

Abstract
The Unified Huntington’s Disease Rating Scale is used to characterize motor impairments and establish motor diagnosis. Little is known about the timing of diagnostic confidence level categories and the trajectory of motor impairments during the prodromal phase. Goals of this study were to estimate the timing of categories, model the prodromal trajectory of motor impairments, estimate the rate of motor impairment change by category, and provide required sample size estimates for a test of efficacy in clinical trials. In total, 1010 gene-expanded participants from the Neurobiological Predictors of Huntington’s Disease (PREDICT-HD) trial were analyzed. Accelerated failure time models were used to predict the timing of categories. Linear mixed effects regression was used to model the longitudinal motor trajectories. Age and length of gene expansion were incorporated into all models. The timing of categories varied significantly by gene expansion, with faster progression associated with greater expansion. For the median expansion, the third diagnostic confidence level category was estimated to have a first occurrence 1.5 years before diagnosis, and the second and first categories were estimated to occur 6.75 years and 19.75 years before diagnosis, respectively. Motor impairments displayed a nonlinear prodromal course. The motor impairment rate of change increased as the diagnostic confidence level increased, with added acceleration for higher progression scores. Motor items can detect changes in motor impairments before diagnosis. Given a sufficiently high progression score, there is evidence that the diagnostic confidence level can be used for prodromal staging. Implications for Huntington’s disease research and the planning of clinical trials of efficacy are discussed.
Keywords: movement disorders, Huntington’s disease, neurodegenerative disease, predictive testing, cohort studies
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by the trinucleotide expansion of cytosine-adenine-guanine (CAG) in exon 1 of the huntingtin (HTT) gene.1 HD is associated with severe motor, cognitive, and psychiatric impairments that typically develop in adulthood.2
Although HD onset is characterized by a tripartite of symptoms and signs, diagnosis is based on motor impairments, and endpoints for clinical trials focus mainly on motor signs.3,4 The main measure of motor impairments is the motor assessment section of the Unified Huntington’s Disease Rating Scale (UHDRS),5 which is administered by a trained examiner. The first part of the motor section consists of 31 items in 15 domains of motor impairment, each rated on a 5-point scale ranging from 0 (normal) to 4 (severest impairment). The motor items are distributed among oculomotor functioning (6 items), chorea (7 items), dystonia (5 items), bradykinesia (11 items), and rigidity (2 items).6 In many studies of HD, interest is in the sum of all 31 items, which is referred to as the total motor score (TMS).2,7-9
The second part of the motor assessment section consists of the diagnostic confidence level (DCL), which is a single item with a 5-category ordinal rating scale. The examiner selects a response to the question, “To what degree are you confident that this person meets the operational definition of the unequivocal presence of an otherwise unexplained extrapyramidal movement disorder (eg, chorea, dystonia, bradykinesia, rigidity) in a subject at risk for Huntington’s disease?” The rating categories are 0 (normal; no abnormalities), 1 (nonspecific motor abnormalities; < 50% confidence), 2 (motor abnormalities that may be signs of HD; 50%-89% confidence), 3 (motor abnormalities that are likely signs of HD; 90%-98% confidence), and 4 (motor abnormalities that are unequivocal signs of HD; ≥ 99% confidence).
Despite widespread use of the DCL and the TMS, many questions remain regarding their ability to track progression, especially in the prodromal period of HD (ie, in the years before DCL = 4). The response format of the DCL suggests that the item can be used as a type of progression staging, but little is known about the timing of the occurrences of the categories. The trajectory of the TMS is well characterized for later stages of HD (ie, the years after first DCL = 4)10-13; however, the nature of the prodromal TMS trajectory is unclear. There is also lack of information regarding the relation between the DCL and the TMS. For example, given a cross-section of progression, there is scarce information regarding what constitutes typical TMS levels for DCL categories. Furthermore, it is possible that individuals remain for a time within a DCL category, and it is of interest to examine how the TMS changes during this time.
The issues raised above can be addressed with longitudinal data that span the prodromal phase of HD. Such data are provided by the Neurobiological Predictors of Huntington’s Disease (PREDICT-HD) study.2,14 Using the PREDICT-HD database, the current study had the following aims: First, examine the timing of DCL categories in progression. Second, examine change in motor impairment (TMS and the motor factors) from the prodromal period (DCL < 4) through the manifest period (DCL = 4). Third, estimate typical mean levels of rates of annual change in motor impairments for DCL categories. Fourth, compute estimated required sample sizes for a hypothetical clinical trial of efficacy examining change in TMS. Age and CAG length are important factors in motor onset and are incorporated into the methods used to address the goals.
Participants and Methods
Participants
Participants were N = 1010 individuals who voluntarily underwent genetic testing and were found to have a CAG repeat length > 36 (mean = 42.35, median = 42, minimum = 37, maximum = 62). A subset of N = 21 participants were diagnosed at study entry. Another subset of N = 204 “converters” received a diagnosis at some point after study entry.
Enrollment was at sites in the United States, Australia, the United Kingdom, Canada, Germany, and Spain. Institutional review boards at each participating site approved the study, and each participant signed an informed consent. At study enrollment, participants were required to be at least 18 years old. Exclusion criteria included history of a significant developmental cognitive disorder, other central nervous system disease or injury, evidence of an unstable medical or psychiatric illness (including substance abuse), a pacemaker or metallic implants, prescribed antipsychotic medication in the last 6 months, or phenothiazine derivative antiemetic medication in the last 3 months. For additional details, see Paulsen et al.2
Clinical Assessments
Participants underwent detailed motor, cognitive, psychiatric, and functional evaluations at baseline and annually thereafter. Participants had data for the TMS and DCL for at least one visit up to a maximum of 10 visits. There was a small amount of missing data (approximately 1%), consisting of unanswered TMS items, which were assigned a value of zero (“normal”). Preliminary analysis (not presented) indicated that missing values did not appear to cause outliers or other unexpected values. There were 88 motor examiners, and the mean number of examinations per examiner was 49.43 (standard deviation = 61.99); and the quartiles (Q) were Q1 = 2, Q2 = 16, and Q3 = 76. In addition, 60.83% of participants had the same rater throughout, 23.64% had two raters, and 15.53% had three or more raters. Findings did not change based on the number of raters or examinations. At study entry, the site coordinator responded to question 81 of the UHDRS: “Does the motor rater know the participant’s gene status? (0 = no, 1 = yes).” Gene mutation status was reported as be known (UHDRS question 81 = “yes”) for 69% of participants. The findings did not vary based on blinding.
