

Abstract
Objectives
Elafin, an endogenous serine protease inhibitor, modulates colonic inflammation. We investigated the role of elafin in celiac disease (CD) using human small intestinal tissues and in vitro assays of gliadin deamidation. We also investigated potential beneficial effects of elafin in a mouse model of gluten sensitivity.
Methods
Epithelial elafin expression in the small intestine of patients with active CD, treated CD and controls without CD was determined by immunofluorescence. Interaction of elafin with human tissue transglutaminase-2 (TG-2) was investigated in vitro. The 33-mer peptide, a highly immunogenic gliadin peptide, was incubated with TG-2 and elafin at different concentrations. The degree of deamidation of the 33-mer peptide was analyzed by liquid chromatography-mass spectrometry. Elafin was delivered to the intestine of gluten-sensitive mice using a recombinant Lactococcus lactis vector. Small intestinal barrier function, inflammation, proteolytic activity, and zonula occludens-1 (ZO-1) expression were assessed.
Results
Elafin expression in the small intestinal epithelium was lower in patients with active CD compared to control patients. In vitro, elafin significantly slowed the kinetics of the deamidation of the 33-mer peptide to its more immunogenic form. Treatment of gluten-sensitive mice with elafin delivered by the L. lactis vector normalized inflammation, improved permeability and maintained ZO-1 expression.
Conclusions
The decreased elafin expression in small intestine of patients with active CD, the reduction of 33-mer peptide deamidation by elafin, coupled to the barrier enhancing and anti-inflammatory effects observed in gluten sensitive mice, suggest this molecule may have pathophysiological and therapeutic importance in gluten-related disorders.
Keywords: Gluten-sensitivity, elafin, celiac disease, proteolytic therapy
INTRODUCTION
Celiac disease (CD) is triggered by the ingestion of gluten in genetically susceptible individuals. Gluten has been proposed to contribute to health issues other than CD (1–4). Currently, there are no pharmacological treatments available for patients with CD or other gluten-related disorders. A life-long adherence to a gluten-free diet (GFD) is required after the diagnosis of CD is confirmed. However, a GFD is associated with high non-adherence rates (5, 6). Gluten is a common ingredient in many food items, making complete removal of gluten from the diet challenging (7, 8). Although effective in inducing clinical remission of CD, following a strict GFD may have a negative impact on lifestyle and quality of life (9–11).
Elafin is a human serine protease inhibitor that has potent inhibitory capacity against various forms of elastases as well as proteinase-3 (12–14). Elafin is expressed throughout the epithelium of the gastrointestinal tract and its expression and induction is decreased in patients with inflammatory bowel disease (15–17). However, it is unknown whether elafin expression is altered and whether it plays a role in chronic inflammatory disorders of the proximal small intestine, such as CD.
Elafin has been identified as a substrate for the cross-linking activity of tissue transglutaminase (TG-2)(18, 19). The discovery that TG-2 plays an important role in CD by introducing negatively charged residues in gluten peptides through deamidation, thereby increasing binding affinity to HLA-DQ2/DQ8 molecules, is one of the recent milestones in CD pathogenesis (20). Given the importance of TG-2 to the pathogenesis of CD, its inhibition is an attractive therapeutic target. TG-2, however, is found systemically and has important functions in a number of other biological processes including wound healing, cell migration, cell death, and inflammation (21). Whether the inhibition of TG-2 is reversible and local to the small intestine are important considerations for a CD targeted therapy. The ability of elafin to inhibit deamidation of gliadin peptides by TG-2 has not yet been tested. The small intestinal expression profile of this immunomodulatory protease inhibitor is unknown in CD. Thus, we first investigated the expression of elafin in patients with active CD and one year after a GFD. Patients undergoing gastroduodenal endoscopy in whom CD was excluded, were included as controls. The potential interaction of elafin and human TG-2, an enzyme with an established role in CD pathogenesis, was tested using 33-mer peptide deamidation assays. Finally, the anti-inflammatory and barrier enhancing properties of elafin in the proximal small intestine were investigated in an animal model of gluten-sensitivity.
METHODS
Elafin expression in human small intestinal mucosa
Human small intestinal biopsies used in this study were randomly selected from a series of subjects participating in previous studies (22, 23). The baseline clinical characteristics of subjects are shown in Table 1. The CD diagnosis was based on the concomitant presence of histological damage in the first and second duodenal portions (Marsh IIIa or greater) and a positive CD serology (IgA anti-Transglutaminase 2; Inova Inc. San Diego; CA; USA). Histology was investigated in biopsy specimens of patients at the moment of the diagnosis (n=10), in the same patients after one year of a GFD (n=9) and in patients with dyspepsia, peptic ulcer, gastroesophageal reflux disease or IBS, where the diagnosis of CD was excluded (n=10).
Table 1.
