

Abstract
Recently, we demonstrated the utility of optical fluorometry to detect a change in the redox status of mitochondrial autofluorescent coenzymes nicotinamide adenine dinucleotide (NADH) and oxidized form of flavin adenine dinucleotide 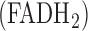 (FAD), as a measure of mitochondrial function in isolated perfused rat lungs (IPL). The objective of this paper was to utilize optical fluorometry to evaluate the effect of rat exposure to hyperoxia (
(FAD), as a measure of mitochondrial function in isolated perfused rat lungs (IPL). The objective of this paper was to utilize optical fluorometry to evaluate the effect of rat exposure to hyperoxia (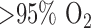 for 48 h) on lung tissue mitochondrial redox status of NADH and FAD in a nondestructive manner in IPL. Surface NADH and FAD signals were measured before and after lung perfusion with perfusate containing rotenone (ROT, complex I inhibitor), potassium cyanide (KCN, complex IV inhibitor), and/or pentachlorophenol (PCP, uncoupler). ROT- or KCN-induced increase in NADH signal is considered a measure of complex I activity, and KCN-induced decrease in FAD signal is considered a measure of complex II activity. The results show that hyperoxia decreased complex I and II activities by 63% and 55%, respectively, when compared to lungs of rats exposed to room air (normoxic rats). Mitochondrial complex I and II activities in lung homogenates were also lower (77% and 63%, respectively) for hyperoxic than for normoxic lungs. These results suggest that the mitochondrial matrix is more reduced in hyperoxic lungs than in normoxic lungs, and demonstrate the ability of optical fluorometry to detect a change in mitochondrial redox state of hyperoxic lungs prior to histological changes characteristic of hyperoxia.
for 48 h) on lung tissue mitochondrial redox status of NADH and FAD in a nondestructive manner in IPL. Surface NADH and FAD signals were measured before and after lung perfusion with perfusate containing rotenone (ROT, complex I inhibitor), potassium cyanide (KCN, complex IV inhibitor), and/or pentachlorophenol (PCP, uncoupler). ROT- or KCN-induced increase in NADH signal is considered a measure of complex I activity, and KCN-induced decrease in FAD signal is considered a measure of complex II activity. The results show that hyperoxia decreased complex I and II activities by 63% and 55%, respectively, when compared to lungs of rats exposed to room air (normoxic rats). Mitochondrial complex I and II activities in lung homogenates were also lower (77% and 63%, respectively) for hyperoxic than for normoxic lungs. These results suggest that the mitochondrial matrix is more reduced in hyperoxic lungs than in normoxic lungs, and demonstrate the ability of optical fluorometry to detect a change in mitochondrial redox state of hyperoxic lungs prior to histological changes characteristic of hyperoxia.
Keywords: NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), flavin adenine dinucleotide 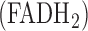 , Nicotinamide Adenine Dinucleotide (NADH), lung surface fluorometry, mitochondrial redox
, Nicotinamide Adenine Dinucleotide (NADH), lung surface fluorometry, mitochondrial redox
I. Introduction
High oxygen  therapy (hyperoxia) is a necessary treatment of low blood
therapy (hyperoxia) is a necessary treatment of low blood 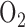 in adult and pediatric patients with acute lung injury (ALI) [1]–[3]. This treatment is effective in restoring blood
in adult and pediatric patients with acute lung injury (ALI) [1]–[3]. This treatment is effective in restoring blood 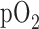 to a level which sustains vital organ metabolic requirements. However, prolonged exposure to high
to a level which sustains vital organ metabolic requirements. However, prolonged exposure to high 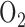 concentrations
concentrations 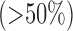 causes lung injury [3]–[7]. Further complicating this situation is the fact that the time frame over which hyperoxic lung injury develops is difficult to predict due to the wide variation between patient tolerance/susceptibility [8]. Thus, a minimally invasive method to detect pulmonary injury in an individual patient exposed to high fractions of
causes lung injury [3]–[7]. Further complicating this situation is the fact that the time frame over which hyperoxic lung injury develops is difficult to predict due to the wide variation between patient tolerance/susceptibility [8]. Thus, a minimally invasive method to detect pulmonary injury in an individual patient exposed to high fractions of 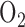 in real time is highly desirable.
in real time is highly desirable.
Rat exposure to 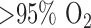 is a well-documented model of hyperoxic lung injury and human ALI [5], [8]–[10]. Previous studies have suggested that mitochondrial dysfunction is a cardinal feature of hyperoxic lung injury [11]–[16]. Although much work has been done in cell cultures and tissue homogenates, studies probing key tissue mitochondrial functions and the effect of oxidant injury in intact lungs in real-time are limited [11], [12], [17]. Because indices of mitochondrial function of in situ tissue are often different than those of tissue homogenates, measurements of indices of oxidative phosphorylation in intact tissue for comparison to those of isolated mitochondria are important.
is a well-documented model of hyperoxic lung injury and human ALI [5], [8]–[10]. Previous studies have suggested that mitochondrial dysfunction is a cardinal feature of hyperoxic lung injury [11]–[16]. Although much work has been done in cell cultures and tissue homogenates, studies probing key tissue mitochondrial functions and the effect of oxidant injury in intact lungs in real-time are limited [11], [12], [17]. Because indices of mitochondrial function of in situ tissue are often different than those of tissue homogenates, measurements of indices of oxidative phosphorylation in intact tissue for comparison to those of isolated mitochondria are important.
Recently, we demonstrated the utility of optical fluorometry (Fig. 1) to detect a change in the redox status of lung mitochondrial autofluorescent coenzymes NADH (Nicotinamide Adenine Dinucleotide) and FAD (oxidized form of Flavin Adenine Dinucleotide 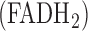 ), in isolated perfused rat lungs [18]. NADH and
), in isolated perfused rat lungs [18]. NADH and 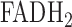 (Flavin Adenine Dinucleotide) are mitochondrial metabolic coenzymes, and are the primary electron carriers in oxidative phosphorylation. The oxidation of these two via the mitochondrial electron transport chain involves the transport of protons from mitochondrial complexes I, III, and IV into the mitochondrial intermembrane space (Fig. 2). This creates a proton gradient, which, along with the presence of adenosine diphosphate (ADP), yields the production of the cell's basic unit of energy, adenosine triphosphate (ATP). This process accounts for approximately 85% of ATP production in lung tissue [19]. Therefore, a change in the redox state of the electron transport chain, and thus NADH and
(Flavin Adenine Dinucleotide) are mitochondrial metabolic coenzymes, and are the primary electron carriers in oxidative phosphorylation. The oxidation of these two via the mitochondrial electron transport chain involves the transport of protons from mitochondrial complexes I, III, and IV into the mitochondrial intermembrane space (Fig. 2). This creates a proton gradient, which, along with the presence of adenosine diphosphate (ADP), yields the production of the cell's basic unit of energy, adenosine triphosphate (ATP). This process accounts for approximately 85% of ATP production in lung tissue [19]. Therefore, a change in the redox state of the electron transport chain, and thus NADH and 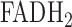 , is a quantitative marker of lung tissue mitochondrial bioenergetics, and hence mitochondrial function [10], [20].
, is a quantitative marker of lung tissue mitochondrial bioenergetics, and hence mitochondrial function [10], [20].
Fig. 1.