Statistical Methods
The first goal of estimating the timing of the DCL categories was addressed by using a Weibull accelerated failure time (AFT) model.15 The same AFT model was used with four different events: first occurrence of DCL = 1, 2, 3, or 4. The log time to each DCL category was predicted separately, with age as the time metric and with CAG and CAG-squared as the predictors. CAG-squared was included because of evidence that the relation between time of onset and CAG is nonlinear.16,17 The outcome for each participant was treated as left-censored, right-censored, or interval-censored. The age at first occurrence for each category was the predicted value from the fitted model stratified by CAG, and the distance between the predicted values was computed for the different categories. The standard error of prediction was used to compute 95% confidence intervals (CIs).
The second aim of examining motor impairment trajectories was addressed using only the converters. For each converter, time in the study (in years) was anchored to the first occurrence of a DCL = 4 (diagnosis). This was accomplished by subtracting the year at first occurrence from each year (zero indicated the time of diagnosis). The trajectory of the TMS and motor factors was estimated using cubic splines in linear mixed effects regression (LMER).18,19 The TMS and each motor factor were modeled separately. Time predictors were cubic splines with the quartiles of time as the knots, and a random effect was included for each spline term plus the intercept. Age and CAG were incorporated into the analysis using the age-CAG product (CAP)20 as a predictor. The CAP is computed as CAP = (age at study entry) × (CAG – 33.66) and is similar to the burden score reported by Penney et al.21 The CAP is a purported index of the cumulative toxicity of mutant huntingtin at the time of study entry. For reference, CAP ≥ 368 denotes a “high” probability of converting in the near future after study entry, 290 < CAP < 368 denotes a “medium” probability, and CAP ≤ 290 denotes a “low” probability.
The third aim concerned estimating the level and slope of motor impairments for DCL categories. All gene-expanded participants were included, except for those with a baseline diagnosis (21 excluded patients). LMER was used again, with predictors being dummy variables for time-varying DCL categories and CAP. To help inform future clinical trials, the zero point of time was anchored to study entry, so that time represented years in the study. All possible interactions among CAP, the dummy variables, and time were included as predictors. Six correlated random effects were specified, one for each DCL category dummy variable and time. Because of the relevance for planning clinical trials of efficacy, emphasis was on the hypothesis test of zero slope. Efficacy trials require a detectible change in the untreated group to have the potential for demonstrating a treatment effect. Detectible change was defined as a slope significantly different from zero.
Finally, the required sample size for clinical trials of efficacy was computed based on LMER models. Single-arm sample size was computed for the test of equal TMS slopes for hypothetical placebo and treatment groups. Additional details are presented in the Appendix.
Results
Table 1 provides demographic information for the larger sample and subsamples of converters and non-converters. Converters had substantially higher mean CAP than non-converters.
TABLE 1.
Demographic information for the sample
| Converters, N = 204 | Non-converters, N = 806 | Combined, N = 1010 | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Variable | Mean | SD | Mean | SD | Mean | SD |
| Agea | 43.99 | 10.26 | 40.13 | 9.94 | 41.17 | 10.17 |
| Education | 14.14 | 2.59 | 14.58 | 2.63 | 14.46 | 2.63 |
| CAG repeat length | 43.24 | 2.78 | 42.01 | 2.30 | 42.34 | 2.50 |
| CAPb | 402.14 | 76.58 | 322.58 | 72.55 | 343.70 | 81.59 |
| Womenc | 0.66 | 0.47 | 0.61 | 0.49 | 0.63 | 0.48 |
The age indicated is the age at study entry.
Age-CAG product (CAP) is computed as: CAP = (age at study entry) × (CAG – 33.66).
Values indicate the proportion of women.
Abbreviations: SD, standard deviation; CAG, cytosine-adenine-guanine.
The results of the DCL timing analysis are illustrated in Figure 1, which depicts the predicted year before diagnosis of a DCL category (color-coded) as a function of CAG, with 95% CIs. Although the entire CAG distribution was used for the analysis, the quartiles of the gene-positive CAG distribution were used for graphing, which were Q1 = 41, Q2 = 42, and Q3 = 44. The horizontal axis indicates the years to diagnosis, with diagnosis occurring at year zero (marked by a solid vertical line). CAG-squared was statistically significant in the prediction of all DCL categories, but its effect was greatest for DCL = 4 (P < 0.001). For the median CAG = 42, DCL = 3 was predicted to occur 1.50 years before diagnosis (95% CI, 0.23-2.77), DCL = 2 was predicted to occur 6.75 years before diagnosis (95% CI, 5.62-7.88), and DCL = 1 was predicted to occur 19.74 years before diagnosis (95% CI, 18.65-20.83). Figure 1 reflects the significance of CAG on the timing of the categories. For CAG = 41, the predicted occurrence of the categories was further from diagnosis. For CAG = 44, the predicted occurrence of the categories was closer to diagnosis, and the 95% CI for DCL = 3 overlapped with DCL = 4 (the vertical line at zero is contained in the CI).
FIG. 1.
