

Abstract
The presence of nitrogen atoms in most chiral pharmaceutical drugs has motivated the development of numerous strategies for the synthesis of enantioenriched amines. Current methods are based on the multi-step transformation of pre-functionalized allylic electrophiles into chiral allylic amines. The enantioselective allylic amination of unactivated olefins represents a more direct and attractive strategy. We report the enantioselective synthesis of ent-sitagliptin via an allylic amination of an unactivated terminal olefin.
Keywords: asymmetric catalysis, amination, drugs, rearrangement, palladium
Nitrogen atoms are key components of more than 80 percent of FDA approved pharmaceutical drugs.1,2 Their presence in small molecules leads to desirable medicinal properties, including improved solubility under physiological conditions, favorable polar surfaces, and hydrogen-bonding interactions with amino acid residues. As a result, many powerful chemical methods have been developed for the incorporation of nitrogen atoms into small molecules, profoundly impacting the discovery of new drugs.3
Chiral amines represent an important sub-class of medicinally relevant nitrogen-containing molecules.4 For example, sitagliptin (1) is an FDA-approved DPP-4 inhibitor for the treatment of Type II diabetes (Scheme 1).5,6 Elegant enantioselective methods have been developed for the synthesis of chiral amines 4 from allylic alcohols and other allylic electrophiles, such as allylic halides 3.7
Scheme 1.

Strategies for the conversion of unactivated olefins into chiral allylic amines
We were interested in developing an alternative approach to access chiral amines 4, which involves the use of a chiral catalyst to directly convert unfunctionalized olefins 2 through an allylic amination.8 Unsaturated hydrocarbons such as 2 are ideal substrates for chemical synthesis, because they are inexpensive and abundant components of petrochemical feedstock.9 However, olefins are also challenging substrates for asymmetric catalysis, because it is difficult to selectively transform a single C–H bond into a C–N bond in the presence of several sterically and electronically similar C–H bonds.
We recently reported a palladium-catalyzed enantioselective allylic amination of unactivated terminal olefins via an ene reaction/[2,3]-rearrangement.10 Herein, we describe the application of this approach to the enantioselective synthesis of ent-sitagliptin (1).
Several elegant approaches to sitagliptin have been reported in the literature.6 Most notably, Merck has developed multiple enantioselective routes to this compound.6a–d,g In our retrosynthetic analysis of ent-sitagliptin, we envisioned that the target molecule 1 could arise from α-amino acid 5. This intermediate could be generated from allylic amine derivative 6 through a series of functional group interconversions. Enantioenriched allylic amine 6 was a suitable retron for our recently developed catalytic enantioselective allylic amination of unactivated olefins. This strategy revealed trifluorophenyl butene 7 as a starting point for our synthetic efforts.
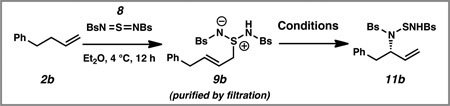
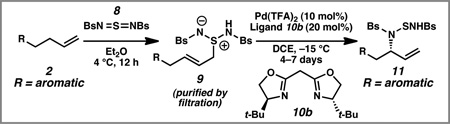
In our initial report, we described a catalytic enantioselective intermolecular allylic amination of unactivated terminal olefins that are substituted with aliphatic substituents (2a→11a, Scheme 3).10 This transformation proceeds through a two-step ene reaction/[2,3]-rearrangement, which was inspired by seminal reports of Sharpless, Kresze, and Katz for the conversion of olefins into racemic allylic amines.11 In the first step, an unactivated olefin 2a reacts with sulfurdiimide reagent 8 through a hetero-ene reaction. The resulting zwitterion 9 a is subjected to a palladium-catalyzed enantioselective [2,3]-rearrangement to furnish the desired allylic amine 11a.
Scheme 3.