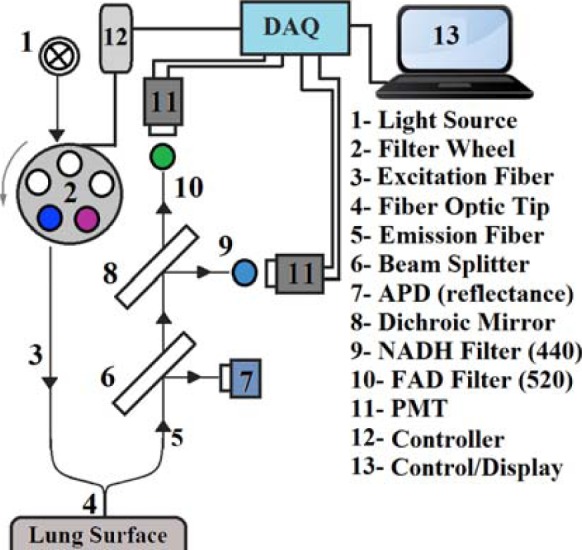
Schematic of the Fluorometer.
Fig. 2.
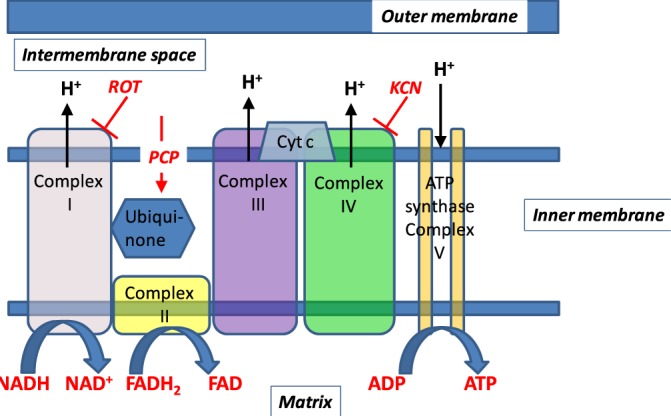
Schematic representation of subunits of mitochondrial oxidative phosphorylation complexes. Hydrogen ions are transported from the mitochondrial matrix across the inner mitochondrial membrane into the intermembrane space by complexes I, III, and IV. The movement of hydrogen ions down the electrochemical gradient is coupled to the phosphorylation of adenosine diphosphate (ADP) to form adenosine triphosphate (ATP) by complex V. Electrons from the autofluorescent reducing agent, nicotine adenine dinucleotide (NADH), move from complex I through ubiquinone to complex III and then complex IV via cytochrome c (Cyt c). Electrons from succinate, another reducing agent, enter the respiratory chain through flavin adenine dinucleotide (FAD), which is covalently linked to complex II of the respiratory chain. Like NADH, the reduced form of FAD (FADH) is autofluorescent. Rotenone (ROT) and potassium cyanide (KCN) inhibit complex I and IV, respectively. Pentachlorophenol (PCP) is a protonophore which increases membrane proton conductivity, disrupts the proton gradient across the membrane, and as a result uncouples mitochondrial electron transport chain from phosphorylation.
The objective of this paper was to utilize optical fluorometry to evaluate the effect of rat exposure to hyperoxia (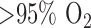 for 48 hours) on lung tissue mitochondrial redox status in a nondestructive manner in intact lungs, with a long term goal of understanding the role of mitochondrial dysfunction in the pathogenesis of lung
for 48 hours) on lung tissue mitochondrial redox status in a nondestructive manner in intact lungs, with a long term goal of understanding the role of mitochondrial dysfunction in the pathogenesis of lung 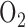 toxicity as well as developing a diagnostic modality of oxygen toxicity.
toxicity as well as developing a diagnostic modality of oxygen toxicity.
II. Materials and Methods
A. Materials
Fatty-acid free bovine serum albumin (Standard Powder, BSA) was purchased from Serologicals Corp. (Gaithersburg, MD). All other reagent grade chemicals were purchased from Sigma Chemical Company.
B. Fluorometer
A schematic for the fluorometer device used in these studies is shown in Fig. 1. The operation of this device has been described previously [18]. Briefly, excitation light is generated from a mercury arc lamp and delivered through a liquid light guide to a filter wheel, where the appropriate excitation wavelength can be selected. On the other side of this filter wheel is one leg of a bifurcated fiber bundle with a distal end of 3.2 mm inner diameter consisting of 158 (79 for excitation and 79 for emission) randomly distributed fibers, each 200 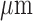 in diameter, which is brought into contact with the tissue with minimal pressure on the lung. The power of the light emitted from the head of the probe is 1
in diameter, which is brought into contact with the tissue with minimal pressure on the lung. The power of the light emitted from the head of the probe is 1 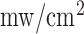 for NADH and 4
for NADH and 4 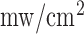 for FAD. The other leg of the bifurcated fiber bundle delivers light to the detection optics where it is collimated and split using a beam splitter. Half of the light is then incident on an avalanche photodiode for detection of reflected light. The other half of the light then passes through a dichroic mirror to separate the NADH and FAD channels. In either of the channels, the light is then filtered to select the emitted fluorescence and is finally incident on a photomultiplier tube. To optimally excite the fluorophores of interest, NADH and FAD, using the mercury arc lamp, the excitation filters used were centered at 370 nm and 452 nm, with bandwidths of 40 nm and 50 nm, respectively. Finally, the filters used to detect the emitted fluorescence are centered at 460 and 520 nm, respectively, each with a bandwidth of 40 nm.
for FAD. The other leg of the bifurcated fiber bundle delivers light to the detection optics where it is collimated and split using a beam splitter. Half of the light is then incident on an avalanche photodiode for detection of reflected light. The other half of the light then passes through a dichroic mirror to separate the NADH and FAD channels. In either of the channels, the light is then filtered to select the emitted fluorescence and is finally incident on a photomultiplier tube. To optimally excite the fluorophores of interest, NADH and FAD, using the mercury arc lamp, the excitation filters used were centered at 370 nm and 452 nm, with bandwidths of 40 nm and 50 nm, respectively. Finally, the filters used to detect the emitted fluorescence are centered at 460 and 520 nm, respectively, each with a bandwidth of 40 nm.
C. Animals
For normoxic (control) lung studies, adult male Sprague-Dawley rats (Charles River; 300–350 g) were exposed to room air. For the hyperoxic lung studies, age matched rats were housed in a Plexiglass chamber maintained at 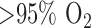 for 48 hours (hyperoxic) [21]. The total gas flow was 3.5 liters/min and the chamber
for 48 hours (hyperoxic) [21]. The total gas flow was 3.5 liters/min and the chamber 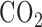 was maintained at
was maintained at 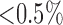 . The temperature within the chamber was 20–22°C. Every other day, the rats were weighed, and their cage, food, water, and
. The temperature within the chamber was 20–22°C. Every other day, the rats were weighed, and their cage, food, water, and  absorbent were changed. All rats were kept on a 12:12-h light-dark cycle. A total of 17 normoxic and 16 hyperoxic rats were studied. All animal experiments were performed under the approval of Institutional Animal Care and Use Committees of the Zablocki Veterans Affairs Medical Center and Marquette University (Milwaukee, WI) and in compliance with the National Research Council's Guide for the Care and Use of Laboratory Animals. For the hyperoxic rats, the optical imaging and other studies described below were conducted immediately following the exposure period.
absorbent were changed. All rats were kept on a 12:12-h light-dark cycle. A total of 17 normoxic and 16 hyperoxic rats were studied. All animal experiments were performed under the approval of Institutional Animal Care and Use Committees of the Zablocki Veterans Affairs Medical Center and Marquette University (Milwaukee, WI) and in compliance with the National Research Council's Guide for the Care and Use of Laboratory Animals. For the hyperoxic rats, the optical imaging and other studies described below were conducted immediately following the exposure period.