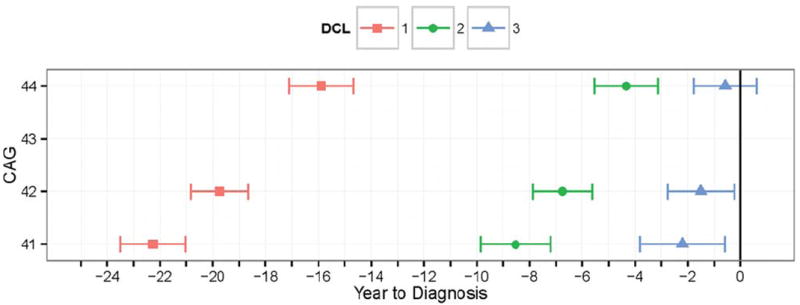
The predicted year of diagnostic confidence level (DCL) category occurrence is illustrated with 95% confidence interval as a function of cytosine-adenine-guanine (CAG) repeat length. A vertical line denotes the time of motor diagnosis (DCL=4).
Converter trajectories of motor impairments are indicated in Figure 2. Individual empirical curves are the jagged gray lines, and the fitted spline curves stratified by CAP are the smooth colored lines. The data thinned over time because the study is ongoing, and many of the individuals depicted in the graphs do not yet have many follow-up visits. The data thinning is reflected in the decreasing acceleration at the extreme right-hand side of each graph. Intercept (starting level) varied significantly by CAP for all motor outcomes (all P < 0.001). The motor trajectories were not statistically different by CAP strata for chorea and rigidity, but they were significantly different for the remaining variables in Figure 2 (all remaining P < 0.001). Although the entire CAP distribution was used in the analysis, curves for the quartiles of the converters were used for graphing (Q1 = 360, Q2 = 405, and Q3 = 450). Negative values on the horizontal axis indicate years to diagnosis, and positive values indicate years after diagnosis (0 = time of diagnosis). The top left graph depicts the TMS trajectory, and the remaining graphs illustrate the trajectories of the motor factors.
FIG. 2.
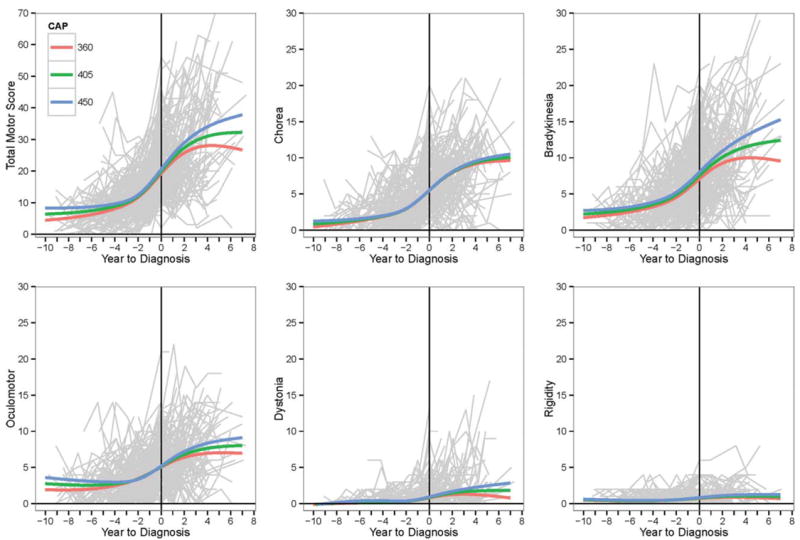
Trajectories of motor impairments for converters are illustrated as a function of age-CAG product (CAP). Light gray lines are the empirical trajectories of the participants, and colored lines are the fitted spline model curves. A vertical line denotes the time of motor diagnosis (diagnostic confidence level=4).
Table 2 lists the LMER results from the analysis examining change in motor impairments by CAP and DCL category. The results are presented only for TMS, but the motor factors had similar patterns. The column headed “Slope” in Table 2 lists the estimated mean rate of annual change for the TMS (along with the standard error). The column headed “Mean TMS at year 2” lists the predicted mean TMS after 2 years in the study. The 2-year point corresponds with the power analysis discussed in the Appendix. Although the entire CAP distribution was used in the analysis, Table 2 shows results only for limited values, including the quartiles (Q1 = 285, Q2 = 350, and Q3 = 400). The rate of change was statistically greater than zero for DCL = 3 or 4, regardless of CAP. CAP = 290 was the minimum that produced slope significance for DCL = 2 at the P < 0.05 level, and CAP = 310 was the minimum for the P < 0.01 level. As indicated in the “Mean TMS at year 2” column, all mean estimates were statistically greater than zero at the P < 0.001 level.
TABLE 2.
Linear mixed effects regression results: total motor score slopes and mean at year 2 by age-CAG product and diagnostic confidence level
| Mean (standard error) | |||
|---|---|---|---|
|
| |||
| CAP | DCL | Slope | Mean TMS at year 2 |
| 285 | 0 | 0.0381 (0.0485) | 1.0632 (0.1023)a |
| 285 | 1 | 0.0919 (0.0494) | 4.8179 (0.135)a |
| 285 | 2 | 0.1638 (0.0902) | 8.4232 (0.3209)a |
| 285 | 3 | 0.5396 (0.1567)a | 11.213 (0.6121)a |
| 285 | 4 | 1.227 (0.1897)a | 13.4854 (1.1873)a |
|
| |||
| 290 | 0 | 0.0414 (0.0475) | 1.088 (0.1012)a |
| 290 | 1 | 0.0894 (0.0479) | 4.8739 (0.1315)a |
| 290 | 2 | 0.1732 (0.0871)b | 8.5255 (0.3112)a |
| 290 | 3 | 0.5542 (0.1517)a | 11.3388 (0.5938)a |
| 290 | 4 | 1.2811 (0.1848)a | 13.5992 (1.1537)a |
|
| |||
| 310 | 0 | 0.0544 (0.0459) | 1.1876 (0.0996)a |
| 310 | 1 | 0.0793 (0.0435) | 5.0978 (0.1205)a |
| 310 | 2 | 0.211 (0.0762)c | 8.9347 (0.2763)a |
| 310 | 3 | 0.6129 (0.1329)a | 11.8418 (0.5244)a |
| 310 | 4 | 1.4974 (0.1655)a | 14.0545 (1.0242)a |
|
| |||
| 350 | 0 | 0.0806 (0.0529) | 1.3867 (0.1114)a |
| 350 | 1 | 0.0593 (0.0443) | 5.5457 (0.1169)a |
| 350 | 2 | 0.2865 (0.0663)a | 9.753 (0.234)a |
| 350 | 3 | 0.7302 (0.105)a | 12.848 (0.4117)a |
| 350 | 4 | 1.9301 (0.1321)a | 14.9651 (0.8016)a |
|
| |||
| 400 | 0 | 0.1133 (0.0745) | 1.6356 (0.1462)a |
| 400 | 1 | 0.0342 (0.0607) | 6.1055 (0.1462)a |
| 400 | 2 | 0.3809 (0.0817)a | 10.776 (0.2531)a |
| 400 | 3 | 0.8768 (0.1014)a | 14.1056 (0.3621)a |
| 400 | 4 | 2.4709 (0.108)a | 16.1032 (0.6554)a |
P < 0.001.