Allylic amination of 4-aryl butene substrates
As a model system for our first step in the synthesis of ent-sitagliptin (7→6, Scheme 2), we examined phenyl butene 2b, which lacked the three fluoride substituents in the aromatic ring of olefin 7 (Scheme 3). We anticipated that these carefully optimized palladium-catalyzed conditions for the generation of allylic amines 11 a with aliphatic substitution at the homoallylic position would be suitable for the conversion of phenyl butene 2b to allylic amine 11b with aromatic substitution at the homoallylic position. To our dismay, in the presence of 10 mol% Pd(TFA)2 and 12 mol% ligand 10a, allylic amine 11b was generated in only 4% enantiomeric excess (ee).
Scheme 2.

Retrosynthetic analysis of sitagliptin
Although it still not known why terminal olefins with aromatic substitution in the homoallylic position do not behave well in our previously optimized conditions, we decided to re-optimize every reaction parameter in the palladium-catalyzed [2,3]-rearrangement step for model substrate phenyl butene 2b (Table 1). We confirmed that zwitterion 9b did not undergo a thermal [2,3]-rearrangement in the absence of a palladium catalyst at 4 °C (entry 1). As discussed above, subjection of zwitterion 9b to the previously optimized conditions with ligand 10a resulted in almost racemic product (entry 2). After a survey of other bisoxazoline and bisoxazolinyl-pyridine ligands Pd(OAc)2, bisoxazoline 10b emerged as a more effective ligand than bisoxazoline 10a (entries 3–4), producing the desired product in 75% yield and 79% ee (entry 4).12 We also examined a series of palladium sources (entries 5–8), and Pd(TFA)2 proved to be the most promising (entry 8). We could now isolate the desired product in 80% yield and 85% ee. Through a screen of several solvents,12 we identified DCE as the optimal medium for the reaction (entry 9). Finally, by lowering the reaction temperature to −15 °C and increasing the loading of ligand 10b to 20 mol%, we generated allylic amination product 11b in 94% isolated yield (from phenyl butene 2b) and 93% ee (entry 10).
Table 1.
Optimization of Enantioselective Allylic Amination
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Metal Catalyst (10 mol%) |
Ligand (12 mol%) |
Solvent (0.13M) |
Temp (°C) |
Time (d) |
Yielda | ee (%) |
| 1 | – | – | – | 4 | 0.5 | < 5 | – |
| 2 | Pd(TFA)2 | 10a | CH2Cl2 | 4 | 2 | 91 | 4 |
| 3 | Pd(OAc)2 | 10a | MeOH | 4 | 0.5 | 71 | 19 |
| 4 | Pd(OAc)2 | 10b | CH2Cl2 | 4 | 0.5 | 75 | 79 |
| 5 | Pd2(dba)3 | 10b | CH2Cl2 | 4 | 2 | 60 | 0 |
| 6 | PdCl2(CH3CN)2 | 10b | CH2Cl2 | 4 | 2 | 63 | 6 |
| 7 | Pd(acac)2 | 10b | CH2Cl2 | 4 | 2 | 73 | 0 |
| 8 | Pd(TFA)2 | 10b | CH2Cl2 | 4 | 2 | 80 | 85 |
| 9 | Pd(TFA)2 | 10b | DCE | 4 | 2 | 94 | 91 |
| 10 | Pd(TFA)2 | 10bb | DCE | –15 | 7 | 94 | 93 |
Isolated yield for 2 steps.
20 mol% ligand 10b used.
With optimal conditions in hand, we converted a series of 4-aryl butene substrates 2 to enantioenriched allylic amine derivatives 11 (Table 2).13 This reaction was tolerant of electron-withdrawing and electron-donating substituents. In all cases, the allylic amination products 11 were isolated in synthetically useful yields with enantiomeric excesses.
Table 2.
Substrate Scope of Enantioselective Allylic Amination
 | ||||
|---|---|---|---|---|
| Entry | Olefin | Product | Yielda | ee (%) |
| 1 | 94 | 93 | ||
| 2 | 89 | 90 | ||
| 3 | 79 | 90 | ||
| 4 | 83 | 95 | ||
| 5 | 84 | 81 | ||
Isolated yield for 2 steps.