D. Isolated Perfused Rat Lung Preparation
The isolated perfused lung preparation allows for manipulation of lung tissue mitochondrial redox state without disrupting the multi-cellular environment of the lung through the addition of metabolic inhibitor(s) to the recirculating perfusate and/or alteration of the composition of the ventilation gas [18].
As previously described, rats were anesthetized with pentobarbital sodium (40 mg/kg body wt. i.p.), the chest opened and heparin (0.7 IU/g body wt.) injected into the right ventricle [18]. The pulmonary artery and the trachea were cannulated, and the pulmonary venous outflow was accessed via a cannula in the left atrium. The heart-lung was removed from the chest and attached to a ventilation and perfusion system. The control perfusate contained (in mM) 4.7 KCl, 2.51 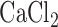 , 1.19
, 1.19 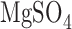 , 2.5
, 2.5 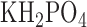 , 118 NaCl, 25
, 118 NaCl, 25 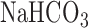 , 5.5 glucose, and 3% fatty-acid free bovine serum albumin (BSA) [22]. The perfusion system was primed (Masterflex roller pump) with the control perfusate maintained at 37°C and equilibrated with 15%
, 5.5 glucose, and 3% fatty-acid free bovine serum albumin (BSA) [22]. The perfusion system was primed (Masterflex roller pump) with the control perfusate maintained at 37°C and equilibrated with 15% 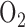 , 6%
, 6% 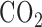 , balance
, balance 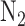 resulting in perfusate
resulting in perfusate 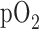 ,
, 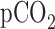 and pH of
and pH of 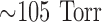 , 40 Torr, and 7.4, respectively. Initially, control perfusate was pumped through the lung until it was evenly blanched and venous effluent was clear of blood. The lung was ventilated (40 breaths/min) with end-inspiratory and end-expiratory pressures of
, 40 Torr, and 7.4, respectively. Initially, control perfusate was pumped through the lung until it was evenly blanched and venous effluent was clear of blood. The lung was ventilated (40 breaths/min) with end-inspiratory and end-expiratory pressures of 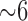 and 3 mm Hg, respectively, with the above gas mixture. The pulmonary arterial pressure was referenced to atmospheric pressure at the level of the left atrium and monitored continuously during the course of the experiments. The venous effluent pressure was atmospheric pressure.
and 3 mm Hg, respectively, with the above gas mixture. The pulmonary arterial pressure was referenced to atmospheric pressure at the level of the left atrium and monitored continuously during the course of the experiments. The venous effluent pressure was atmospheric pressure.
E. Experimental Protocols
Lung surface fluorescence measurements: The fluorometer was used in a dark room to minimize stray-light effects [18]. Surface fluorescence was then measured by placing the fiber optic probe against the pleural surface of the right lobe. For a given lung, NADH and FAD surface fluorescence signals were first acquired under resting conditions (lung perfused with control perfusate and ventilated with 6% 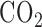 balance
balance 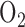 ventilation gas), and then following the addition of one or more of the following agents (rotenone, potassium cyanide, and pentachlorophenol) to the recirculation perfusate. Rotenone (20
ventilation gas), and then following the addition of one or more of the following agents (rotenone, potassium cyanide, and pentachlorophenol) to the recirculation perfusate. Rotenone (20 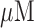 ) was used to inhibit mitochondrial complex I activity (Fig. 2), which would be expected to increase NADH signal by reducing the chain upstream from complex I. As a mitochondrial complex IV inhibitor (Fig. 2), potassium cyanide (KCN, 2 mM) would be expected to reduce the chain upstream from complex IV and hence increase NADH signal, and decrease FAD signal from succinate dehydrogenase (complex II) Uncoupled mitochondrial condition was achieved by the addition of pentachlorophenol (PCP, mitochondrial uncoupler; 3 mM) to the recirculating perfusate. As a protonophore, PCP should oxidize the chain and hence decrease NADH signal and increase FAD signal (Fig. 2). For some of the lungs, KCN was added after the initial addition of rotenone or PCP to the recirculating perfusate. The above concentrations of rotenone, KCN, and PCP were chosen to achieve maximal inhibition or uncoupling and hence maximal changes in NADH and/or FAD signal based upon published reports [11], [18].
) was used to inhibit mitochondrial complex I activity (Fig. 2), which would be expected to increase NADH signal by reducing the chain upstream from complex I. As a mitochondrial complex IV inhibitor (Fig. 2), potassium cyanide (KCN, 2 mM) would be expected to reduce the chain upstream from complex IV and hence increase NADH signal, and decrease FAD signal from succinate dehydrogenase (complex II) Uncoupled mitochondrial condition was achieved by the addition of pentachlorophenol (PCP, mitochondrial uncoupler; 3 mM) to the recirculating perfusate. As a protonophore, PCP should oxidize the chain and hence decrease NADH signal and increase FAD signal (Fig. 2). For some of the lungs, KCN was added after the initial addition of rotenone or PCP to the recirculating perfusate. The above concentrations of rotenone, KCN, and PCP were chosen to achieve maximal inhibition or uncoupling and hence maximal changes in NADH and/or FAD signal based upon published reports [11], [18].
At the end of the above imaging protocol, a subset of normoxic and hyperoxic lungs were fixed inflated with paraformaldehye, paraffin embedded, sectioned in 5 micron thick whole mounts, then stained with hematoxylin and eosin (H&E) staining.
F. Fluorescent Signal Processing
The signal measured by the fluorometer contains a time shared sequence of NADH and FAD pulses collected from the surface of the lung. Superimposed over the signal for NADH or FAD is lung ventilation noise. To remove this noise, the trend of the data was first calculated by extracting the maximum value of each pulse. This trend was then smoothed out using a fourth order median filter followed by a fourth order moving average filter. The resulting NADH and FAD signals were then normalized by dividing each by its baseline value (signal level in the absence of any metabolic inhibitor or uncoupler) [18].
G. Complex I and II Assays
For a separate sets of normoxic and hyperoxic lungs the activities of mitochondrial complex I and II were determined as previously described [21], [22]. Briefly, lungs were isolated and washed free of blood with perfusate containing (in mM) 4.7 KCl, 2.51 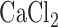 , 1.19
, 1.19 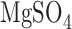 , 2.5
, 2.5 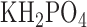 , 118 NaCl, 25
, 118 NaCl, 25 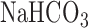 , 5.5 glucose, and 2.5% Ficoll. Lungs were then removed from the perfusion system, weighed, minced, and homogenized with buffer (pH 7.2) containing (in mM) 225 mannitol, 75 sucrose, 5 3-[N-morpholino] propanesulfonic acid, 20 ethylene glycol-bis (B-aminoethyl ether)-N,N,N',N'-tetraacetic acid, 2% fatty-acid free BSA, and 0.02 ml per ml protease inhibitor cocktail set III (Calbiochem, La Jolla, CA), utilizing a Polytron tissue homogenizer. Lung homogenates were centrifuged at 1,500 g for 5 min at 4 °C, and the resulting supernatants were centrifuged again at 13,000 g for 30 min at 4 °Cto obtain a crude mitochondrial fraction (P2). The P2 fractions were washed twice by resuspension in 8 ml ice-cold homogenization buffer without BSA and then centrifuged (13,000 g for 20 min at 4°C). The final P2 fractions were resuspended in 1-ml BSA-free homogenization buffer. Mitochondrial complex I (NADH dehydrogenase) activity
, 5.5 glucose, and 2.5% Ficoll. Lungs were then removed from the perfusion system, weighed, minced, and homogenized with buffer (pH 7.2) containing (in mM) 225 mannitol, 75 sucrose, 5 3-[N-morpholino] propanesulfonic acid, 20 ethylene glycol-bis (B-aminoethyl ether)-N,N,N',N'-tetraacetic acid, 2% fatty-acid free BSA, and 0.02 ml per ml protease inhibitor cocktail set III (Calbiochem, La Jolla, CA), utilizing a Polytron tissue homogenizer. Lung homogenates were centrifuged at 1,500 g for 5 min at 4 °C, and the resulting supernatants were centrifuged again at 13,000 g for 30 min at 4 °Cto obtain a crude mitochondrial fraction (P2). The P2 fractions were washed twice by resuspension in 8 ml ice-cold homogenization buffer without BSA and then centrifuged (13,000 g for 20 min at 4°C). The final P2 fractions were resuspended in 1-ml BSA-free homogenization buffer. Mitochondrial complex I (NADH dehydrogenase) activity 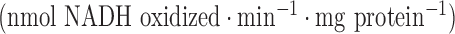 was determined as the difference between the rates of NADH oxidation in the presence and absence of rotenone over the linear portion of the reaction progress curve as we have previously described [21]. Mitochondrial complex II (succinate-coenzyme Q reductase) activity was determined as the difference between the rates of reduction of the artificial electron acceptor 2,6-dichlorophenolindophenol (DCPIP) in the presence and absence of thenoyltrifluoroacetone (TTFA, complex II inhibitor) over the linear portion with succinate as donor [22]. The protein concentrations were determined colorimetrically as previously described [21].