P < 0.01.
P < 0.05.
Abbreviations: TMS, total motor score; CAP, age-CAG product; DCL, diagnostic confidence level.
Table 3 indicates the estimated required sample size (in boldface) for a hypothetical 2-year clinical trial of efficacy with measurements every 6 months. The single-arm sizes are for testing the null hypothesis of equal TMS rate of change over time for placebo and treatment groups (see Appendix). Minimum CAP indicates sampling all available participants with CAP ≥ minimum CAP, and dropout refers to the rate in each group (see Appendix).
TABLE 3.
Required sample size (boldface) for a hypothetical clinical trial of efficacya
| Parameter Estimates | Effect Size | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Dropout, % | Min CAP | Type I error, % | βS | G11 | G12 | G22 | σ2e | 30% | 40% | 50% | 60% | 70% |
| 0 | 290 | 5 | 1.56 | 21.40 | 4.45 | 3.20 | 16.54 | 552 | 310 | 199 | 138 | 101 |
| 0 | 350 | 5 | 2.04 | 25.96 | 5.61 | 3.67 | 19.39 | 377 | 212 | 136 | 94 | 69 |
| 0 | 400 | 5 | 2.81 | 32.11 | 6.56 | 5.26 | 24.63 | 263 | 148 | 95 | 66 | 48 |
| 0 | 290 | 10 | 1.56 | 21.40 | 4.45 | 3.20 | 16.54 | 402 | 226 | 145 | 101 | 74 |
| 0 | 350 | 10 | 2.04 | 25.96 | 5.61 | 3.67 | 19.39 | 275 | 155 | 99 | 69 | 51 |
| 0 | 400 | 10 | 2.81 | 32.11 | 6.56 | 5.26 | 24.63 | 192 | 108 | 69 | 48 | 35 |
|
| ||||||||||||
| 10 | 290 | 5 | 1.56 | 21.40 | 4.45 | 3.20 | 16.54 | 591 | 332 | 213 | 148 | 109 |
| 10 | 350 | 5 | 2.04 | 25.96 | 5.61 | 3.67 | 19.39 | 404 | 227 | 145 | 101 | 74 |
| 10 | 400 | 5 | 2.81 | 32.11 | 6.56 | 5.26 | 24.63 | 281 | 158 | 101 | 70 | 52 |
| 10 | 290 | 10 | 1.56 | 21.40 | 4.45 | 3.20 | 16.54 | 431 | 242 | 155 | 108 | 79 |
| 10 | 350 | 10 | 2.04 | 25.96 | 5.61 | 3.67 | 19.39 | 295 | 166 | 106 | 74 | 54 |
| 10 | 400 | 10 | 2.81 | 32.11 | 6.56 | 5.26 | 24.63 | 205 | 115 | 74 | 51 | 38 |
|
| ||||||||||||
| 20 | 290 | 5 | 1.56 | 21.40 | 4.45 | 3.20 | 16.54 | 636 | 358 | 229 | 159 | 117 |
| 20 | 350 | 5 | 2.04 | 25.96 | 5.61 | 3.67 | 19.39 | 435 | 245 | 157 | 109 | 80 |
| 20 | 400 | 5 | 2.81 | 32.11 | 6.56 | 5.26 | 24.63 | 303 | 170 | 109 | 76 | 56 |
| 20 | 290 | 10 | 1.56 | 21.40 | 4.45 | 3.20 | 16.54 | 464 | 261 | 167 | 116 | 85 |
| 20 | 350 | 10 | 2.04 | 25.96 | 5.61 | 3.67 | 19.39 | 317 | 178 | 114 | 79 | 58 |
| 20 | 400 | 10 | 2.81 | 32.11 | 6.56 | 5.26 | 24.63 | 221 | 124 | 79 | 55 | 41 |
The single-arm sample size is for a treatment–placebo slope difference analyzed with linear mixed effects regression. Sample size is listed as a function of percentage of dropout, minimum CAP, Type I error rate, estimated parameters, and effect size. Details are provided in the Appendix.
Abbreviations: Min, minimum; CAP, age-CAG product.
Discussion
The results of this analysis provide information concerning the timing of DCL categories in HD progression and the nature of change in motor impairments. There is evidence the DCL and the UHDRS motor items can detect changes in motor impairments during the HD prodromal period. Age and CAG repeat length were significant both for the timing of DCL categories and for change in motor impairments. Greater CAG length was associated with more rapid DCL progression, and higher CAP was associated with faster change in motor impairments.