Patient population
| Characteristics | NC Controls (n=11) | Active CD (n=10) | 1 yr GFD (n=9) |
|---|---|---|---|
| Age (mean+/− SD) | 48 (+/−15) | 40 (+/−18) | 47 (+/−15) |
| Gender (F/M) | 10/1 | 9/1 | 8/1 |
| IgA TG-2; cut-off 20 U/mL) | 11/11 negative | 10/10 positive | 9/9 negative |
| Biopsy (Marsh’s modified) | 11/11 Marsh 0 | 10/10 positive (>IIIa) | 7/9 Marsh 0; 1/9 Marsh I; 1/9 Marsh II |
NC; non-celiac, CD; celiac disease, GFD; gluten-free diet, TG-2; tissue transglutaminase
Five-micrometer sections were deparaffinized and treated with a buffer solution for heat-induced epitope retrieval (pH 6.0). Slides were incubated overnight at 4°C with anti-elafin (Santa Cruz Biotechnology, Montrouge, France) and anti-cytokeratin 18 (CK18; Santa Cruz) antibodies. Anti-CK18 was used as a marker for epithelial cells. Slides were mounted and nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI) Fluorescent mounting medium (Invitrogen, Burlington, ON, Canada) and analyzed on confocal microscope Zeiss LSM710 (Carl Zeiss, Toronto, ON, Canada). For relative quantification by immunofluorescence staining, four different fields of acquisitions for one patient were taken with the same microscope settings in a blinded fashion. We have converted images from mucosal epithelial marker (CK18) to a binary mask in order to obtain a final image representing only the specific staining of elafin in the mucosa. Elafin fluorescence intensity was then quantified in this image using ImageJ software (NIH, Bethesda, MD, USA) and was reported as arbitrary unit per surface of tissue. Each plots represent the mean fluorescence intensity of four different acquisition fields of the same patient. Data was represented as fold increase of signal intensity quantified in control patients.
Inhibition of deamidation of 33-mer peptide by elafin
The 33-mer peptide (25 μM, alpha2-gliadin, 33mer (aa 56–88) Zedira, Darmstadt, Germany, diluted in 50 mM phosphate buffer, pH 7.5) was incubated with 1 μM TG-2 (0.59 U/mg, purity > 90 %, Zedira, Darmstadt, Germany) at 37 °C in 100 mM Tris/HCl buffer, pH 7.6 with 10 mM CaCl2. The TG-2-catalyzed deamidation of 33-mer served as a control reaction in the assay. In the competitive reaction, 25, 50 and 100 μM elafin (human, recombinant, purity >90%, Sigma-Aldrich, Saint Louis, MO, USA) was mixed with the peptide before starting the enzymatic reaction. A negative control with insulin (human, recombinant, purity >98%, Sigma-Aldrich) and a positive control with tridegin (recombinant, purity >95%, Zedira) were prepared each in a 1:1 molar ratio with the 33-mer. The reaction was inactivated at different time points (0, 5, 15, 30, 60 min) by the addition of 25 mM iodoacetamide. All reactions were performed in triplicates. The analysis of the samples was performed using a liquid chromatography (LC) system consisting of Rheos Allegroquaternary pump, C18-column (Hypersil Gold, 100 × 1 mm, 1.9 μm) and XCalibur control software (Thermo Fisher Scientific Inc, San Jose, CA, USA). Samples were injected (10 μL) with a flow rate of 50 μL/min, 95 % water (+0.1 % v/v formic acid) as an initial value for 1 min followed by a linear gradient of ACN (+0.1 % v/v formic acid) over 19 min (95 – 5 % v/v). The LC was directly connected to an LTQ XL linear quadrupole Ion Trap (Thermo Fisher Scientific Inc.) equipped with an electrospray ion source. Data were acquired by full MS. Given that deamidation yields in a mass increase of 1 Da only, the untreated and deamidated peptide overlap in the mass spectrum. Therefore the centroid mass determination was used for the quantification of deamidation (Eq. 1).
| Eq. 1 |
where mi and Ii are the masses and intensities of individual signals, respectively (24). The total deamidation degree was determined by calculating the difference of centroid masses of TG-2-reacted and untreated peptide.
Animal model of gluten sensitivity
Eight- to 10-week-old male NOD AB° DQ8 (NOD/DQ8) transgenic mice were used for experiments (25). Mice were weaned and maintained on a low-fat (4.4%), gluten-free diet, purchased from Harlan Laboratories and bred in a conventional, specific pathogen-free colony at McMaster University. All experiments were conducted with approval from the McMaster University Animal Care Committee.
Mice were orally sensitized with a peptic-tryptic digest of gliadin (PT-gliadin), prepared as described previously (26). Mice were gavaged with 500 μg PT-gliadin plus 25 μg cholera toxin (Sigma-Aldrich) once per week for 3 weeks (27). Non-sensitized controls were gavaged with 25 μg cholera toxin only. Following sensitization, mice were challenged with gliadin (2 mg/mouse) dissolved in 0.02 M acetic acid by intragastric gavage once a week for two weeks.
Mucosal delivery of elafin
We chose a Lactococcus lactis (L. lactis) vector as mucosal delivery system for elafin, as this approach was extensively validated in previous mouse models of colitis (17). Briefly, an elafin-producing recombinant strain of L. lactis, was used as a bacterial vector to deliver elafin to the intestine. L lactis carrying the empty plasmid vector (Ll-WT) was used as wild type control (17). All bacteria were suspended in PBS containing 15% glycerol. Twenty-four hours following oral sensitization with PT-gliadin and cholera toxin, NOD/DQ8 mice were treated with Ll-WT, Ll-E (109 CFU) or PBS/glycerol by oral gavage, daily for 15 days. Mice received gliadin challenges (2 mg/mouse) on day 7 and 15 of L. lactis treatment.