was determined as the difference between the rates of NADH oxidation in the presence and absence of rotenone over the linear portion of the reaction progress curve as we have previously described [21]. Mitochondrial complex II (succinate-coenzyme Q reductase) activity was determined as the difference between the rates of reduction of the artificial electron acceptor 2,6-dichlorophenolindophenol (DCPIP) in the presence and absence of thenoyltrifluoroacetone (TTFA, complex II inhibitor) over the linear portion with succinate as donor [22]. The protein concentrations were determined colorimetrically as previously described [21].
H. Statistical Evaluation of Data
Data are presented as 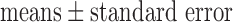 (SE) unless otherwise stated. Statistical comparisons were carried out in each group using paired and unpaired
(SE) unless otherwise stated. Statistical comparisons were carried out in each group using paired and unpaired 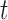 -test as appropriate for design, with
-test as appropriate for design, with 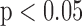 as the criterion for statistical significance.
as the criterion for statistical significance.
III. Results
Rats did not display any sign of distress after 48 hours of exposure to 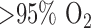 . Over the course of this exposure period, the rats experienced a small
. Over the course of this exposure period, the rats experienced a small  , but significant loss in body weight (pre- and post-body weights of
, but significant loss in body weight (pre- and post-body weights of 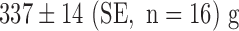 and
and 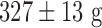 , respectively). Histology revealed preserved lung morphology after 48 hours of hyperoxic exposure (Fig. 3).
, respectively). Histology revealed preserved lung morphology after 48 hours of hyperoxic exposure (Fig. 3).
Fig. 3.
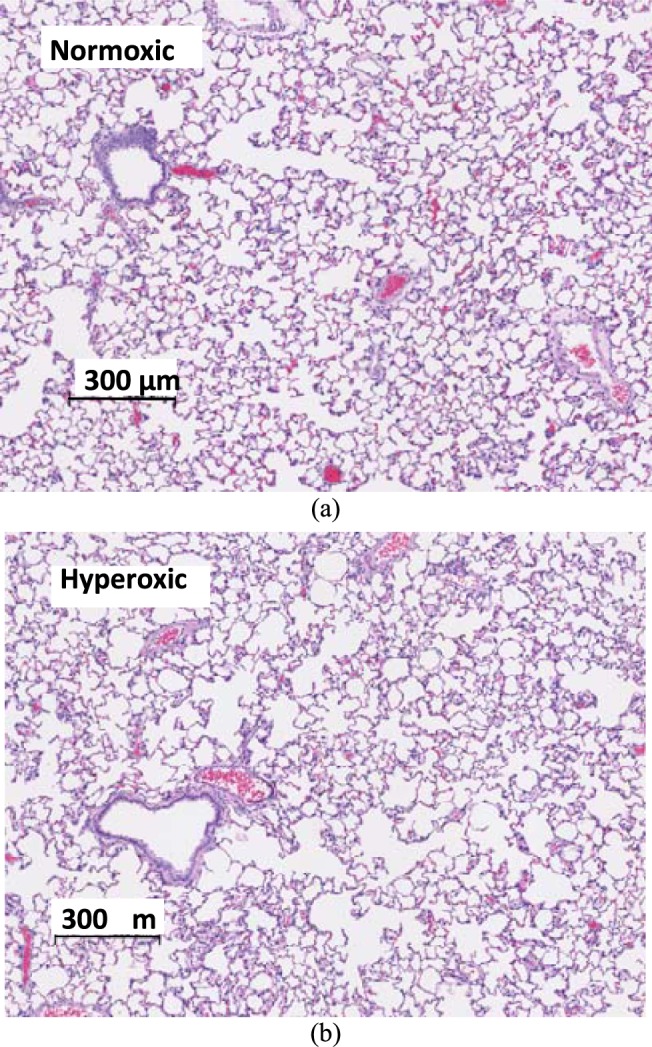
Histology: Images of H&E section of normoxic (a) vs. hyperoxic (b) lungs. After 48 hours of exposure, lungs show no evidence of neutrophilic infiltrates, pulmonary edema, or vascular smooth muscle thickening, which are characteristic of hyperoxic injury sustained after rat exposure for 60 hours or longer.
For both normoxic and hyperoxic lungs, the response of both NADH and FAD lung surface signals to lung perfusion with rotenone, KCN, or PCP appeared within a minute of adding the chemical to the perfusate reservoir (Figs. 4 and 5). An increase (from baseline) in NADH fluorescence signal indicates reduction of the mitochondrial matrix (Fig. 2), whereas an increase in the FAD fluorescence signal indicates oxidation of the electron transport chain. In this paper, the change in NADH signal in the presence of rotenone or KCN is considered a measure of mitochondrial complex I activity, and the change in the FAD signal in the presence of KCN is considered a measure of mitochondrial complex II activity.
Fig. 4.
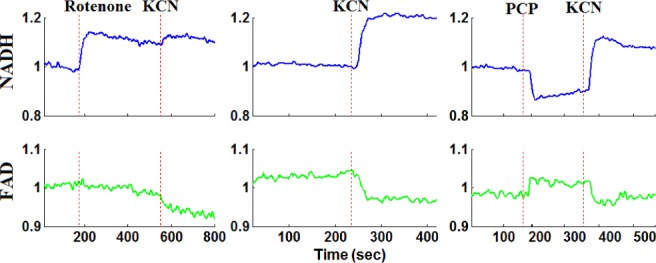
Representative fluorometer response to perfusion with chemical inhibitors and uncouplers in a normoxic lung.
Fig. 5.
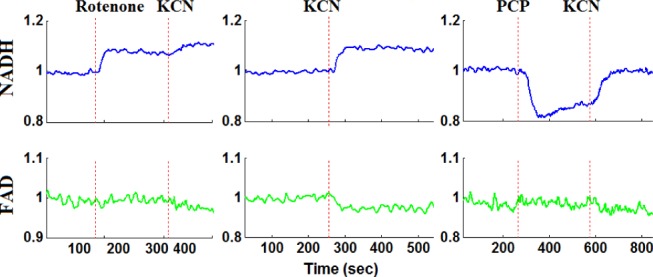
Representative fluorometer response to perfusion with chemical inhibitors and uncouplers in a hyperoxic lung.