Trajectories of motor impairments were characterized by nonlinear change in the prodromal phase (see Fig. 2). The more targeted analysis of TMS change while in the same DCL category provides an indication of how annual rates of change in motor impairments vary by DCL and CAP (see Table 2). There was no significant change associated with DCL = 0 (normal) or DCL = 1 (nonspecific motor abnormalities) regardless of CAP. DCL = 2 (possible signs of HD; 50%-89% confidence) showed significance for a minimum CAP = 290 or 300, depending on the desired level of significance. DCL = 3 (likely signs of HD; 90%-98% confidence) and DCL = 4 (unequivocal signs of HD; ≥ 99% confidence) showed significance regardless of CAP, suggesting that a clinical examination with corroborative history of HD may be a sufficient entry criterion.
The finding that motor impairment slopes increased with DCL provides evidence that the stage of disease is related to the rate of progression, even during prodromal HD. The pattern of progression for our results is consistent with a long-term slow build-up of impairments that ultimately gives rise to an accelerated trajectory of deterioration that varies by CAP (see Fig. 2). This pattern has been verified in other cohorts of HD patients.5,10,11,13,22 For example, Mahant and colleagues observed that motor impairments did not increase at a constant rate in their longitudinal study of diagnosed patients with early through late HD.12 Individuals were stratified based on initial TMS, and it was found that the rate of yearly TMS change increased with initial TMS for ranges of values similar to those observed in our study (ie, TMS ∈ [0,77]). Similar results have been found using CAG length rather than TMS,23-25 and there is evidence that other domains, such as cognitive impairments, also may change as a function of stage.26
The results have implications for the classification of progression at study entry. In HD research, it is common to use DCL = 4 as the definition of diagnosis or manifest HD. By complement, DCL < 4 defines the prodromal, pre-diagnosis, or pre-manifest phase. In some studies, a different DCL cutoff is used; for example, DCL < 2 for pre-manifest HD and DCL ≥ 2 for manifest HD.27 In other studies, several values are used to define multiple groups, such as manifest (DCL = 4), pre-manifest with abnormalities (DCL = 2 or 3), and pre-manifest with minimal signs (DCL = 0 or 1).26,28,29
If the goal of classification is to distinguish individuals who are actively deteriorating from those who are not, then our results suggest that DCL = 4 is assigned relatively late. Figure 2 and Table 2 provide evidence of active and meaningful motor decline for DCL = 3 and perhaps even for DCL = 2, provided the CAP is sufficiently large. Cutoffs for phases of activity might be DCL ≤ 2 versus DCL ≥ 3 when the entire CAP range is considered, or DCL ≤ 1 versus DCL ≥ 2 when CAP ≥ 290 is considered (see Table 2).
The findings have implications for the planning of clinical trials of efficacy, with the outcome variable being motor impairments and the TMS in particular. The slope results from Table 2 indicate a possible minimum CAP that might be considered should a researcher want to recruit patients with the greatest likelihood of active motor decline. Recruitment based on DCL might be problematic because of the unreliability of the rating at the individual level. In ancillary analysis (results not presented), we have observed that progression through the DCL categories is not necessarily monotonic, with some patients even dropping over time after an initial value of DCL = 4. In contrast, the CAP can be highly reliable provided that age is accurately reported and CAG length is laboratory-verified.20 Therefore, we recommend using CAP as the primary consideration in recruitment. Table 3 is a resource for planning clinical trials of efficacy if the CAP of recruits can be computed. Sample sizes are provided for comparing the rate of change (slope) of the treatment and placebo groups under various non-informative dropout scenarios. Graphical evidence (not presented) suggests that non-informative dropout is a reasonable assumption for the planning of a clinical trial based on the PREDICT-HD database.30
Finally, the sensitivity to change of the TMS has been questioned for use with prodromal individuals.31,32 Our results indicate that the TMS is sensitive in the prodromal period in that it can detect statistically and substantively significant change.
Acknowledgments
Funding agencies: This research is supported by the National Institutes of Health (NIH), the National Institute of Neurological Disorders and Stroke (NS040068), the CHDI Foundation, Inc. (A3917), Cognitive and Functional Brain Changes in Preclinical Huntington’s Disease (HD) (5R01NS054893), 4D Shape Analysis for Modeling Spatiotemporal Change Trajectories in Huntington’s (1U01NS082086), Functional Connectivity in Pre-manifest Huntington’s Disease (1U01NS082083), and Basal Ganglia Shape Analysis and Circuitry in Huntington’s Disease (1U01NS082085). The publication was supported by the National Center for Advancing Translational Sciences and the NIH through grant 2 UL1 TR000442-06. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Appendix
Required sample size for slope difference
Estimates of sample size required for a randomized clinical trial of efficacy were based on power formulas for LMER33,34. Sample size was calculated for the endpoint of TMS rate of change (slope) over a 2 year study with observations every 6 months (baseline, 6, 12, 28, and 24 months). The design was a two-arm Placebo (P) and Treatment (T) group study, and dropout was assumed to be non-informative (or ignorable). The evaluation of efficacy is defined as the test of the null hypothesis that the TMS rates of change in the P and T groups are equal. The null hypothesis can be evaluated based on parameter estimates in LMER.
Suppose Yij is the TMS value for the ith participant (i = 1, …, N) at the jth month (j = 1, …, ni), and tij denotes time in months. Assuming linear change over time, the LMER model can be written as the following,
| (A1) |
where gi is a dummy variable for group (gi = 1 if the participant is in the T group, and 0 otherwise). In Equation A1, the βs are the fixed effects, with βI being the P group intercept and βS being the P group slope; βΔI is the difference in the intercepts among the groups, and βΔI is the difference among the slopes. The bs are random effects, and e is random error. We make the typical assumptions, . The prime object of inference is βΔS, as this is the difference in TMS longitudinal change of the T and P groups. The null hypothesis of equality of T and P slopes is H0: βΔS = 0, and can be evaluated with a Z-test. The Z statistic is and leads to the rejection of H0 when |Z| > Z1-α, with the latter being the 100(1-α)th quantile of the standard normal distribution (single tailed).
