mice). ChR2R denotes the ChR2 variant with the H134R mutation. ChETATR denotes the ChR2 variant with dual E123T/H134R mutations.
Tauoff was measured for cortical fast spiking interneurons expressing ChR2R (mean = 10.4 ms) or ChETATR (mean = 4.6 ms) (see Figure 5C). A direct comparison of Tauoff values measured for ChR2R-expressing cortical pyramidal neurons versus ChR2R-expressing cortical fast spiking interneurons revealed a 2.5 fold difference, demonstrating that kinetic properties of ChR variants are highly dependent on cell type (see Note 13).
Figure 4.
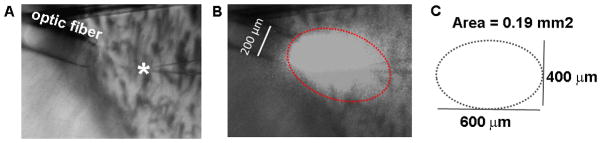
Positioning of laser-coupled optical fibers for focalized laser stimulation in brain slices.(A) Example positioning of a 200 μm core optic fiber directly over a recorded striatal neuron (asterisk).(B) Each laser pulse illuminates an ellipse surrounding the target neuron.(C) Estimated area of illumination for determining power output as a function of area (irradiance).
Figure 5.

Comparative analysis of ChR kinetic variants in distinct cell types. Measurement of the kinetics of channel closure (Tauoff) following a brief 2-ms light stimulation was performed using whole-cell voltage clamp (−70 mV) as a sensitive assay for screening novel ChR kinetic variants.(A) Experiment #1: comparison of Tauoff measured in cortical pyramidal neurons with transgenic expression of ChR2R or VChR1. Scale bars: 200 pA, 100 ms. Experiment #2: comparison of Tauoff measured in cortical pyramidal neurons expressing the novel variants ChETAARC or oChIEFAC. Scale bars: 50 pA, 100 ms.(B) Combined data for pyramidal neurons.(C) Comparison of Tauoff measured in cortical fast spiking interneurons with transgenic expression of ChR2R (VGAT-ChR2R-EYFP line 8) or ChETATR (R26-2XChETATR/Pvalb-IRES-Cre). Scale bars: 200pA, 10 ms.(D) Summary data comparing Tauoff measured from either cortical fast-spiking interneurons or cortical pyramidal neurons both expressing ChR2R. Intrinsic cell type differences influence measured kinetic properties and thus preclude comparative analysis across cell-types.
3.5 Improved methods for visually-guided patch clamp recording of ChR2-expressing neurons in slices
An important consideration for patch clamp analysis in adult brain slices is the fluorescent labeling of cellular populations under investigation. In many cases it is not practical to target defined neuronal subsets without the aid of a fluorescent label, particularly when the targeted population is rare, highly distributed, or intermingled with morphologically identical but functionally distinct subsets. In addition, for optogenetics-based investigations fluorescent reporters are important to identify neurons with functional expression of the optogenetic probe. Fortunately many transgenic animals and viral expression strategies have been developed to label diverse neuronal populations and allow access for patch clamp recordings and monitoring or manipulation of neuronal activity.
In the case of ChR expression strategies, the majority of the available viral vectors and transgenic lines were developed for expression of ChR variants as fusions with EYFP (ChR2-EYFP) with fewer studies using the ChR2-mCherry fusion design (21). Due to the strong trafficking of ChR to the plasma membrane the physically linked fluorescent reporter is also confined to this domain and is largely excluded from the cell body. This scenario leads to difficulty in identifying transgene expressing cells for patch clamp recording in slices (or in vivo), especially when the labeled population is relatively dense. The resulting signal is often a dense, fluorescent neuropil with no clearly discernible cell bodies (see Figure 6A and 6B). A preferable expression strategy would be one in which physically separate but equal levels of opsin and fluorophore are produced in the labeled populations. This can be accomplished by incorporating viral 2A linkers, as was first demonstrated in the mouse acute slice preparation following in vivo injections of AAV-Synapsin-ChR2-2A-tDimer and AAV-Synapsin-NpHR-2A-Venus into the hippocampus (22). Despite the demonstrated utility of this approach, the 2A expression strategy has been slow to be widely adopted, with only a few examples in recent studies (23–25). We recently developed a transgenic mouse line for optogenetics-based research incorporating the 2A expression strategy for Cre-dependent expression of ChETATR and a physically separate tdTomatofluorophore (19). Using this line (when crossed to an appropriate Cre driver line) we were able to demonstrate improved visualization of ChR-expressing neurons by virtue of the cytosolic tdTomato fluorescence readily detected at the cell body (see Figure 6C and 6D). In addition, because the 2A peptide linker segregates with the upstream encoded protein, it was still possible to visualize the membrane targeted ChR by immunostaining using a commercially available anti-2A antibody (see Figure 6D). In essence, the 2A sequences are highly useful as epitope tags.