For normoxic lungs (Fig. 4 and Table I), lung perfusion with rotenone(complex I inhibitor) reduced the mitochondrial matrix (ETC) upstream from complex I resulting in an increase in NADH signal by 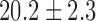 (SE) %, with no effect on FAD signal. Lung perfusion with KCN (complex IV inhibitor) reduced the ETC resulting in a
(SE) %, with no effect on FAD signal. Lung perfusion with KCN (complex IV inhibitor) reduced the ETC resulting in a 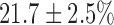 increase in NADH and
increase in NADH and 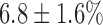 decrease in FAD. Lung perfusion with PCP, which uncoupled ETC from phosphorylation, decreased NADH signal by
decrease in FAD. Lung perfusion with PCP, which uncoupled ETC from phosphorylation, decreased NADH signal by 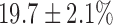 with no effect on FAD signal. The addition of KCN to PCP-treated lungs reversed the effect of PCP on the redox status of the ETC, increasing NADH signal by
with no effect on FAD signal. The addition of KCN to PCP-treated lungs reversed the effect of PCP on the redox status of the ETC, increasing NADH signal by 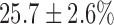 and decreasing FAD signal by
and decreasing FAD signal by 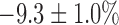 . In the presence of rotenone, KCN resulted in a small but significant decrease
. In the presence of rotenone, KCN resulted in a small but significant decrease 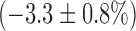 in lung surface FAD signal and increase
in lung surface FAD signal and increase 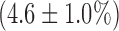 on NADH signal as compared to values in the presence of rotenone only.
on NADH signal as compared to values in the presence of rotenone only.
Table I. The Effects of Metabolic Inhibitors and Uncoupler on the Lung Tissue Surface FAD and NADH Fluorescence Signals of Normoxic and Hyperoxic Rats.
| Inhibitor/ Uncoupler | FAD % | NADH % | ||
|---|---|---|---|---|
| Normoxic | Hyperoxic | Normoxic | Hyperoxic | |
| Rotenone | 0.2 ± 0.2 | −0.3 ± 0.3 | 20.2 ± 2.3 | 7.5 ± 1.0* |
| KCN | −6.8 ± 1.6 | −5.2 ± 0.5 | 21.7 ± 2.5 | 9.2 ± 1.2* |
| PCP | 1.7 ± 0.8 | −2.3 ± 1.4* | −19.7 ± 2.1 | −20.8± 1.2 |
| PCP + KCN | −9.3 ± 1.0 | −4.2 ± 0.7 * | 25.7 ± 2.6 | 9.0 ± 1.0* |
| Rotenone + KCN | 0.8–3.3 ± | −3.8 ± 0.6 | 4.6 ± 1.0 | 2.8 ± 1.2 |
Values are mean ± SE. For the (PCP + KCN) and (rotenone + KCN) treatments, the percentage change due to additional treatment with KCN is calculated relative to the new baseline in the presence of PCP or rotenone, respectively. For normoxic lungs, 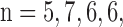 and 5 for rotenone, potassium cyanide (KCN), pentachlorophenol (PCP), PCP + KCN, and rotenone + KCN, respectively. For hyperoxic lungs
and 5 for rotenone, potassium cyanide (KCN), pentachlorophenol (PCP), PCP + KCN, and rotenone + KCN, respectively. For hyperoxic lungs 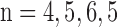 , and 4 for rotenone, KCN, PCP, PCP + KCN, and rotenone + KCN, respectively.
, and 4 for rotenone, KCN, PCP, PCP + KCN, and rotenone + KCN, respectively.
* indicates value significantly different from the corresponding normoxic value (t-test 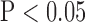 ).
).
For hyperoxic lungs (Fig. 5, Table I), lung perfusion with rotenone increased NADH signal by 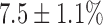 , which is 63% lower than in normoxic lungs, with no effect on FAD signal. Lung perfusion with KCN increased NADH by
, which is 63% lower than in normoxic lungs, with no effect on FAD signal. Lung perfusion with KCN increased NADH by 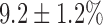 , which is 58% lower than in normoxic lungs. KCN effect on FAD signal in hyperoxic lungs was not significantly different from that in normoxic lungs. Lung perfusion with PCP had the same qualitative and quantitative effects on NADH and FAD signals as in normoxic lungs. The addition of KCN to PCP-treated hyperoxic lungs increased NADH by
, which is 58% lower than in normoxic lungs. KCN effect on FAD signal in hyperoxic lungs was not significantly different from that in normoxic lungs. Lung perfusion with PCP had the same qualitative and quantitative effects on NADH and FAD signals as in normoxic lungs. The addition of KCN to PCP-treated hyperoxic lungs increased NADH by 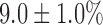 , which is 65% smaller than that in normoxic lungs. Furthermore, KCN decreased FAD signal by
, which is 65% smaller than that in normoxic lungs. Furthermore, KCN decreased FAD signal by 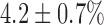 , which is 55% smaller than in normoxic lungs. These results are consistent with a more reduced chain upstream from complex I and II in hyperoxic lungs as compared to normoxic lungs, and suggest a decrease in complex I and II activities in hyperoxic lungs.
, which is 55% smaller than in normoxic lungs. These results are consistent with a more reduced chain upstream from complex I and II in hyperoxic lungs as compared to normoxic lungs, and suggest a decrease in complex I and II activities in hyperoxic lungs.
Because the data in Table I suggested that the hyperoxic exposure decreased complex I and II activities, we measured the activities of complex I and II in the P2 fractions obtained from lung homogenates. Table II shows that complex I and II activities normalized to protein were  and 63% lower, respectively, in P2 fractions derived from hyperoxic than those of normoxic lungs.
and 63% lower, respectively, in P2 fractions derived from hyperoxic than those of normoxic lungs.
Table II. Mitochondrial Complex I and Complex II Activity Measured in p2 Fractions of Lung Homogenate.
| Complex I (nmol/min/mg protein) | Complex II (nmol/min/mg protein) | |
|---|---|---|
| Normoxic | 37.6 ± 3.2 | 79.1 ± 5.9 |
| Hyperoxic | 8.6 ± 0.7* | 28.5 ± 2.3* |
Values are mean ± SE. 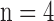 and 3 for normoxic and hyperoxic lungs, respectively.
and 3 for normoxic and hyperoxic lungs, respectively.
*indicates value significantly different from the corresponding normoxic value 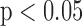 ; t-test).
; t-test).
IV. Discussion
This paper demonstrates the utility of optical fluorescent imaging for evaluating the effect of subacute rat exposure to hyperoxia on the redox state of lung tissue mitochondrial electron transport chain (ETC) in a non-destructive manner in isolated perfused lungs. The results suggest a hyperoxia-induced decrease in complex I and II activities, and demonstrate the ability of this approach to detect a change in mitochondrial redox state in the early phase of hyperoxic lung injury, when histology is not different from that of normoxic rats.
A. NADH Signal and Complex I Activity
Surface fluorometry results establish that rat exposure to 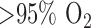 for 48 hours decreased the change in NADH signal in the presence of KCN or rotenone by 58% and 63%, respectively, as compared to those of normoxic lungs. A similar decrease (65%) in NADH signal was also measured following the addition of KCN to PCP-treated hyperoxic lungs as compared to normoxic lungs. Rotenone- or KCN-induced change in NADH signal is a measure of complex I activity. Thus these result suggest that the ETC upstream from complex I is more reduced in intact hyperoxic lungs than in normoxic lungs, and that complex I activity is lower in hyperoxic lungs. This is consistent with the 77% decrease in mitochondrial complex I activity per mg protein which we measured in P2 fractions derived from hyperoxic lungs as compared to normoxic lung.
for 48 hours decreased the change in NADH signal in the presence of KCN or rotenone by 58% and 63%, respectively, as compared to those of normoxic lungs. A similar decrease (65%) in NADH signal was also measured following the addition of KCN to PCP-treated hyperoxic lungs as compared to normoxic lungs. Rotenone- or KCN-induced change in NADH signal is a measure of complex I activity. Thus these result suggest that the ETC upstream from complex I is more reduced in intact hyperoxic lungs than in normoxic lungs, and that complex I activity is lower in hyperoxic lungs. This is consistent with the 77% decrease in mitochondrial complex I activity per mg protein which we measured in P2 fractions derived from hyperoxic lungs as compared to normoxic lung.