The LMER model of A1 can be written more generally as
where Xi is the design matrix of the fixed effects, β is the vector of fixed effects, Zi is the design matrix of the random effects, b is the vector of random effects, and ei is the vector of random errors. In this context, Xi for a person with no dropout has dimensions 5 × 4 with the first column being a vector of 1s, the second column a vector of time values (0, 6, 12, 18, 24), the third column a vector of gi values, and the last column a vector of gi × time values. An individual is defined as a dropout if their row dimension is less than 5.
TABLE A1.
Dropout proportions (π) for computing required sample size.
| Dropout Condition | |||
|---|---|---|---|
|
| |||
| Missing Visits | 0% | 10% | 20% |
| 0 | 1.00 | 0.90 | 0.80 |
| 1 | 0 | 0.04 | 0.08 |
| 2 | 0 | 0.03 | 0.06 |
| 3 | 0 | 0.02 | 0.04 |
| 4 | 0 | 0.01 | 0.02 |
The required sample size for the Z-test depends on the variance of the responses,
Vi is 5 × 5 for no dropout, but has reduced dimension under dropout similar to Xi. Suppose α is the type I error rate (set to 0.05 or 0.10), and 1 − γ is power (set to 0.80). Then the required sample size for a single arm (half the total sample size) is
| (A2) |
where l refers to a specific dropout pattern, Xkl is the design matrix for the kth group with the lth dropout pattern, and πkl is the proportion of participants with the lth missing pattern in the kth group, and the πkl sum to one among the dropout patterns within a group. The 4,4 subscript indicates the element in the 4th row and 4th column of the resulting matrix. When there is no dropout, π11 = π21 = 1, and the right hand expression in the numerator is , where the subscripts 1 and 2 represent the P and T groups.
Three dropout scenarios were considered: no dropout (0%), 10%, and 20%. The values pertain to the percentage of participants who dropped out in each group, not the total missing data. Dropout was identical for the P and T groups (π1l = π2l = πl), and the dropout proportions, πl, tapered to create a scenario in which dropout was slight very earlier in the hypothetical study, but increased over time. Five dropout patterns were considered, no dropout, missing last visit, missing last two visits, missing last three visit, and missing last four visits. Table A1 shows the πl used for the three dropout conditions.
The variance components of Equation A2 can be estimated using the PREDICT-HD database. However, PREDICT-HD is not a treatment study and all geneexpanded patients are considered as members of the untreated placebo group in this context (not to be confused with gene-negative patients), with mean slope βS. The object of inference, βΔS, can be estimated as the proportion reduction of the placebo group slope under a hypothetical treatment, expressed as βΔS = ψβS, where ψ is the proportion reduction in the placebo slope. Sample size was estimated for ψ = 0.30, 0.40, 0.50, 0.60, 0.70. The last value, representing a 70% improvement by treatment, was the approximate percentage group difference in change over 12 weeks in a HD randomized clinical trial of tetrabenazine,34,35 which is one of the few studies to show statistically significant efficacy.36 The range of effects is also consistent with clinical trials of other neurodegenerative diseases, such as Multiple Sclerosis.37 Table 3 presents parameter estimate for βS, the unique elements of G (g11, g12, g22), and σ2e. These values can be used along with different design matrices, dropout proportions, etc., to provide estimates for a wide variety of clinical trial scenarios.
We assumed a single-tailed statistical test, 80% power, and a 2-year study with measurements taken at baseline and every half-year, t = 0, 6, 12, 18, 24 months. The variance components were estimated using the PREDICT-HD participants in the range of CAP = [minimum CAP, 846], with the latter value being the maximum value in the sample. Different dropout scenarios, including differential dropout by group, can be created by altering the design matrices of Equation A2.
PREDICT-HD Investigators, Coordinators, Motor Raters, Cognitive Raters
Stephen Cross, Patricia Ryan, Eric A. Epping, and Stacie Vik (University of Iowa, Iowa City, Iowa, USA); Edmond Chiu, Joy Preston, Anita Goh, Stephanie Antonopoulos, and Samantha Loi (St. Vincent’s Hospital, The University of Melbourne, Kew, Victoria, Australia); Phyllis Chua and Angela Komiti (The University of Melbourne, Royal Melbourne Hospital, Melbourne, Australia); Lynn Raymond, Joji Decolongon, Mannie Fan, and Allison Coleman (University of British Columbia, Vancouver, British Columbia, Canada); Christopher A. Ross, Mark Varvaris, and Nadine Yoritomo (John Hopkins University, Baltimore, Maryland, USA); William M. Mallonee and Greg Suter (Hereditary Neurological Disease Centre, Wichita, Kansas, USA); Ali Samii and Alma Macaraeg (University of Washington and VA Puget Sound Health Care System, Seattle, Washington, USA); Randi Jones, Cathy Wood-Siverio, and Stewart A. Factor (Emory University School of Medicine, Atlanta, Georgia, USA); Roger A. Barker, Sarah Mason, and Natalie Valle Guzman (Cambridge Centre for Brain Repair, Cambridge, UK); Elizabeth McCusker, Jane Griffith, Clement Loy, and David Gunn (Westmead Hospital, Sydney, Australia); Michael Orth, Sigurd Süβmuth, Katrin Barth, Sonja Trautmann, Daniela Schwenk, and Carolin Eschenbach (University of Ulm, Ulm, Germany); Kimberly Quaid, Melissa Wesson, and Joanne Wojcieszek (Indiana University School of Medicine, Indianapolis, IN); Mark Guttman, Alanna Sheinberg, and Albie Law (Centre for Addiction and Mental Health, University of Toronto, Markham, Ontario, Canada); Susan Perlman and Brian Clemente (UCLA Medical Center, Los Angeles, California, USA); Michael D. Geschwind, Sharon Sha, and Gabriela Satris (University of California San Francisco, California, USA); Tom Warner and Maggie Burrows (National Hospital for Neurology and Neurosurgery, London, UK); Anne Rosser, Kathy Price, and Sarah Hunt (Cardiff University, Cardiff, Wales, UK); Frederick Marshall, Amy Chesire, Mary Wodarski, and Charlyne Hickey (University of Rochester, Rochester, New York, USA); Peter Panegyres, Joseph Lee, Maria Tedesco, and Brenton Maxwell (Neurosciences Unit, Graylands, Selby-Lemnos & Special Care Health Services, Perth, Australia); Joel Perlmutter, Stacey Barton, and Shineeka Smith (Washington University, St. Louis, Missouri, USA); Zosia Miedzybrodzka, Daniela Rae, and Mariella D’Alessandro (Clinical Genetics Centre, Aberdeen, Scotland, UK); David Craufurd, Judith Bek, and Elizabeth Howard (University of Manchester, Manchester, UK); Pietro Mazzoni, Karen Marder, and Paula Wasserman (Columbia University Medical Center, New York, New York, USA); Rajeev Kumar, Diane Erickson, and Breanna Nickels (Colorado Neurological Institute, Englewood, Colorado, USA); Vicki Wheelock, Lisa Kjer, Amanda Martin, and Sarah Farias (University of California Davis, Sacramento, California, USA); Wayne Martin, Pamela King, Marguerite Wieler, and Satwinder Sran (University of Alberta, Edmonton, Alberta, Canada); and Oksana Suchowersky, Anwar Ahmed, Stephen Rao, Christine Reece, Alex Bura, Lyla Mourany, and Jagan Pallai (Cleveland Clinic Foundation, Cleveland, Ohio, USA).