Measurement of small intestinal permeability
Twenty-four hours following the final gliadin challenge, in vitro intestinal permeability was assessed by Ussing chamber technique as previously described (World Precision Instruments, Sarasota, FL, USA) (27–29). Two sections of jejunum from each mouse were assessed. Tissue conductance (the passive permeability to ions) and mucosal-to-serosal flux of the paracellular probe 51Cr-EDTA were used to assess barrier function and paracellular permeability as previously described (30). 51Cr-EDTA flux was calculated as the average over a 2-h period and expressed as %hot/cm2/h.
Evaluation of small intestinal inflammation
Cross-sections of the proximal small intestine were fixed in 10% buffered formalin, embedded in paraffin, and stained with H&E for histologic evaluation by light microscopy (Olympus, Richmond Hill, ON, Canada) as previously described (27). Paraffin sections were used to preserve tissue morphology and orientation. Immunostaining for CD3+ cells was performed on paraffin sections to detect the presence of intraepithelial lymphocytes (IELs) in sections of the proximal small intestine as described previously (29, 31). Slides were viewed in a blinded fashion by light microscopy under 200X magnification. IELs per 20 enterocytes in five randomly chosen villus tips were counted according to the previously described methods and expressed as IEL/100 enterocytes (29).
ZO-1 expression in small intestine
Staining for the tight junction protein zonula occludens-1 (ZO-1) was performed on jejunal sections. Sections were incubated for 2 h at room temperature with an anti-ZO-1 (primary) antibody (Invitrogen), washed and secondary antibody was applied for 2 h at room temperature. Slides were then mounted and coverslipped with Prolong Gold Anti-fade with 4′6-diamidino-2-phenylindole (DAPI; Invitrogen) and staining assessed in a blinded fashion using a fluorescent microscope (Olympus). A total of 10 images were obtained for each slide and the mean fluorescence intensity (MFI) of five identically sized regions of interest (ROI) was determined for each image using Image J Software (NIH). Each ROI was centered on the epithelial border. The MFI for the negatively stained sections was then subtracted from that of positively stained sections.
Trypsin-like and elastase-like activities in small intestine
A section of the jejunum from non-sensitized NOD/DQ8 mice (n=5) was collected using a 4 mm biopsy punch and transferred into a 48 well plate (BD Bioscience, Mississauga ON, Canada) containing RPMI 1640 enriched with 10% FBS, 100 U/ml penicillin, 100 U/ml streptomysin, and 2 mM L-glutamine. Wells were supplemented with 100 μg/ml of one the following gliadin peptides: 19-mer (p31-49), 20-mer (p120-139), 33-mer (p56-89; ProImmune, Sarasota, FL, USA). Media alone was used for negative controls. Plates were incubated for 3 hours at 37°C with 95% O2 and 5% CO2. Following incubation, the tissue section was removed and frozen at −80°C. Supernatants were collected and stored at −80°C. Trypsin and elastase-like activity was measured in organ culture supernatant, biopsies, as well as intestinal samples from non-sensitized and gliadin-sensitized mice as previously described (32).
Statistical analysis
Statistical analysis was conducted using GraphPad Prism software (Version 4, 2004, CA, USA). Statistical significance was determined by one-way ANOVA followed by Bonferroni’s multiple comparison tests. Significance was reported for p-values <0.05 at a 95% confidence level. All data are presented as mean ± standard error of mean (SEM).
RESULTS
Elafin expression is reduced in the small intestine of patients with active celiac disease
We tested whether elafin expression is altered in small intestinal biopsies of patients with CD. Biopsies were collected from the duodenum and jejunum of patients with active CD (on a gluten-containing diet), treated CD patients (on a GFD for at least one year), and control patients (gastrointestinal complaints in which CD has been excluded). Biopsies were then stained for elafin expression by immunofluorescence. Elafin expression in the epithelium, as determined by co-staining with CK18, was lower in patients with active CD compared to control patients (Figure 1A). This reduction was confirmed by determining the MFI of elafin expression in areas of tissue staining with the CK18 marker. Elafin intensity was significantly lower in active CD patients compared to control patients (Figure 1B). Elafin levels in treated CD were intermediate between untreated CD and controls but were not statistically different from either.
Figure 1.

Decreased elafin expression in patients with active celiac disease (CD). Biopsies were obtained from patients in whom CD was excluded (controls), patients with treated CD (1 year gluten-free diet (GFD); remission) and patients with active CD and were stained for elafin (red) expression. (A) Representative immunofluorescence figures are shown. Nuclei labelled with 4′6-diamidino-2-phenylindole (DAPI; blue). (B) Quantification of fluorescence intensity, expressed as a fold increase to control patients. Each dot represents the mean fluorescent value of 4 different areas of one patient and the line represents the mean of each group. *p<0.05.
Inhibition of deamidation of 33-mer peptide in vitro
Elafin has been identified as a substrate for TG-2 (19). Using a competitive enzymatic assay, we therefore tested elafin’s inhibiting effect on the deamidation of 33-mer gliadin peptide, which served as a model immunogenic substrate. The addition of elafin significantly slowed the kinetics of the deamidation of the 33-mer peptide to its more immunogenic form (Figure 2). The inhibiting effect of elafin was found to be dose and time-dependent, but relatively modest compared to tridegin, the positive control (33). When used at a concentration which was 4-fold higher than tridegin, the deamidation of 33-mer was approximately 50% less effective after 30 min incubation. As expected, the negative protein control, insulin, did not show any inhibition of TG-2 activity (Figure 2).
Figure 2.