Figure 6.

Improved visualization of ChR2-expressing neurons for targeted patch clamp recordings in brain slices using viral P2A linkers.(A) The challenge of identifying ChR2-EYFP expressing neurons is examined in acute striatal brain slices from Ai32/D1-Cre mice. This line has strong expression of the ChR2-EYFP transgene in roughly half of all striatal medium spiny neurons. The ChR2-EYFP gene fusion is localized to the cell membrane and produces a dense fluorescent neuropil with little signal from cell bodies. A recorded neuron is shown (asterisk) along with an inset of recorded photocurrent, thus confirming the identity as a D1+ MSN.(B) A recorded MSN in a nearby region had no photocurrent and was presumed D1 negative (D1−/D2+ MSNs account for the other half of the MSN population). The recorded neurons were indistinguishable on the basis of morphology or live EYFP fluorescence.(C) Analysis of native tdTomato fluorescence together with double immunostaining with anti-2A (indicating localization of ChR2) and DARPP32 (indicating all MSNs in the striatum region) demonstrates the unambiguous identification of ChR2 expressing neurons with the opsin-2A-XFP expression strategy. The example shown here is from our Cre-inducible ChETA-P2A-tdTomato reporter line crossed to the RGS9-Cre driver mice for labeling a subset of striatal MSNs.(D) The use of the viral P2A linker (green sequence) allows for physical uncoupling of opsin and fluorophore, and the 2A epitope tag can then be used to track the localization of the membrane-targeted opsin protein. The cytosolic fluorophore, in this case tdTomato, fills the entire cell body. Scale bars: 20 μm in panels A and B, 50 μm in panel C.
3.6 Avoiding unintended photoactivation during cell selection and targeting
An addition important issue caused by the widespread use of ChR-EYFP fusions is the unintended photoactivation of transgene expressing neurons while searching for neurons to record from in acute brain slices. This is due to the extensive overlap of the ChR activation spectrum with the excitation range for EYFP. Thus, visual identification of EYFP expression in a targeted neuron requires a transient but strong photoactivation of the targeted neuron and the surrounding transgene expressing neurons in the area of blue light illumination. This activation may induce alterations in cellular or synaptic function lasting variable durations, and perhaps in some cases leading to irreversible changes.
In order to circumvent this issue it is important to select judicious pairings of opsin and fluorophore. For example, our Cre-inducible ChETATR knock-in mouse line allows for expression of ChETATR and a physically separate tdTomatofluorophore (19). By using a custom designed tdTomato filter set with only minimal overlap of the ChETA activation range we were able to titrate the excitation intensity to a point where visual identification of tdTomato positive neurons was easily accomplished without inducing neuronal firing. In contrast, a typical Texas red filter set was not suitable for this purpose as even low intensity light still induced robust neuronal firing (see Figure 7). It is important to note that direct fusion of ChR2 to tdTomato was reported to impair ChR2 function to some extent in the Ai27 mouse line (20). The 2A expression strategy overcomes this issue; however, it may still be advantageous to explore additional opsin-fluorophore combinations. Although tdTomato was selected in our work due to its bright fluorescence, we have found that the more red-shifted mKate2 is ideal for most applications in combination with ChR2 variants. With a suitable filter set the ChR2-P2A-mKate2 or ChR2-mKate2 pairing allows for virtual elimination of photocurrents during fluorophore visualization.
Figure 7.