Executive Committee
Jane S. Paulsen, Principal Investigator; Eric A. Epping, Jeffrey D. Long, Hans J. Johnson, Jeremy H. Bockholt, and Kelsey Montross.
Scientific Consultants
Brain: Jean Paul Vonsattel and Carol Moskowitz (Columbia University Medical Center).
Cognitive: Deborah Harrington (University of California, San Diego); Tamara Hershey (Washington University); Holly Westervelt (Rhode Island Hospital/Alpert Medical School of Brown University); Megan M. Smith, and David J. Moser (University of Iowa).
Functional: Janet Williams and Nancy Downing (University of Iowa).
Imaging: Hans J. Johnson (University of Iowa); Elizabeth Aylward (Seattle Children’s Research Institute); Christopher A. Ross (Johns Hopkins University); Vincent A. Magnotta (University of Iowa); and Stephen Rao (Cleveland Clinic, Cleveland, OH.).
Psychiatric: Eric A. Epping (University of Iowa); David Craufurd (University of Manchester).
Core Sections
Biostatistics: Jeffrey D. Long, Ji-In Kim, James A. Mills, Ying Zhang, Dawei Liu, Wenjing Lu, and Spencer Lourens (University of Iowa).
Ethics: Cheryl Erwin (McGovern Center for Health, Humanities and the Human Spirit); Eric A. Epping and Janet Williams (University of Iowa); Martha Nance (University of Minnesota).
Biomedical Informatics: Jeremy H. Bockholt and Ryan Wyse (University of Iowa).
Footnotes
Relevant conflicts of interest: Nothing to report.
financial disclosures
Full financial disclosures and author roles may be found in the online version of this article.
References
- Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington’s disease decades before diagnosis: the PREDICT-HD Study. J Neurol Neurosurg Psychiatry. 2008;79:874–880. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonelli RM, Hofmann P. A systematic review of the treatment studies in Huntington’s disease since 1990. Expert Opin Pharmacother. 2007;8:141–153. doi: 10.1517/14656566.8.2.141. [DOI] [PubMed] [Google Scholar]
- Armstrong MJ, Miyasaki JM. Evidence-based guideline: pharmacologic treatment of chorea in Huntington disease: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2012;79:597–603. doi: 10.1212/WNL.0b013e318263c443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington Study Group. Unified Huntington’s Disease Rating Scale: reliability and-consistency. Mov Disord. 1996;11:136–142. doi: 10.1002/mds.870110204. [DOI] [PubMed] [Google Scholar]
- Marder K, Zhao H, Myers RH, et al. Rate of functional decline in Huntington’s disease. Neurology. 2000;54:452–458. doi: 10.1212/wnl.54.2.452. [DOI] [PubMed] [Google Scholar]
- Puri BK, Leavitt BR, Hayden MR, et al. Ethyl-epa in Huntington disease: a double-blind, randomized, placebo-controlled trial. Neurology. 2005;65:286–292. doi: 10.1212/01.wnl.0000169025.09670.6d. [DOI] [PubMed] [Google Scholar]
- Solomon AC, Stout JC, Weaver M, et al. Ten year rate of longitudinal change in neurocognitive and motor function in prediagnosis Huntington disease. Mov Disord. 2008;23:1830–1836. doi: 10.1002/mds.22097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witjes-Ane MN, Mertens B, van Vugt JP, Bachoud-Levi AC, van Ommen GJ, Roos RA. Longitudinal evaluation of “presymptomatic” carriers of Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2007;19:310–317. doi: 10.1176/jnp.2007.19.3.310. [DOI] [PubMed] [Google Scholar]
- Meyer C, Landwehrmeyer B, Schwenke C, et al. Rate of change in early Huntington’s disease: a clinicometric analysis. Mov Disord. 2012;27:118–124. doi: 10.1002/mds.23847. [DOI] [PubMed] [Google Scholar]
- Kirkwood SC, Su JL, Conneally P, et al. Progression of symptoms in the early and middle stages of Huntington disease. Arch Neurol. 2001;58:273–278. doi: 10.1001/archneur.58.2.273. [DOI] [PubMed] [Google Scholar]
- Mahant N, McCusker EA, Byth K, et al. Huntington’s disease: clinical correlates of disability and progression. Neurology. 2003;61:1085–1092. doi: 10.1212/01.wnl.0000086373.32347.16. [DOI] [PubMed] [Google Scholar]
- Siesling S, van Vugt JP, Zwinderman KA, et al. Unified Huntington’s Disease Rating Scale: a follow-up. Mov Disord. 1998;13:915– 919. doi: 10.1002/mds.870130609. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Hayden M, Stout JC, et al. Preparing for preventive clinical trials: the PREDICT-HD Study. Arch Neurol. 2006;63:883– 890. doi: 10.1001/archneur.63.6.883. [DOI] [PubMed] [Google Scholar]
- Kalbfleisch JD, Prentice RL. The Statistical Analysis of Failure Time Data. 2. Hoboken, NJ: John Wiley & Sons, Inc.; 2002. [Google Scholar]