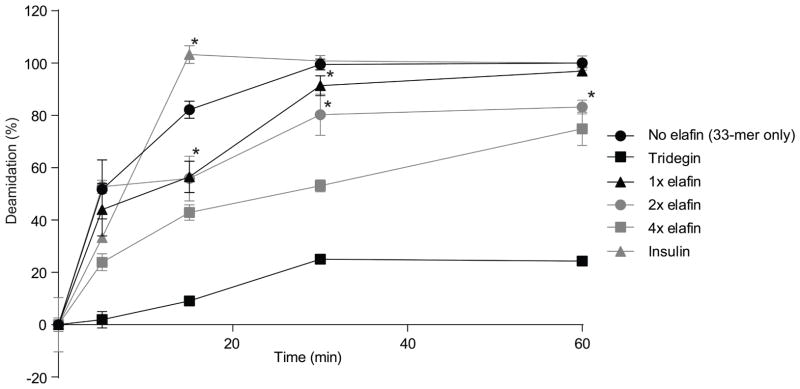
Deamidation of 33-mer in the competitive reactions. 33-mer peptide (25 μM) was incubated with tissue transglutaminase-2 (TG-2) (1 μM) that served as a control reaction. For the competitive reactions, 25 (x1) or, 50 (x2), or 100 μM (x4) elafin was added. Tridegin and insulin were used as positive and negative controls, respectively. The degree of deamidation was analyzed by liquid chromatography-mass spectrometry (LC-MS). Data are represented as mean ± SEM (n=3). *p<0.05 vs control reaction (no elafin), all data points of the enzymatic reaction with 100 μM (x 4) elafin and tridegin are statistically significant (p<0.02) vs. control reaction (no elafin).
Ll-elafin reduces gliadin-induced IEL infiltration in gliadin-sensitized NOD/DQ8 mice
We then tested the therapeutic potential of elafin in gliadin-sensitized NOD/DQ8 mice, a mouse model of gluten sensitivity, using L. lactis to deliver elafin (Ll-E) to the small intestine. In agreement with previous findings, (5, 27) sensitization and acute gliadin challenge increased IELs in the jejunum (Figure 3). Ll-E treatment normalized IEL counts, whereas Ll-WT had no effect (Figure 3).
Figure 3.

L. lactis expressing elafin(Ll-E) treatment attenuates gliadin-induced intraepithelial lymphocytosis in sensitized NOD/DQ8 mice. (A) CD3+-stained sections of the proximal small intestine. Original magnification x20. Black arrows indicate intraepithelial lymphocytes (IELs). (B) Quantification of CD3+ cells in villi tips, expressed as IEL per 100 enterocytes (n=8–12 per group). White bars represent non-sensitized animals, grey bars represent sensitized animals. Data are represented as mean ± SEM. **p<0.01, ***p<0.001. Non-sensitized (NS); Lactococcus lactis wild type (Ll-WT); L. lactis expressing elafin (Ll-E).
Ll-elafin reduces intestinal barrier dysfunction and preserves zonula occludens-1 distribution following gliadin challenge in sensitized NOD/DQ8 mice
Gliadin-sensitization and acute gliadin challenge increased small intestinal paracellular permeability as indicated by in vitro 51Cr-EDTA flux in NOD/DQ8 mice (Figure 4A). Gliadin challenge in sensitized mice also increased tissue conductance (Figure 4B). Sensitized mice treated with Ll-E had lower 51Cr-EDTA flux and tissue conductance compared to sensitized animals receiving either PBS-glycerol or Ll-WT (Figure 4A, B).
Figure 4.

L. lactis expressing elafin(Ll-E) therapy protects NOD/DQ8 mice from gliadin-induced increases in paracellular permeability and preserves zonula occludens-1 (ZO-1) distribution. Sections of small intestine were mounted in Ussing chambers and (A) 51Cr-EDTA flux (% Hot Sample/h/cm2; n=8–11 per group) and (B) tissue conductance (mS/cm2; n=4–7 per group) was measured 24 h after the final gliadin challenge. (C) Sections of the proximal small intestine were stained for ZO-1 (green) expression. Nuclei labelled with 4′6-diamidino-2-phenylindole (DAPI; blue). Original magnification x20. White arrows indicate strong immunofluorescence at the apical junctional complex; arrowheads indicate patchy expression. (D) Mean fluorescence intensity (MFI) of ZO-1 staining in proximal small intestinal sections was determined (n=3–4 per group). MFI was corrected for background fluorescence. White bars represent non-sensitized animals, grey bars represent sensitized animals. Data are represented as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001. Lactococcus lactis wild type (Ll-WT); L. lactis expressing elafin (Ll-E).
We investigated a previously unexplored effect of elafin on ZO-1 distribution in the small intestine of gluten-sensitive mice. Staining for ZO-1 was decreased in gliadin sensitized mice compared to controls. The expression of ZO-1 was patchy with most of the expression being limited to the villus base and crypts. The distribution of ZO-1 normalized in sensitized mice treated with Ll-E (Figure 4C). We confirmed this reduction in ZO-1 staining by determining the MFI (Figure 4D). Sensitized animals treated with PBS-glycerol had significantly lower ZO-1 MFI than non-sensitized controls. Treatment of sensitized animals with Ll-E normalized ZO-1 MFI (Figure 4D).