Avoiding photoactivation while searching for ChR2-expressing cells.(A–C) The excitation/emission spectra of various optical filter sets are plotted together with the activation spectrum for ChETA. Considerable overlap with ChETA activation is observed for the excitation range using EYFP (A) and Texas red (B) filter sets but not with a custom tdTomato filter set (C). (D) Raw traces of maximal photocurrents evoked by 1-s epifluorescent illumination (blue line) using the various filter sets for a ChETA-P2A-tdTomato expressing neuron recorded in whole-cell voltage clamp.(E) Cell-attached recordings demonstrating light-evoked spiking with the various filter sets at full and reduced light intensity. Complete elimination of spiking was only achieved with the custom tdTomato filter set at reduced light intensity.
3.7 Strategies for further improving the quality of adult brain slices
The adult brain slice method we have described has been successfully implemented in a variety of experimental contexts for analysis of diverse brain regions and cell types. However, we would encourage adopters to view this method as a work in progress, and we believe there is still substantial room for systematic improvement. As a case in point, we have observed that mature adult brain slices experience high levels of oxidative stress due in large part to rapid depletion of cellular antioxidants including ascorbate and reduced glutathione (GSH). This can lead to lipid peroxidation, neuronal membrane rigidity, and tissue deterioration. There appears to be a nonuniform susceptibility to this form of oxidative damage, for example, CA1 and CA3 pyramidal neurons are particularly vulnerable, making patch clamp recording of these cells difficult in brain slices from adult and aging animals in spite of the protective recovery method.
The specific restoration of intracellular pools of neuronal GSH (e.g. supplementation with the cell-permeable GSH-ethyl ester) is highly effective at curbing deterioration and prolonging slice viability under these circumstances. Thus, we have been able to further improve the NMDG recovery method by devising strategies for stimulating de novo synthesis of glutathione during acute brain slice preparation and incubation. This is most readily accomplished by adding the inexpensive GSH precursor N-acetyl-L-cysteine (NAC, 5–12 mM) to the NMDG aCSF and HEPES holding aCSF formulas, but not the recording aCSF (see Note 14). NAC is cell-permeable and has been shown to specifically increase neuronal glutathione levels in brain tissue (26). Within 1–2 hours of slice preparation we are able to observe notable improvements in the general appearance of neurons and in the ease of patch clamp recording, and the slices are able to be maintained in a healthy state for extended time periods.
Although these more advanced methods are not absolutely required for successful adult brain slice patch clamp recordings (as demonstrated by the specific application we have described in this chapter), we include this information in hopes of providing more options to extend the versatility of our method for particularly challenging applications. Glutathione restoration is highly effective at maintaining healthy brain slices but may not be appropriate in every experimental context, e.g. investigations focusing on oxidative stress in the aging brain. On the other hand, without implementing the NMDG protective recovery method together with glutathione restoration strategy, targeted patch clamp analysis in brain slices from very old animals is prohibitively challenging.
Acknowledgments
This work was supported in part by a National Alliance for Research on Schizophrenia and Depression: The Brain and Behavior Research Foundation Young Investigator Award to J.T.T., and U.S. National Institutes of Health Ruth L. Kirschstein National Research Service Awards to J.T.T. (F32-MH084460).
Footnotes
Addition of the NMDG powder will make the solution very alkaline. When making NMDG aCSF it is important to carefully titrate the pH to 7.3–7.4 with concentrated hydrochloric acid. This step should ideally be performed prior to addition of divalent cations to avoid precipitation. In addition, the HEPES and thiourea plus ascorbate were included as critical components to reduce edema and oxidative damage during slicing, recovery and extended slice incubation (27,28). Due to the presence of the HEPES it is necessary to elevate NaHCO3 in order to maintain the proper pH with constant carbogenation.
Solutions can be stored at 4°C for one week without causing any detrimental impact on experiments. However, the pH and solution color should be carefully monitored to ensure stability. If Na-ascorbate is included the color will gradually change to a yellowish-orange hue over time, and this is a clear indication that the solution needs to be discarded. Many people prefer to use concentrated stock solutions for brain slice work. Given the difficulty in preserving healthy slices derived from adult animals and the numerous potential sources for failure, it is strongly recommended to prepare solutions fresh each day and avoid using stock solutions.