The measured hyperoxia-induced change in complex I redox state (Table II) could be due to a change in complex I protein and/or change in rate of NADH production. The latter could result from impairment to the Krebs cycle and/or change in cytoslic 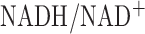 which could affect mitochondrial NADH level via the malate-aspartate shuttle. However, the hyperoxia-induced change in complex I redox state is most likely due to a change in complex I protein since for the complex I assay (P2 fraction), the NADH concentration used was the same for both normoxic and hyperoxic lung mitochondrial preparations (P2 fractions). The results of this assay (Table II) show that the measured hyperoxia-induced change in complex I activity is qualitatively and quantitatively consistent with the hyperoxia-induced change in complex I redox state measured on the surface of the lung (Table I).
which could affect mitochondrial NADH level via the malate-aspartate shuttle. However, the hyperoxia-induced change in complex I redox state is most likely due to a change in complex I protein since for the complex I assay (P2 fraction), the NADH concentration used was the same for both normoxic and hyperoxic lung mitochondrial preparations (P2 fractions). The results of this assay (Table II) show that the measured hyperoxia-induced change in complex I activity is qualitatively and quantitatively consistent with the hyperoxia-induced change in complex I redox state measured on the surface of the lung (Table I).
The sulfhydryl-containing Krebs cycle enzymes pyruvate and alpha-ketoglutarate dehydrogenase and their coenzymes CoA and lipoic acid are sensitive to oxidative stress [23]. Previous studies evaluated the effect of rat exposure to 100% 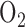 for 24 hrs on these and other Krebs cycle enzymes [15], [23], [24]. Gardner et al. [24] reported a 73% decrease in lung aconitase activity after 24 hrs of rate exposure to 100%
for 24 hrs on these and other Krebs cycle enzymes [15], [23], [24]. Gardner et al. [24] reported a 73% decrease in lung aconitase activity after 24 hrs of rate exposure to 100% 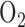 . This decrease is consistent with the increase in citrate level (aconitase substrate) in lung tissue under the same exposure conditions as reported by Bassett et al. [23]. This impairment to the Krebs cycle could affect NADH supplied to the electron transport chain and in turn complex I redox state. However it is not known whether this impairment to aconitase activity persists after 48 hrs of exposure to 100%
. This decrease is consistent with the increase in citrate level (aconitase substrate) in lung tissue under the same exposure conditions as reported by Bassett et al. [23]. This impairment to the Krebs cycle could affect NADH supplied to the electron transport chain and in turn complex I redox state. However it is not known whether this impairment to aconitase activity persists after 48 hrs of exposure to 100% 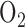 or whether it is sufficient to account for the measured hyperoxia-induced change in complex I redox state. In another study, Bassett et al. [15] reported that rat exposure to 100%
or whether it is sufficient to account for the measured hyperoxia-induced change in complex I redox state. In another study, Bassett et al. [15] reported that rat exposure to 100% 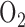 for 24 hrs had no effect on the activities of the Krebs cycle enzymes succinate dehydrogenase, isocitrate dehydrogenase or
for 24 hrs had no effect on the activities of the Krebs cycle enzymes succinate dehydrogenase, isocitrate dehydrogenase or 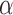 -glycerophosphate dehydrogenase. In the present study we report a 64% decrease in succinate dehydrogenase (complex II) activity after 48 hrs of exposure to 100%
-glycerophosphate dehydrogenase. In the present study we report a 64% decrease in succinate dehydrogenase (complex II) activity after 48 hrs of exposure to 100% 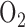 . Additional studies would be needed to evaluate the effect of rat exposure to 100%
. Additional studies would be needed to evaluate the effect of rat exposure to 100% 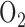 for 48 hrs on the activities of Krebs cycle enzymes isocitrate dehydrogenase,
for 48 hrs on the activities of Krebs cycle enzymes isocitrate dehydrogenase, 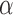 -glycerophosphate dehydrogenase, and aconitase to determine the potential contribution of impaired production of NADH by the Krebs cycle to the measured change in complex I redox state.
-glycerophosphate dehydrogenase, and aconitase to determine the potential contribution of impaired production of NADH by the Krebs cycle to the measured change in complex I redox state.
Fisher AB [16] reported that rat exposure to 100% 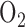 for 48 hrs increased lung lactate production rate (78%) and lactate to pyruvate ratio (108%). Since lactate to pyruvate ratio is proportional to cytosolic
for 48 hrs increased lung lactate production rate (78%) and lactate to pyruvate ratio (108%). Since lactate to pyruvate ratio is proportional to cytosolic 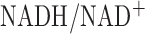 , the increase in lactate to pyruvate ratio suggests an increase in cytosolic NADH transported to the mitochondria matrix via the malate-aspartate shuttle. This shuttle provide more cytosolic NADH to mitochondria when the cytosolic
, the increase in lactate to pyruvate ratio suggests an increase in cytosolic NADH transported to the mitochondria matrix via the malate-aspartate shuttle. This shuttle provide more cytosolic NADH to mitochondria when the cytosolic 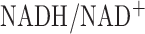 is higher than in the mitochondrial matrix. Thus the hyperoxia-induced increase in cytosolic
is higher than in the mitochondrial matrix. Thus the hyperoxia-induced increase in cytosolic 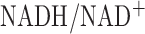 could contribute to the measured change in complex I redox state in hyperoxic lungs.
could contribute to the measured change in complex I redox state in hyperoxic lungs.
Rat exposure to hyperoxia had no effect on the change in NADH signal in the presence of PCP as compared to that in normoxic lungs as a measure of the coupling between ETC and phosphorylation. This observation implies that rat treatment with hyperoxia (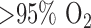 for 48 hours) did not alter the coupling between respiration and phosphorylation in lung tissue despite the apparent decrease in complex I activity. However, since the ETC upstream from complex I is more reduced in hyperoxic lungs than in normoxic lungs, one would expect a larger PCP-induced decrease in NADH signal in hyperoxic lung. Currie et al. demonstrated that rat exposure to 100%
for 48 hours) did not alter the coupling between respiration and phosphorylation in lung tissue despite the apparent decrease in complex I activity. However, since the ETC upstream from complex I is more reduced in hyperoxic lungs than in normoxic lungs, one would expect a larger PCP-induced decrease in NADH signal in hyperoxic lung. Currie et al. demonstrated that rat exposure to 100% 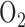 for 48 hours decreased ADP-stimulated
for 48 hours decreased ADP-stimulated 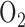 consumption (state 3) by
consumption (state 3) by 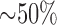 with a-ketoglutarate as an NAD-linked substrate [25]. Together these results support a decreased tightness of coupling between respiration and phosphorylation in hyperoxic lungs as compared to normoxic lungs.
with a-ketoglutarate as an NAD-linked substrate [25]. Together these results support a decreased tightness of coupling between respiration and phosphorylation in hyperoxic lungs as compared to normoxic lungs.