The new reaction conditions were utilized for the enantioselective allylic amination of trifluorophenyl butene 7 for the synthesis of ent-sitagliptin (Scheme 4). To our delight, trifluorophenyl butene 7 was smoothly converted to allylic amine derivative 6 through a hetero-ene reaction followed by a palladium-catalyzed enantioselective [2,3]-rearrangement. The resulting allylic amination product 6 was treated with methanolic K2CO3, which unveiled allylic sulfonamide 12 in 78% yield (for the three steps) and 93% ee. Hydroboration and extensive oxidation of olefin 12 generated α-amino acid 5. Coupling with amine 13 and subsequent deprotection of the sulfonamide furnished ent-sitagliptin (1).
Scheme 4.

Enantioselective synthesis of ent-sitagliptin
In conclusion, our synthesis of ent-sitagliptin highlights the potential utility of enantioselective allylic amination as an economically efficient and environmentally benign alternative for the production of pharmaceutical drugs. We anticipate that this method will be useful for the synthesis of other nitrogen-containing pharmaceutical agents from inexpensive and abundant unactivated olefins.
Supplementary Material
Acknowledgment
Financial support was provided by the W. W. Caruth, Jr. Endowed Scholarship, the Robert A. Welch Foundation (Grant I-1748), the National Institutes of Health (1R01GM102604-01), the Chilton Foundation Fellowship (H.B.), the University of Texas System Chemistry and Biology Training Grant (L.B.), and the Sloan Research Fellowship (U.K.T.).
Biographies
![]()
Uttam K. Tambar moved from India to the United States when he was four years old. He received his A.B. degree from Harvard University in 2000 and his Ph.D. from the California Institute of Technology in 2006 with Professor Brian Stoltz. After he completed his NIH Postdoctoral Fellowship at Columbia University with Professor James Leighton in 2009, he began his independent research career at UT Southwestern Medical Center in Dallas. He is currently an Assistant Professor in the Biochemistry Department and a W. W. Caruth, Jr. Scholar in Biomedical Research. The Tambar lab is interested in asymmetric catalysis, natural product synthesis, and medicinal chemistry.
Hongli Bao received her B.S. degree in Chemistry from the University of Science & Technology of China in 2002. She obtained her Ph.D. from the joint program of the Shanghai Institute of Organic Chemistry and the University of Science & Technology of China in 2008 with Professor Kuiling Ding and Professor Tianpa You. She joined the Tambar lab in 2009, and she is interested in developing metal catalyzed enantioselective [2,3]-rearrangements. Hongli is the recipient of the UT Southwestern Chilton Postdoctoral Fellowship in Biochemistry.
Liela Bayeh received her B.S. degree in Biochemistry from Baylor University in 2011 and has since moved on to UT Southwestern Medical Center in Dallas to complete her Ph.D. studies. Since joining the Tambar lab in 2012, Liela’s research has primarily focused on asymmetric catalysis and medicinal chemistry.
Footnotes
Supporting Information for this article is available online at http://www.thieme-connect.com/ejournals/toc/synlett.
Primary Data for this article are available online at http://www.thieme-connect.com/ejournals/toc/synlett and can be cited using the following DOI: (number will be inserted prior to online publication).
References
- 1.(a) Henkel T, Brunne RM, Müller H, Reichel F. Angew. Chem. Int. Ed. 1999;38:643. doi: 10.1002/(SICI)1521-3773(19990301)38:5<643::AID-ANIE643>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]; (b) Hili R, Yudin AK. Nat. Chem. Bio. 2006;2:284. doi: 10.1038/nchembio0606-284. [DOI] [PubMed] [Google Scholar]
- 2.The structures of more than 1400 FDA-approved small molecule drugs were downloaded from DrugBank (http://www.drugbank.ca/) and analyzed manually for the presence of nitrogen atoms. See Supporting Information.