Freshly cut brain slices will detach from the surrounding agarose more easily when the low melt agarose type I-B is dissolved in phosphate buffered saline rather than water. Gently collecting and then expelling the slices with a wide mouth cut-off plastic Pasteur pipet is generally sufficient to dislodge the agarose. Repeat as necessary to free the slices.
The top of the line slicers will have a ‘zero-z’ feature, meaning that the displacement of the cutting blade in the z-axis during blade vibration can be tuned to nominal zero. Some machines do not offer the ability to tune z-axis deflection, but nonetheless empirically exhibit suitably low values. We have used the Leica VT1000, the Leica VT1200S with vibro-check, and the Precisionary Instruments Compresstome VF-200 all with excellent results. There are several features of the Compresstome VF-200 that make this slicer particularly well-suited for acute brain slice from adult and aging animals:(1) The speed of slicing is about two times faster than other models due to the stability provided by agarose embedding.(2) This machine also allows uniform thin sections (~100–150 μm) to be prepared from live tissue, which may be advantageous when examining densely myelinated regions where visualization is more difficult.(3) The Compresstome VF-200 has a very small footprint and can be easily moved or transferred between rooms or lab sites. (4) The cost of the Compresstome VF-200 is about half the cost of the top-of-the-line machines.
The temperature of the transcardial perfusion and slicing solution is not as critical when using NMDG aCSF in this protocol. The NMDG aCSF formulation is particularly effective at shutting down metabolic activity of the brain tissue. When using less protective formulations (e.g. sucrose aCSF or standard aCSF) it is much more crucial to chill the solutions to ~2–4°C prior to use. A recent study demonstrated that warm slicing solution is advantageous for functional preservation of some cell types in brain slices derived from adult animals (29). In our experience room temperature NMDG aCSF is highly effective and obviates prolonged preparation times for cooling. If chilled solution is preferred, we advise cooling to 2–4°C. Partially frozen slushy aCSF (as is commonly used for juvenile brain slice) was observed to be detrimental for adult brain tissue.
The exact duration of the recovery period was critical for obtaining the optimal balance between morphological and functional preservation of the brain slices, and the timing of this recovery step exhibited clear temperature dependence, as indicated above based on extensive empirical testing. Proper implementation of this brief protective recovery step using our NMDG-based aCSF formula greatly reduced initial neuronal swelling during rewarming and enabled routine preparation of healthy acute brain slices for targeted whole-cell recordings from mature adult and aging mice.
A protective recovery step of ≤ 12 min is suitable for diverse brain regions and adult ages. However, the use of the NMDG protective recovery method is not optimized for animals younger than 5–6 weeks of age. We observe that it is difficult to completely wash out the NMDG aCSF in juvenile slices following the short protective recovery step, which often leads to difficulty in forming giga-ohm seals. Thus, the application of the NMDG protective recovery method for brain slices from animals younger than 5–6 weeks old requires either shorter recovery duration (e.g. ≤ 5 min) or using a 50/50 mix of NMDG aCSF to standard aCSF solution. The procedures we developed are intended to facilitate patch clamp analysis in brain slices from adult and aging animals and we are not advocating the application towards juvenile animals. The standard sucrose cutting method is perfectly suitable for work with juvenile animal brain slices.
In our experience it is highly desirable to have a 900 nm IR filter rather than the more common 775 nm IR filter. In adult brain the myelination is more developed than for juvenile brain, which poses a challenge for clear visualization of neurons located deep in adult brain slices. In some instances it is desirable to be able to see clear neuronal profiles at depths up to 50–100 μm into the slice. This is most easily accomplished with 900 nm IR-DIC optics. Proper alignment for Kohler illumination is also important for clear visualization. For a step by step guide for IR-DIC optimization please see (30).
In our experience the morphological appearance of neuronal membranes under IR-DIC optics is well correlated with ease of patch clamping and physiological state of the neurons. This is in excellent agreement with other patch clamp studies in adult brain slices where the morphology of neurons was tracked by time lapse imaging prior to patch recordings (2). With high quality optics and repeated effort it becomes very easy to target healthy neurons based on visual criteria alone. However, additional objective criteria must still be applied to ensure adequate recovery of normal electrophysiological properties for each recorded neuron.