There is ample evidence that increased production of reactive oxygen species (ROS) is a major pathophysiological factor in the genesis of hyperoxic lung injury [3], [26]–[28]. Thus, one strategy that cells may have evolved to protect against hyperoxic lung injury is to mitigate the activities of ROS sources [12], [29]–[31]. Mitochondrial electron transport complex I is a major source of ROS [3], [27], [32]. Moreover, studies have shown that the rate of ROS formation at complex I in endothelial cells increased with an increase in 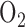 tension, and that complex I inactivation using rotenone decreased ROS generation in sheep pulmonary microvascular endothelial cells exposed to hyperoxia (100%
tension, and that complex I inactivation using rotenone decreased ROS generation in sheep pulmonary microvascular endothelial cells exposed to hyperoxia (100% 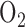 for 30 min) [27], [32], [33]. Thus the measured decrease in complex I could be an adaptive mechanism by the cells to decrease the rate of ROS formation at complex I under hyperoxic conditions.
for 30 min) [27], [32], [33]. Thus the measured decrease in complex I could be an adaptive mechanism by the cells to decrease the rate of ROS formation at complex I under hyperoxic conditions.
On the other hand, the measured decrease in complex I activity could represent hyperoxia-induced injury leading to increases in the rate of ROS formation. Mitochondrial DNA (mDNA) is highly sensitive to ROS [34]. Hyperoxia-induced increase in the rate of ROS formation could damage mDNA and as a result compromise complex I activity since 7 out of 45 subunits of complex I are encoded by mDNA [34], [35]. Hyperoxia-induced increase in ROS formation could also cause direct damage to complex I activity by oxidizing cardiolipin, which is sensitive to ROS [36], [37]. This phospholipid is important for the function of complex I [38]. In addition, oxidation of this lipid could lead to increase in the loss of electrons at complex I and in the rate of mitochondrial superoxide formation at complex I [36], [37].
Ratner et al. demonstrated that exposure of neonatal mice to hyperoxia (75% 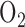 for 72 hours) decreases complex I activity in lung homogenates by
for 72 hours) decreases complex I activity in lung homogenates by 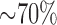 , and that this decrease compromises mitochondrial oxidative phosphorylation and contributes to alveolar development arrest [7]. Fisher AB [16] showed that rat exposure to 100%
, and that this decrease compromises mitochondrial oxidative phosphorylation and contributes to alveolar development arrest [7]. Fisher AB [16] showed that rat exposure to 100% 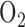 for 48 hours had no effect on lung tissue ATP content as compared to normoxic lungs, although there was an increase (105%) in lactate/pyruvate ratio indicating an increase in ATP production via glycolysis. He suggested that the increased lactate/pyruvate ratio could be due to demand for glycolytic ATP and/or decrease in ATP production via oxidative phosphorylation which is compensated for by an increase in ATP production via glycolysis.
for 48 hours had no effect on lung tissue ATP content as compared to normoxic lungs, although there was an increase (105%) in lactate/pyruvate ratio indicating an increase in ATP production via glycolysis. He suggested that the increased lactate/pyruvate ratio could be due to demand for glycolytic ATP and/or decrease in ATP production via oxidative phosphorylation which is compensated for by an increase in ATP production via glycolysis.
B. FAD Signal and Complex II Activity
Compared to NADH, the measured change in the FAD signal in response to lung treatment with the metabolic inhibitors was relatively small for normoxic and hyperoxic lungs (Table I). One reason for this observation could be lower lung tissue concentration of FAD as compared to NADH. In fact, the FAD baseline signal is small relative to the NADH signal as evidenced by the difference in signal amplification for the NADH and FAD channels (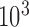 for NADH compared to
for NADH compared to 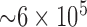 for FAD) [18].
for FAD) [18].
Lung treatment with KCN, which reduces the ETC upstream from complex IV, should reduce complex II and as a result decrease FAD signal. Just as a change in NADH tracks complex I activity, a change in FAD signal reflect complex II activity. Rat exposure to hyperoxia had no significant effect on the change in FAD signal in the presence of KCN as compared to that in lungs from normoxic rats. However, the change in FAD signal following the addition of KCN to PCP-treated lungs was 56% lower in hyperoxic lungs than normoxic lungs. It could be that lung treatment with PCP, which simulates the flow of reducing equivalents through the chain, fully oxidized the chain and as a result exposed the difference in the capacities of complex II between normoxic and hyperoxic lungs. Regardless, this apparent decrease in complex II activity is consistent with the 63% decrease in mitochondrial complex II activity per mg protein in P2 fractions derived from hyperoxic lungs as compared to normoxic lung (Table II).
The measured hyperoxia-induced change in FAD signal in the presence of  could be due to a direct oxidant-induced inhibition of complex II or indirect inhibition of complex II by impairment of Krebs cycle enzymes/coenzymes that result in a decrease in succinate concentration or increase in the concentration of oxaloacetate (OAA). Complex II activity is inhibited by a high level of OAA. However the results of the mitochondrial isolates (P2 fractions) suggest that the hyperoxia-induced change in complex II redox state is most likely due to impairment of complex II itself since for this assay the concentrations of succinate and the electron acceptor (DCPIP) used were the same for the mitochondrial isolates from both normoxic and hyperoxic lungs and should be in excess of what is required for maximal function of complex II. The measured decrease in complex II activity using this assay (Table II) is qualitatively and quantitatively consistent with the measured hyperoxia-induced decrease in FAD signal in the presence of
could be due to a direct oxidant-induced inhibition of complex II or indirect inhibition of complex II by impairment of Krebs cycle enzymes/coenzymes that result in a decrease in succinate concentration or increase in the concentration of oxaloacetate (OAA). Complex II activity is inhibited by a high level of OAA. However the results of the mitochondrial isolates (P2 fractions) suggest that the hyperoxia-induced change in complex II redox state is most likely due to impairment of complex II itself since for this assay the concentrations of succinate and the electron acceptor (DCPIP) used were the same for the mitochondrial isolates from both normoxic and hyperoxic lungs and should be in excess of what is required for maximal function of complex II. The measured decrease in complex II activity using this assay (Table II) is qualitatively and quantitatively consistent with the measured hyperoxia-induced decrease in FAD signal in the presence of 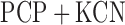 as a measure of complex II activity (Table I).
as a measure of complex II activity (Table I).
Complex II is the only ETC complex that is completely encoded by nuclear DNA [39]. Hence, damage to mDNA due to hyperoxia-induced increase in the rate of ROS formation should have no effect on complex II. Since electrons channeled through complex II produce 4-fold more mitochondrial superoxide than electrons channeled through complex I [40], the apparent decrease in complex II activity could be a means by the cells to reduce ROS formation under hyperoxic conditions.
Additional studies are needed to evaluate the effect of the depression in complex I and complex II activities observed in hyperoxic lungs on ROS production at complex I and III and mitochondrial bioenergetics, and to determine whether this depression is an injury or an adaptive response to the hyperoxic environment.
C. Sources of NADH and FAD Surface Fluorescent Signals
Our assumption in this paper is that changes in the mitochondrial pool of NADH is a key contributor to the measured changes in the NADH fluorescence signal since the metabolic inhibitors we used target the mitochondrial electron transport chain [18]. Another source of NADH signal is the cytosolic pool of NADH. Fisher et al. [41] demonstrated that rat lung treatment with KCN increased lung lactate/pyruvate by 4-fold. In this paper, lung perfusion with KCN increased lung surface NADH signal by 22% in normoxic lungs. Since lactate/pyruvate ratio reflects cytosolic 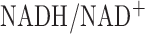 [42], [43], this suggests that cytosolic NADH did not contribute significantly to the measured lung surface NADH signal.
[42], [43], this suggests that cytosolic NADH did not contribute significantly to the measured lung surface NADH signal.