- 3.(a) Jiang L, Buchwald SL. Metal-Catalyzed Cross-Coupling Reactions. Ed. 2. Vol. 2. Weinheim: Wiley-VCH; 2004. pp. 699–760. [Google Scholar]; (b) Hartwig JF. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. New York: Wiley-Interscience; 2002. p. 1051. [Google Scholar]; (c) Davies HML, Long MS. Angew. Chem. Int. Ed. 2005;44:3518. doi: 10.1002/anie.200500554. [DOI] [PubMed] [Google Scholar]; (d) Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem. Rev. 2007;107:5318. doi: 10.1021/cr068006f. [DOI] [PubMed] [Google Scholar]; (e) Surry DS, Buchwald SL. Chem. Sci. 2011;2:27. doi: 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Collet F, Dodd RH, Dauban P. Chem. Commun. 2009:5061. doi: 10.1039/b905820f. [DOI] [PubMed] [Google Scholar]; (g) Davies HM, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farina V, Reeves JT, Senanayake CH, Song JJ. Chem. Rev. 2006;106:2734. doi: 10.1021/cr040700c. [DOI] [PubMed] [Google Scholar]
- 5.For reviews on sitagliptin, see: Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Diabetes Care. 2006;29:2632. doi: 10.2337/dc06-0703. Thornberry NA, Weber AE. Curr. Top. Med. Chem. 2007;7:557. doi: 10.2174/156802607780091028. Miller SA, St OEL. Ann. Pharmacother. 2006;40:133. Desai AA. Angew. Chem. Int. Ed. 2011;50:1974. doi: 10.1002/anie.201007051.
- 6.For recent syntheses of sitagliptin, see: Hansen KB, Balsells J, Dreher S, Hsiao Y, Kubryk M, Palucki M, Rivera N, Steinhuebel D, Armstrong JD, III, Askin D, Grabowski EJJ. Org. Process Res. Dev. 2005;9:634. Clausen AM, Dziadul B, Cappuccio KL, Kaba M, Starbuck C, Hsiao Y, Dowling TM. Org. Process Res. Dev. 2006;10:723. Hansen KB, Hsiao Y, Xu F, Rivera N, Clausen A, Kubryk M, Krska S, Rosner T, Simmons B, Balsells J, Ikemoto N, Sun Y, Spindler F, Malan C, Grabowski EJJ, Armstrong JD. J. Am. Chem. Soc. 2009;131:8798. doi: 10.1021/ja902462q. Steinhuebel D, Sun Y, Matsumura K, Sayo N, Saito T. J. Am. Chem. Soc. 2009;131:11316. doi: 10.1021/ja905143m. Zeng LL, Ding YJ, Zhang GC, Song HR, Hu WH. Chin. Chem. Lett. 2009;20:1397. Liu F, Yu W, Ou W, Wang X, Ruan L, Li Y, Peng X, Tao X, Pan X. J. Chem. Res. 2010;34:230. Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes GJ. Science. 2010;329:305. doi: 10.1126/science.1188934. Davies SG, Fletcher AM, Lv L, Roberts PM, Thomson JE. Tetrahedron Lett. 2012;53:3052. Fistikci M, Gundogdu O, Aktas D, Secen H, Sahin MF, Altundas R, Kara Y. Tetrahedron. 2012;68:2607.
- 7.For excellent discussions of the synthesis of allylic amines by means of catalytic asymmetric allylic amination from functionalized substrates, see: Hartwig JF, Stanley LM. Acc. Chem. Res. 2010;43:1461. doi: 10.1021/ar100047x. Trost BM, Zhang T, Sieber JD. Chem. Sci. 2010;1:427. Evans PA, Robinson JE, Nelson JD. J. Am. Chem. Soc. 1999;121:6761–6762. Lafrance M, Roggen M, Carreira EM. Angew. Chem. Int. Ed. 2012;51:3470. doi: 10.1002/anie.201108287. and references therein.