Gliadin challenge does not affect small intestinal trypsin-like or elastase-like proteolytic activity
To investigate potential mechanisms of action of elafin, we measured trypsin- and elastase-like activity in small intestinal tissues from sensitized and non-sensitized NOD/DQ8 mice treated. The levels of trypsin-like proteases were similar in sensitized and non-sensitized mice, and were not affected by elafin treatment (Figure 5A). Elastase-like activity was undetectable (data not shown). Proteolytic activity was also tested small intestinal tissue from non-sensitized and sensitized mice stimulated in vitro with the gliadin peptides 33-mer, 20-mer and 19-mer. Elastase-like activity was undetectable (data not shown) and trypsin-like activity was not significantly affected in either the small intestinal biopsies or culture supernatant following in vitro stimulation with the gliadin fragments (Figure 5B, C).
Figure 5.

Gliadin sensitization and challenge do not alter trypsin-like activity in NOD/DQ8 mice in vivo or in vitro. (A) Sections of proximal small intestine were collected 24 h after final gliadin challenge. Levels of trypsin-like proteases were quantified in tissue homogenates and expressed as U/mg protein (n=8–11 per group). (B, C) Proximal small intestinal biopsies were collected from sensitized and non-sensitized NOD-DQ8 mice and stimulated for 3 h in vitro with gliadin peptides. Trypsin-like activity was quantified in the (B) culture supernatant and (C) biopsies and expressed as U/mL and U/mg protein respectively. White bars represent non-sensitized animals, grey bars represent sensitized animals. Data are represented as mean ± SEM. Lactococcus lactis wild type (Ll-WT); L. lactis expressing elafin (Ll-E).
DISCUSSION
CD is one of the most common gastrointestinal diseases, affecting about 1% of the Caucasian population, and its incidence has substantially risen in the last two decades (34, 35). The only treatment is a life-long adherence to a GFD, which is costly and difficult to follow, leading to high non-adherence rates (6). In CD, gluten-derived peptides are recognized by HLA-DQ2 or HLA-DQ8 heterodimers on antigen-presenting cells, which trigger a pro-inflammatory gluten-specific T cell response (36). Through the process of deamidation, TG-2 activity plays an important role by introducing negatively charged residues on gluten-peptides. This enhances recognition of these gluten-peptides by the HLA-DQ2 or -DQ8 molecules, which are efficiently recognized by T cells (36, 37). However, the factors that drive TG-2 activation in patients with CD are not completely understood (37, 38).
Elafin is a potent endogenous human serine protease inhibitor, and its levels have been found to be reduced in the colon of patients with IBD (15, 17). In this study we show that patients with active CD also have reduced elafin expression within the small intestinal epithelium compared to patients in whom CD was excluded. After 1 year of GFD, elafin expression was somewhat higher than in active CD patients, but was not statistically different from non-celiac controls. The reduced elafin expression in patients with active CD may reflect the presence of enteropathy and its loss may contribute to enhanced inflammation in CD. In fact, expression of other enzymes in the small intestine, such as cytochrome P450, CYP3A, was found to correlate with the degree of enteropathy, raising the possibility of using these markers as an indication of villous health (39, 40). Reduced expression of elafin in patients with active CD may reflect activity of inflammation or mucosal damage in the small intestine, similar to what has been described in the colon of patients with IBD (15–17).
We then hypothesized that elafin may play a role in gluten-related disorders and tested the interaction of elafin with human TG-2, the enzyme that catalyzes the deamidation of gluten peptides enhancing T-cell recognition (36). Indeed, elafin has recently been identified as a substrate for the cross-linking activity of tissue transglutaminase (19, 41, 42). Our in vitro results demonstrate that the addition of elafin inhibited the deamidation of the digestion-resistant 33-mer gliadin peptide, which is one of the potential triggers of the adaptive immune response in CD (43). The comparison of elafin to tridegin, a known transglutaminase inhibitor, revealed that elafin moderately inhibits TG-2. These data support that elafin interacts with TG-2, raising the possibility that in addition to correlating with mucosal damage, a reduction of elafin in active CD could play a pathogenic role. TG-2 plays important roles in tissue homeostasis and wound healing (21, 44). Thus, the moderate inhibition and small intestinal delivery of elafin could constitute an advantage over systemic, complete and irreversible inhibition, particularly when combined with other potential beneficial effects of elafin. Our in vitro results point at an exciting novel role and specific mechanism of action of elafin in CD that should be further explored.
Elafin has been shown to have anti-inflammatory and barrier modulating properties in models of respiratory, cardiovascular, and colonic inflammation (17, 32, 41, 45). We explored whether elafin administration affected these parameters in the small intestine using a model of gluten sensitivity. We employed a validated delivery method that exploits a food grade strain of L. lactis, to ensure administration of elafin to the gastrointestinal tract in mice (17, 32). Although the quantification of elafin delivered to the intestine was not determined, using GFP-tagged Ll-WT and Ll-E, we have previously demonstrated effective delivery of L. lactis and elafin to the gastrointestinal tract. Elafin was found to cluster around L. lactis, but was also detected at sites remotes from the GFP-tagged L. lactis, suggesting that elafin is secreted in the mucosa in situ(17). We found that Ll-E reduced intraepithelial lymphocytosis and restored barrier function in gliadin sensitized mice. We showed that treatment with Ll-E normalized 51Cr-EDTA flux and conductance in the small intestine suggesting that Ll-E protects the paracellular pathway in the small intestine. Furthermore, increased permeability after gliadin challenge was accompanied by a reduction in ZO-1 protein expression and Ll-E therapy preserved ZO-1 protein distribution in sensitized mice. Overall, our results suggest that there are beneficial effects of Ll-E in this model of gluten-induced small intestinal dysfunction that involve immunomodulatory effects and regulation of barrier and tight junction function. This is consistent with previous reports on the effect of elafin in the colon using animal models of colitis (14, 17, 32). Proteolytic imbalances have been described in patients with IBS, IBD, and in animal models of experimental colitis (17, 32). However, we found no differences in trypsin or elastase-like activity between gliadin sensitized and control mice. In addition, treatment with Ll-E had no effect on small intestinal protease levels. Our findings highlight a difference in the mechanisms of action of elafin in gluten intolerance compared to colitis, which does not involve correction of elastolytic or trypsin activity imbalance in the small intestine.