High K+ bath stimulation was selected to evaluate time-locked functional responding of the largest population of neurons possible. For more refined analysis of functional responding we have also used bath application of low concentrations of glutamate receptor agonists such as NMDA and glutamate, which induce more stochastic patterns of firing with slower onset and lasting for sustained periods.
Other more elegant solutions for light delivery are possible such as directing the optic fiber through the microscope objective or through the patch electrode holder (OptoPatcher). It is also possible to achieve photostimulation of particular regions of single neurons using focal light spots or grids with a 2-photon confocal system. In our screening application a light delivery system with crude spatial resolution was both practical and adequate.
Previous work has established that fast kinetic properties (shorter duration Tauoff values) of ChR variants are correlated with smaller plateau potential size, reduced light sensitivity, and smaller photocurrent amplitude. This evidence supports a theoretical tradeoff between the precision of spiking and the overall efficacy of photoactivation (31). Thus, this protocol we describe for measuring Tauoff can be quite robust for inferring several very important parameters of ChR function. For the sake of brevity we have chosen to present a simple and straightforward optogenetic stimulation protocol. More detailed protocols are required to thoroughly address distinct properties of novel variants (31).
Based on the 2.5 fold difference in Tauoff for ChR2R in cortical pyramidal neurons versus fast spiking interneurons we project a Tauoff of 7.7 ms for ChETAARC and 3.6 ms for oChIEFAC in cortical fast spiking interneurons. This prediction awaits direct experimental validation.
Addition of 12 mM NAC to the NMDG aCSF and HEPES holding aCSF formulas will substantially reduce pH and increase osmolarity. Thus, it is important to re-adjust the pH to 7.3–7.4 and osmolarity to 300–310 mOsm. Alternatively, it is feasible to revise the aCSF formulas to account for NAC addition. We contend that adding NAC fresh immediately before use is preferable to ensure that the NAC is not degraded and retains full potency.
References
- 1.Aghajanian GK, Rasmussen K. Intracellular studies in the facial nucleus illustrating a simple new method for obtaining viable motoneurons in adult rat brain slices. Synapse. 1989;3:331–338. doi: 10.1002/syn.890030406. [DOI] [PubMed] [Google Scholar]
- 2.Moyer JR, Jr, Brown TH. Methods for whole-cell recording from visually preselected neurons of perirhinal cortex in brain slices from young and aging rats. J Neurosci Meth. 1998;86:35–54. doi: 10.1016/s0165-0270(98)00143-5. [DOI] [PubMed] [Google Scholar]
- 3.Bischofberger J, Engel D, Li L, Geiger JR, Jonas P. Patch-clamp recording from mossy fiber terminals in hippocampal slices. Natureprot. 2006;1:2075–2081. doi: 10.1038/nprot.2006.312. [DOI] [PubMed] [Google Scholar]
- 4.Mainen ZF, Maletic-Savatic M, Shi SH, et al. Two-photon imaging in living brain slices. Methods. 1999;18:231–239. doi: 10.1006/meth.1999.0776. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka Y, Furuta T, Yanagawa Y, Kaneko T. The effects of cutting solutions on the viability of GABAergic interneurons in cerebral cortical slices of adult mice. J Neurosci Meth. 2008;171:118–125. doi: 10.1016/j.jneumeth.2008.02.021. [DOI] [PubMed] [Google Scholar]
- 6.Ye JH, Zhang J, Xiao C, Kong JQ. Patch-clamp studies in the CNS illustrate a simple new method for obtaining viable neurons in rat brain slices: glycerol replacement of NaCl protects CNS neurons. J Neurosci Meth. 2006;158:251–259. doi: 10.1016/j.jneumeth.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Dugue GP, Dumoulin A, Triller A, Dieudonne S. Target-dependent use of co-released inhibitory transmitters at central synapses. J Neurosci. 2005;2:6490–6498. doi: 10.1523/JNEUROSCI.1500-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aitken PG, Breese GR, Dudek FF, et al. Preparative methods for brain slices: a discussion. J Neurosci Meth. 1995;59:139–149. doi: 10.1016/0165-0270(94)00204-t. [DOI] [PubMed] [Google Scholar]
- 9.Lipton P, Aitken PG, Dudek FE, et al. Making the best of brain slices: comparing preparative methods. J Neurosci Meth. 1995;59:151–156. doi: 10.1016/0165-0270(94)00205-u. [DOI] [PubMed] [Google Scholar]