- 8.(a) Johannsen M, Jørgensen KA. Chem. Rev. 1998;98:1689. doi: 10.1021/cr970343o. [DOI] [PubMed] [Google Scholar]; (b) Ramirez TA, Zhao B, Shi Y. Chem. Soc. Rev. 2012;41:931. doi: 10.1039/c1cs15104e. [DOI] [PubMed] [Google Scholar]; (c) Collet F, Lescot C, Dauban P. Chem. Soc. Rev. 2011;40:1926. doi: 10.1039/c0cs00095g. [DOI] [PubMed] [Google Scholar]; (d) Zalatan DN, Du Bois J. Top. Curr. Chem. 2010;292:347. doi: 10.1007/128_2009_19. [DOI] [PubMed] [Google Scholar]
- 9.Grubbs RH, editor. Handbook of Metathesis. ume 2. Weinheim: Wiley-VCH; 2003. [Google Scholar]
- 10.Bao H, Tambar UK. J. Am. Chem. Soc. 2012;134:18495. doi: 10.1021/ja307851b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Sharpless KB, Hori T. J. Org. Chem. 1976;41:176. [Google Scholar]; (b) Sharpless KB, Hori T, Truesdale LK, Dietrich CO. J. Am. Chem. Soc. 1976;98:269. [Google Scholar]; (c) Kresze G, Münsterer H. J. Org. Chem. 1983;48:3561. [Google Scholar]; (d) Katz TJ, Shi S. J. Org. Chem. 1994;59:8297. [Google Scholar]; (e) Bruncko M, Khuong T-AV, Sharpless KB. Angew. Chem. Int. Ed. 1996;35:454. [Google Scholar]
- 12.For a more complete account of the optimization of reaction parameters, see Table S1 in the Supporting Information.
- 13.General procedure for the catalytic enantioselective allylic amination: A solution of benzenesulfonyl sulfurdiimide 8 (685 mg, 2 mmol) in Et2O (4 mL, 0.5 M) was cooled to 0 °C and treated with the terminal olefin 4-Phenyl-1-butene (6 mmol, 3 equiv). The reaction was stirred at 4 °C for 12 h. The ene adduct 9, which formed a white precipitate, was purified at room temperature by vacuum filtration, washed with Et2O (20–40 mL), and dried under vacuum. The ene adduct 9 was then suspended in DCE (1,2-dichloroethane, 5 mL) and cooled to −20 °C. The solution was treated with the palladium-ligand complex in DCE (10 mL), which was made by premixing Pd(TFA)2 (10 mol%) and ligand 10b (20 mol%) in DCE (10 mL) and stirring for 30 min at 50 °C. The reaction was warmed to −15 °C and stirred for 7 days. Purification by flash chromatography (20:1 hexanes/ethyl acetate to 5:1 hexanes/ethyl acetate) afforded the product (887 mg, 94 % yield for two steps) as a clear oil. The enantiomeric excess of the product was determined to be 93% by comparison to a sample of the racemate (see HPLC trace below). [α]23D = +93.0° (c = 1.0, CH2Cl2). 1H NMR (500 MHz, CDCl3, 50 °C), δ 7.90 (d, J = 7.5 Hz, 2H), 7.59 (t, J = 7.5 Hz, 1H), 7.52 (t, J = 7.5 Hz, 4H), 7.37 (m, 2H), 7.18-7.10 (m, 6H), 6.95 (s, 1H), 6.10 (bs, 1H), 5.07 (m, 2H), 4.80 (m, 1H), 3.23 (m, 2H). 13C NMR (100 MHz, CDCl3, 50 °C), δ 140.9, 139.6, 137.8, 133.4, 133.3, 129.6, 129.4, 129.1, 128.6, 127.9, 127.3, 126.7, 118.9, 68.2, 39.9. IR (thin film): 3234, 1447, 1352, 1165, 1088, 806 cm−1. HRMS (ESI) calcd for [C22H22N2O4S3Na]+ ([M+Na]+): 497.0634, found 497.0645.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.