While the NOD/DQ8 mouse model does not develop severe atrophy that is characteristic of classical CD, sensitization and challenge induces moderate inflammation (27). It will be important to test the effect of elafin, as well as other potential therapies for CD in a variety of models of gluten sensitivity that express different pathological characteristics and underlying mechanisms, such as IL-15 driven models of CD (46).
In conclusion, the reduced small intestinal elafin expression in patients with active CD coupled to the finding that elafin may compete with gliadin peptides and reduce their deamidation, raise the possibility that loss of this protease inhibitor contributes to CD pathogenesis. The results in the animal model of gluten sensitivity suggest mucosal delivery of elafin restores small intestinal barrier function and inflammation, likely through preservation of tight junction function and anti-inflammatory effects in the small intestine. Due to the combination of specific and non-specific beneficial effects of elafin in our study, we propose its replacement could have potential as adjuvant therapy in gluten-related disorders.
What is current knowledge?
The only treatment for celiac disease (CD) is a life-long adherence to a gluten-free diet (GFD)
A strict GFD is costly and difficult to follow, leading to high non-adherence rates and increased co-morbidities, highlighting the need for alternative treatments.
Elafin is an anti-inflammatory human serine protease inhibitor that has potent inhibitory activity against murine neutrophil elastase and proteinase-3
Elafin is decreased in IBD patients and prevents inflammation in murine models of experimental colitis
The role of elafin in gluten-related disorders is unknown.
What is new here?
Elafin expression is significantly decreased in patients with active CD, compared to patients without CD.
In vitro elafin reduces the deamidation of an immunogenic gliadin peptide, an important step in the autoimmune process of CD.
In an animal model of gluten sensitivity, elafin delivery at the mucosal level by a food grade L. lactis vector prevented gluten-induced barrier dysfunction and inflammation.
The identification of a molecule (elafin) that is altered in the small intestine of active CD patients and that reduces deamidation of gliadin peptide, and that has barrier-enhancing effects in the small intestine, will help increase understanding of the pathophysiology of CD and promote development of new supportive treatments for gluten-related disorders.
Acknowledgments
Heather J Galipeau and Michelle Wiepjes contributed equally to the manuscript. We would like to acknowledge the technical help of Xianxi Huang and Yikang Deng with molecular analysis and of Jean-Jacques Gratadoux for the preparation of bacterial cultures.
FUNDING
This work was funded by a CIHR grant to E.F. Verdu (MOP-123282) and by JA Campbell Young Investigator Awards from the Canadian Celiac Association to M Wiepjes and H Galipeau. E.F. Verdu holds a Canada Research Chair. J.C. Leroux acknowledges financial support from the Swiss National Science Foundation (310030_135732). The elafin project of N. Vergnolle is funded by the Agence Nationale de la Recherche (Elaprob-IBD, Blanc SVSE1-2012), and N. Vergnolle is funded by the European Research Council (ERC-2012-StG_20111109). JP Motta was funded by the Inserm and the Region Midi-Pyrenees. P. Langella and L. Bermudez-Humaran are also involved in the elafin project of N. Vergnolle funded by the Agence Nationale de la Recherche (Elaprob-IBD, Blanc SVSE1-2012). R. Martin is funded by the French FUI (Fond Unique Interministériel; FUI: n°F1010012D), the FEDER (Fonds Européen de Développement Régional; Bourgogne: 34606), the Burgundy Region, the Conseil Général 21 and the Grand Dijon. JA Murray was supported in part by DK71003 and the Mayo Foundation.