- 10.Hille B. The permeability of the sodium channel to organic cations in myelinated nerve. J Gen Physiol. 1971;58:599–619. doi: 10.1085/jgp.58.6.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peca J, Feliciano C, Ting JT, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao S, Ting JT, Atallah HE, et al. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods. 2011;8:745–752. doi: 10.1038/nmeth.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Q, Cichon J, Wang W, et al. Imaging neural activity using Thy1-GCaMP transgenic mice. Neuron. 2012;76:297–308. doi: 10.1016/j.neuron.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ting JT, Feng G. Development of transgenic animals for optogenetic manipulation of mammalian nervous system function: progress and prospects for behavioral neuroscience. Behav Brain res. 2013;255:3–18. doi: 10.1016/j.bbr.2013.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arenkiel BR, Peca J, Davison IG, et al. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron. 2007;54:205–218. doi: 10.1016/j.neuron.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asrican B, Augustine GJ, Berglund K, et al. Next-generation transgenic mice for optogenetic analysis of neural circuits. Fronts neurcirc. 2013;7:160. doi: 10.3389/fncir.2013.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ren J, Qin C, Hu F, et al. Habenula “cholinergic” neurons co-release glutamate and acetylcholine and activate postsynaptic neurons via distinct transmission modes. Neuron. 2011;69:445–452. doi: 10.1016/j.neuron.2010.12.038. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Peca J, Matsuzaki M, et al. High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc Nat Acad Sci. 2007;104:8143–8148. doi: 10.1073/pnas.0700384104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ting JT, Peca J, Daigle TL, et al. Ultrafast optogenetic control of diverse neuronal populations with with cre-inducible ChETA knock-in mice. Soc Neurosci Abs. 2012;208.11 [Google Scholar]
- 20.Madisen L, Mao T, Koch H, et al. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci. 2012;15:793–802. doi: 10.1038/nn.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yizhar O, Fenno LE, Davidson TJ, et al. Optogenetics in neural systems. Neuron. 2011;71:9–34. doi: 10.1016/j.neuron.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Tang W, Ehrlich I, Wolff SB, et al. Faithful expression of multiple proteins via 2A-peptide self-processing: a versatile and reliable method for manipulating brain circuits. J Neurosci. 2009;29:8621–8629. doi: 10.1523/JNEUROSCI.0359-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prakash R, Yizhar O, Grewe B, et al. Two-photon optogenetic toolbox for fast inhibition, excitation and bistable modulation. Nat methods. 2012;9:1171–1179. doi: 10.1038/nmeth.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yonehara K, Balint K, Noda M, et al. Spatially asymmetric reorganization of inhibition establishes a motion-sensitive circuit. Nature. 2011;469:407–410. doi: 10.1038/nature09711. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Tsien RW. pH Tomato, a red, genetically encoded indicator that enables multiplex interrogation of synaptic activity. Nat Neurosci. 2012;15:1047–1053. doi: 10.1038/nn.3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aoyama K, Suh SW, Hamby AM, et al. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- 27.MacGregor DG, Chesler M, Rice ME. HEPES prevents edema in rat brain slices. Neurosci lett. 2001;303:141–144. doi: 10.1016/s0304-3940(01)01690-1. [DOI] [PubMed] [Google Scholar]
- 28.Brahma B, Forman RE, Stewart EE, et al. Ascorbate inhibits edema in brain slices. J Neurochem. 2000;74:1263–1270. doi: 10.1046/j.1471-4159.2000.741263.x. [DOI] [PubMed] [Google Scholar]
- 29.Huang S, Uusisaari MY. Physiological temperature during brain slicing enhances the quality of acute slice preparations. Front Cell Neurosci. 2013;7:48. doi: 10.3389/fncel.2013.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davie JT, Kole MH, Letzkus JJ, et al. Dendritic patch-clamp recording. Nat Prot. 2006;1:1235–1247. doi: 10.1038/nprot.2006.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattis J, Tye KM, Ferenczi EA, et al. Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat Methods. 2012;9:159–172. doi: 10.1038/nmeth.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]