- Langbehn DR, Hayden MR, Paulsen JS, et al. CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:397–408. doi: 10.1002/ajmg.b.30992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Ramos EM, Lee JH, et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012;78:690–695. doi: 10.1212/WNL.0b013e318249f683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro JC, Bates DM. Mixed-Effects Models in S and S-Plus. New York: Springer; 2000. [Google Scholar]
- Long JD. Longitudinal Data Analysis for the Behavioral Sciences Using R. Thousand Oaks, CA: Sage Publications Inc.; 2012. [Google Scholar]
- Zhang Y, Long JD, Mills JA, et al. Indexing disease progression at study entry with individuals at-risk for Huntington disease. Am J Med Genet B Neuropsychiatr Genet. 2011;156:751–763. doi: 10.1002/ajmg.b.31232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penney JJB, Vonsattel JP, MacDonald ME, et al. CAG repeat number governs the development rate of pathology in Huntington’s disease. Ann Neurol. 1997;41:689–692. doi: 10.1002/ana.410410521. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Reilmann R, Roos RAC, et al. Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the Track-HD study: analysis of 24 month observational data. Lancet Neurol. 2012;11:42–53. doi: 10.1016/S1474-4422(11)70263-0. [DOI] [PubMed] [Google Scholar]
- Rosenblatt A, Liang KY, Zhou H, et al. The association of CAG repeat length with clinical progression in Huntington disease. Neurology. 2006;66:1016–1020. doi: 10.1212/01.wnl.0000204230.16619.d9. [DOI] [PubMed] [Google Scholar]
- Rosenblatt A, Kumar BV, Mo A, et al. Age, CAG repeat length, and clinical progression in Huntington’s disease. Mov Disord. 2012;27:272–276. doi: 10.1002/mds.24024. [DOI] [PubMed] [Google Scholar]
- Ravina B, Romer M, Constantinescu R, et al. The relationship between CAG repeat length and clinical progression in Huntington’s disease. Mov Disord. 2008;23:1223–1227. doi: 10.1002/mds.21988. [DOI] [PubMed] [Google Scholar]
- Stout JC, Weaver M, Solomon AC, et al. Are cognitive changes progressive in prediagnostic HD? Cogn Behav Neurol. 2007;20:212–218. doi: 10.1097/WNN.0b013e31815cfef8. [DOI] [PubMed] [Google Scholar]
- Reedeker N, Mast RCVD, Giltay EJ, et al. Hypokinesia in Huntington’s disease co-occurs with cognitive and global dysfunctioning. Mov Disord. 2010;25:1612–1618. doi: 10.1002/mds.23136. [DOI] [PubMed] [Google Scholar]
- Blekher T, Johnson SA, Marshall J, et al. Saccades in presymptomatic and early stages of Huntington disease. Neurology. 2006;67:394–399. doi: 10.1212/01.wnl.0000227890.87398.c1. [DOI] [PubMed] [Google Scholar]
- Marshall J, White K, Weaver M, et al. Specific psychiatric manifestations among preclinical Huntington disease mutation carriers. Arch Neurol. 2007;64:116–121. doi: 10.1001/archneur.64.1.116. [DOI] [PubMed] [Google Scholar]
- Hu C, Sale ME. A joint model for nonlinear longitudinal data with informative dropout. J Pharmacokinet Pharmacodyn. 2002;30:83–103. doi: 10.1023/a:1023249510224. [DOI] [PubMed] [Google Scholar]
- Bechtel N, Scahill RI, Rosas HD, et al. Tapping linked to function and structure in premanifest and symptomatic Huntington disease. Neurology. 2010;75:2150–2160. doi: 10.1212/WNL.0b013e3182020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino AL, Anderson K, Borowsky B, et al. An item response analysis of the motor and behavioral subscales of the Unified Huntington’s Disease Rating Scale in Huntington disease gene expansion carriers. Mov Disord. 2011;26:877–884. doi: 10.1002/mds.23574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallbraith S, Marschner IC. Guidelines for the design of clinical trials with longitudinal outcomes. Control Clin Trials. 2002;23(3):257–273. doi: 10.1016/s0197-2456(02)00205-2. [DOI] [PubMed] [Google Scholar]
- Yi Q, Panzarella T. Estimating sample size for tests on trends across repeated measurements with missing data based on the interaction term in a mixed model. Control Clin Trials. 2002;23:481–496. doi: 10.1016/s0197-2456(02)00223-4. [DOI] [PubMed] [Google Scholar]
- Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2006;66:366–372. doi: 10.1212/01.wnl.0000198586.85250.13. [DOI] [PubMed] [Google Scholar]
- Armstrong MJ, Miyasaki JM. American Academy of Neurololgy. Evidence-based guideline: pharmacologic treatment of chorea in Huntington disease: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2012;79:597–603. doi: 10.1212/WNL.0b013e318263c443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann DR, Jasperse B, Barkhof F, et al. Sample sizes for brain atrophy outcomes in trials for secondary progressive multiple sclerosis. Neurology. 2009;72:595–601. doi: 10.1212/01.wnl.0000335765.55346.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]