References
- 1.Verdu EF. Editorial: Can Gluten Contribute to Irritable Bowel Syndrome. Am J Gastroenterol. 2011;106:516–8. doi: 10.1038/ajg.2010.490. [DOI] [PubMed] [Google Scholar]
- 2.Verdu EF, Armstrong D, Murray JA. Between celiac disease and irritable bowel syndrome: the “no man’s land” of gluten sensitivity. Am J Gastroenterol. 2009;104:1587–94. doi: 10.1038/ajg.2009.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biesiekierski JR, Newnham ED, Irving PM, et al. Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol. 2011;106:508–14. doi: 10.1038/ajg.2010.487. [DOI] [PubMed] [Google Scholar]
- 4.Ludvigsson JF, Leffler DA, Bai JC, et al. The Oslo definitions for coeliac disease and related terms. Gut. 2013;62:43–52. doi: 10.1136/gutjnl-2011-301346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinier M, Fuhrmann G, Galipeau HJ, et al. The copolymer P(HEMA-co-SS) binds gluten and reduces immune response in gluten-sensitized mice and human tissues. Gastroenterology. 2012;142:316–25. doi: 10.1053/j.gastro.2011.10.038. [DOI] [PubMed] [Google Scholar]
- 6.Hall N, Rubin G, Charnock A. Systematic review: adherence to a gluten-free diet in adult patients with coeliac disease. Aliment Pharmacol Ther. 2009;30:315–30. doi: 10.1111/j.1365-2036.2009.04053.x. [DOI] [PubMed] [Google Scholar]
- 7.Lee A, Newman JM. Celiac diet: its impact on quality of life. J Am Diet Assoc. 2003;103:1533–5. doi: 10.1016/j.jada.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 8.Stevens L, Rashid M. Gluten-free and regular foods: a cost comparison. Can J Diet Pract Res. 2008;69:147–50. doi: 10.3148/69.3.2008.147. [DOI] [PubMed] [Google Scholar]
- 9.Hallert C, Sandlund O, Broqvist M. Perceptions of health-related quality of life of men and women living with coeliac disease. Scand J Caring Sci. 2003;17:301–7. doi: 10.1046/j.1471-6712.2003.00228.x. [DOI] [PubMed] [Google Scholar]
- 10.Hallert C, Grännö C, Hulten S, et al. Living with coeliac disease: controlled study of the burden of illness. Scand J Gastroenterol. 2002;37:39–42. doi: 10.1080/003655202753387338. [DOI] [PubMed] [Google Scholar]
- 11.Black J, Orfila C. Impact of coeliac disease on dietary habits and quality of life. J Hum Nutr Diet. 2011;24:582–7. doi: 10.1111/j.1365-277X.2011.01170.x. [DOI] [PubMed] [Google Scholar]
- 12.Ying QL, Simon SR. Kinetics of the inhibition of human leukocyte elastase by elafin, a 6-kilodalton elastase-specific inhibitor from human skin. Biochemistry. 1993;32:1866–74. doi: 10.1021/bi00058a021. [DOI] [PubMed] [Google Scholar]
- 13.Ying QL, Simon SR. Kinetics of the inhibition of proteinase 3 by elafin. Am J Respir Cell Mol Biol. 2001;24:83–9. doi: 10.1165/ajrcmb.24.1.4300. [DOI] [PubMed] [Google Scholar]
- 14.Williams S, Brown T, Roghanian A, et al. SLPI and elafin: one glove, many fingers. Clin Sci. 2006;110:21–35. doi: 10.1042/CS20050115. [DOI] [PubMed] [Google Scholar]
- 15.Schmid M, Fellermann K, Fritz P, et al. Attenuated induction of epithelial and leukocyte serine antiproteases elafin and secretory leukocyte protease inhibitor in Crohn’s disease. J Leukoc Biol. 2007;81:907–15. doi: 10.1189/jlb.0906581. [DOI] [PubMed] [Google Scholar]
- 16.Eriksson A, Jennische E, Flach CF, et al. Real-time PCR quantification analysis of five mucosal transcripts in patients with Crohn’s disease. Eur J Gastroenterol Hepatol. 2008;20:290–6. doi: 10.1097/MEG.0b013e3282f3557c. [DOI] [PubMed] [Google Scholar]
- 17.Motta JP, Bermúdez-Humarán LG, Deraison C, et al. Food-grade bacteria expressing elafin protect against inflammation and restore colon homeostasis. Sci Transl Med. 2012;4:158ra144. doi: 10.1126/scitranslmed.3004212. [DOI] [PubMed] [Google Scholar]
- 18.Steinert PM, Marekov LN. The proteins elafin, filaggrin, keratin intermediate filaments, loricrin, and small proline-rich proteins 1 and 2 are isodipeptide cross-linked components of the human epidermal cornified cell envelope. J Biol Chem. 1995;270:17702–11. doi: 10.1074/jbc.270.30.17702. [DOI] [PubMed] [Google Scholar]
- 19.Baranger K, Zani M, Labas V, et al. Secretory leukocyte protease inhibitor (SLPI) is, like its homologue trappin-2 (pre-elafin), a transglutaminase substrate. PloS one. 2011;6:e20976. doi: 10.1371/journal.pone.0020976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kristiansen C, Madsen L, Fugger L, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4:713–7. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 21.Park D, Choi SS, Ha K. Transglutaminase 2: a multi-functional protein in multiple subcellular compartments. Amino Acids. 2010;39:619–31. doi: 10.1007/s00726-010-0500-z. [DOI] [PubMed] [Google Scholar]
- 22.Sugai E, Moreno ML, Hwang HJ, et al. Celiac disease serology in patients with different pretest probabilities: Is biopsy avoidable? World J Gastroenterol. 2010;16:3144–52. doi: 10.3748/wjg.v16.i25.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugai E, Nachman F, Váquez H, et al. Dynamics of celiac disease-specific serology after initiation of a gluten-free diet and use in the assessment of compliance with treatment. Dig Liver Dis. 2010;42:352–8. doi: 10.1016/j.dld.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 24.Zubarev RA, Demirev PA, Haakansson P, et al. Approaches and limits for accurate mass characterization of large biomolecules. Anal Chem. 1995;67:3793–8. [Google Scholar]
- 25.Marietta E, Black K, Camilleri M, et al. A new model for dermatitis herpetiformis that uses HLA-DQ8 transgenic NOD mice. J Clin Invest. 2004;114:1090–7. doi: 10.1172/JCI21055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas KE, Sapone A, Fasano A, et al. Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: role of the innate immune response in Celiac disease. J Immunol. 2006;176:2512–21. doi: 10.4049/jimmunol.176.4.2512. [DOI] [PubMed] [Google Scholar]
- 27.Galipeau HJ, Rulli NE, Jury J, et al. Sensitization to gliadin induces moderate enteropathy and insulitis in nonobese diabetic-DQ8 mice. J Immunol. 2011;187:4338–46. doi: 10.4049/jimmunol.1100854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Natividad JM, Huang X, Slack E, et al. Host responses to intestinal microbial antigens in gluten-sensitive mice. PLoS one. 2009;4:e6472. doi: 10.1371/journal.pone.0006472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verdu EF, Huang X, Natividad J, et al. Gliadin-dependent neuromuscular and epithelial secretory responses in gluten-sensitive HLA-DQ8 transgenic mice. Am J Physiol Gastrointest Liver Physiol. 2008;294:G217–25. doi: 10.1152/ajpgi.00225.2007. [DOI] [PubMed] [Google Scholar]
- 30.Silva MA, Jury J, Sanz Y, et al. Increased bacterial translocation in gluten-sensitive mice is independent of small intestinal paracellular permeability defect. Dig Dis Sci. 2012;57:38–47. doi: 10.1007/s10620-011-1847-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biagi F, Luinetti O, Campanella J, et al. Intraepithelial lymphocytes in the villous tip: do they indicate potential coeliac disease? J Clin Pathol. 2004;57:835–9. doi: 10.1136/jcp.2003.013607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motta JP, Magne L, Descamps D, et al. Modifying the protease, antiprotease pattern by elafin overexpression protects mice from colitis. Gastroenterology. 2011;140:1272–82. doi: 10.1053/j.gastro.2010.12.050. [DOI] [PubMed] [Google Scholar]
- 33.Finney S, Seale L, Sawyer RT, et al. Tridegin, a new peptidic inhibitor of factor XIIIa, from the blood-sucking leech Haementeria ghilianii. Biochem J. 1997;324:797–805. doi: 10.1042/bj3240797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubio-Tapia A, Ludvigsson JF, Brantner TL, et al. The prevalence of celiac disease in the United States. Am J Gastroenterol. 2012;107:1538–44. doi: 10.1038/ajg.2012.219. [DOI] [PubMed] [Google Scholar]
- 35.Ludvigsson JF, Rubio-Tapia A, van Dyke CT, et al. Increasing incidence of celiac disease in a North American population. Am J Gastroenterol. 2013;108:818–24. doi: 10.1038/ajg.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jabri B, Sollid LM. Tissue-mediated control of immunopathology in coeliac disease. Nat Rev Immunol. 2009;9:858–70. doi: 10.1038/nri2670. [DOI] [PubMed] [Google Scholar]
- 37.Klöck C, DiRaimondo TR, Khosla C. Role of transglutaminase 2 in celiac disease pathogenesis. Semin Immunopathol. 2012;34:513–22. doi: 10.1007/s00281-012-0305-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dafik L, Albertelli M, Stamnaes J, et al. Activation and inhibition of transglutaminase 2 in mice. PloS one. 2012;7:e30642. doi: 10.1371/journal.pone.0030642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson TN, Tanner MS, Taylor CJ, et al. Enterocytic CYP3A4 in a paediatric population: developmental changes and the effect of coeliac disease and cystic fibrosis. Br J Clin Pharmacol. 2001;51:451–60. doi: 10.1046/j.1365-2125.2001.01370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lang CC, Brown RM, Kinirons MT, et al. Decreased intestinal CYP3A in celiac disease: reversal after successful gluten-free diet: a potential source of interindividual variability in first-pass drug metabolism. Clin Pharmacol Ther. 1996;59:41–6. doi: 10.1016/S0009-9236(96)90022-3. [DOI] [PubMed] [Google Scholar]
- 41.Zani ML, Tanga A, Saidi A, et al. SLPI and trappin-2 as therapeutic agents to target airway serine proteases in inflammatory lung diseases: current and future directions. Biochem Soc Trans. 2011;39:1441–6. doi: 10.1042/BST0391441. [DOI] [PubMed] [Google Scholar]
- 42.Guyot N, Zani M, Maurel M, et al. Elafin and its precursor trappin-2 still inhibit neutrophil serine proteinases when they are covalently bound to extracellular matrix proteins by tissue transglutaminase. Biochemistry. 2005;44:15610–8. doi: 10.1021/bi051418i. [DOI] [PubMed] [Google Scholar]
- 43.Shan L, Molberg Ø, Parrot I, et al. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2275–9. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Griffin M. TG2, a novel extracellular protein with multiple functions. Amino Acids. 2012;42:939–49. doi: 10.1007/s00726-011-1008-x. [DOI] [PubMed] [Google Scholar]
- 45.Alam SR, Newby DE, Henriksen PA. Role of the endogenous elastase inhibitor, elafin, in cardiovascular injury: from epithelium to endothelium. Biochem Pharmacol. 2012;83:695–704. doi: 10.1016/j.bcp.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 46.DePaolo RW, Abadie V, Tang F, et al. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature. 2011;471:220–4. doi: 10.1038/nature09849. [DOI] [PMC free article] [PubMed] [Google Scholar]