Unlike the NADH signal which has cytosolic and mitochondrial components, the FAD signal derives only from the mitochondria [18], [44]. Sources of redox-sensitive flavoprotein (FAD) include succinate dehydrogenase (complex II), lipoamide dehydrogenase (LipDH), and electron transfer flavoprotein (ETF) [18], [45], [46]. However, as previously discussed, most of the measured lung surface FAD signal is from complex II flavin [18].
Fisher AB demonstrated that rat exposure to 100% 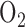 for 48 hours stimulated glycolysis as measured by
for 48 hours stimulated glycolysis as measured by 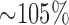 increase in the lung lactate/pyruvate ratio [16]. Since this ratio is a measure of cytosolic
increase in the lung lactate/pyruvate ratio [16]. Since this ratio is a measure of cytosolic 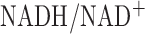 , this suggest that lung tissue cytoplasm is more reduced in hyperoxic lungs than normoxic lungs. Thus, coupled with the results of the present study, the data document that both tissue cytosolic and mitochondrial compartments are more reduced in hyperoxic lungs than normoxic lungs.
, this suggest that lung tissue cytoplasm is more reduced in hyperoxic lungs than normoxic lungs. Thus, coupled with the results of the present study, the data document that both tissue cytosolic and mitochondrial compartments are more reduced in hyperoxic lungs than normoxic lungs.
The hyperoxia-induced decrease in complex I and II activities in the present study represents a relatively early in situ metabolic consequence of hyperoxia in that it precedes effects on lung histology, hemodynamic and functional endpoints observed with rat exposure to  for
for  [8], [47]. Of the few studies evaluating the metabolic consequences of hyperoxia in the 18–48 hr period, a decrease in serotonin clearance and an increase in lactate production have been observed in lungs from rats exposed to 100%
[8], [47]. Of the few studies evaluating the metabolic consequences of hyperoxia in the 18–48 hr period, a decrease in serotonin clearance and an increase in lactate production have been observed in lungs from rats exposed to 100%  for 18 and 36 hrs, respectively [16], [48]. Audi et al. reported that rat exposure to hyperoxia (85%
for 18 and 36 hrs, respectively [16], [48]. Audi et al. reported that rat exposure to hyperoxia (85% 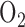 for 48 hours) results in 47% decrease in the capacity of complex I mediated coenzyme
for 48 hours) results in 47% decrease in the capacity of complex I mediated coenzyme 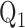 (amphipathic homolog of ubiquinone) reduction on passages through the pulmonary circulation as compared to that in lungs of normoxic rats [11]. Additionally, Klein et al. demonstrated a decrease in the metabolism of prostaglandin
(amphipathic homolog of ubiquinone) reduction on passages through the pulmonary circulation as compared to that in lungs of normoxic rats [11]. Additionally, Klein et al. demonstrated a decrease in the metabolism of prostaglandin 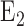 metabolism in intact lungs of rats exposed to
metabolism in intact lungs of rats exposed to 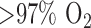 for 36 hrs [49].
for 36 hrs [49].
D. Limitations and Potential Solutions
In the present study and previous studies by us and others [18], [50]–[52], the relative change in the NADH and FAD signals are reported instead of the actual signals which are sensitive to various factors including probe distance from the organ surface, day-to-day variations in light intensity, and PMT gain settings. One approach to overcoming this limitation would be by imaging a phantom of known NADH and FAD concentrations at the end of a given experiment (lung), and then using this information to scale the measured NADH and FAD signals. This would allow us to compare un-normalized signals from different lungs.
The lung surface optical imaging data do not provide information about the specific types of lung cells contributing to the measured NADH and FAD signals, although endothelial cells would be expected to contribute significantly because of their relatively large surface area and fraction of total lung cells [8]. Though determining the contributions of specific lung cell types to the measured signal is potentially important, the global oxidoreductive state of the lung tissue is a highly valuable piece of information irrespective of the individual cell types contributing to the redox ratio.
Another limitation of lung surface optical imaging is that it may not detect deeper than 500 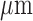 , with a diameter of 3.2 mm, for a volume of
, with a diameter of 3.2 mm, for a volume of 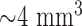 . However, this resolution is more than sufficient for determining the RR of parenchymal tissue which has a thickness (air to plasma) of 1.6
. However, this resolution is more than sufficient for determining the RR of parenchymal tissue which has a thickness (air to plasma) of 1.6 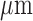 [8]. That said, central lung lesions without pleural extension would not be expected to be detected by surface fluorescence measurements such as those used in the present study.
[8]. That said, central lung lesions without pleural extension would not be expected to be detected by surface fluorescence measurements such as those used in the present study.
V. Clinical and Translational Significance
Over 900,000 adults receive invasive mechanical ventilation (MV) in the United States each year [53], [54]. Many either have acute lung injury (ALI) or have conditions such as shock and severe sepsis that place them at particular risk of ALI [54]–[59]. Treatment with high fractions of oxygen to maintain adequate tissue oxygenation may further exacerbate lung injury. However susceptibilities to lung injury (from hyperoxia or shock) are widely variable from person to person, and there are only crude means of screening for or following these injuries clinically.
The results of this paper suggest that hyperoxia-induced mitochondrial dysfunction occurs prior to the inflammatory phase of lung  toxicity. If so, a change in lung surface mitochondrial redox state measured using optical fluorescence techniques could be used as an index of lung
toxicity. If so, a change in lung surface mitochondrial redox state measured using optical fluorescence techniques could be used as an index of lung 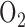 toxicity in patients with ALI requiring high oxygen therapy or to monitor the progression of ALI and its most severe form Acute Respiratory Distress Syndrome (ARDS), one of the most frequent causes of admission to the intensive care unit [60]. The fiber optic probe could be placed on the lung pleural surface through a small thoracotomy incision or a thoracostomy tube in patients with these devices in place. Alternatively, the probe might be introduced through an endotracheal tube, and positioned against airway epithelium. The same probe could be used to evaluate the efficacy of novel or existing interventions on lung tissue mitochondrial redox state and energy homeostasis in real time.
toxicity in patients with ALI requiring high oxygen therapy or to monitor the progression of ALI and its most severe form Acute Respiratory Distress Syndrome (ARDS), one of the most frequent causes of admission to the intensive care unit [60]. The fiber optic probe could be placed on the lung pleural surface through a small thoracotomy incision or a thoracostomy tube in patients with these devices in place. Alternatively, the probe might be introduced through an endotracheal tube, and positioned against airway epithelium. The same probe could be used to evaluate the efficacy of novel or existing interventions on lung tissue mitochondrial redox state and energy homeostasis in real time.
An individual with enhanced susceptibility to ARDS would be a strong candidate for strategies such as scrupulous attention to ventilation with low tidal volumes, deliberate tolerance of lower arterial oxygen or higher carbon dioxide tensions to limit oxygen toxicity or barotrauma, avoidance of transfusions, etc [53]–[59]. These interventions decrease injury but incur additional risks (increased sedation, impairment of vital organ function) that limit their universal and strict application in ALI patients.
The diagnostic and therapeutic monitoring applications of this tool are important not only for ALI/ARDS, but also for other lung conditions including lung cancer which is characterized by mitochondrial impairment (so called Warburg effect) [61], lung transplant related ischemia-reperfusion injury [62], or other animal models of human ALI [63]. Additional applications include diagnostic and therapeutic monitoring of conditions in other organs with high energy flux such as heart ischemia-reperfusion injury and heart failure management, or intraoperative identification of ischemic intestine and others [63]–[66].
Acknowledgment
We appreciate the technical help from R. Bongard, and input from Dr. M. Merker in the C. J. Zablocki VA, Milwaukee, WI.
Biographies

Reyhaneh Sepehr received the B.S. and M.S. degrees in biomedical engineering from the Amirkabir University of Technology, Tehran, Iran, and is currently pursuing the Ph.D. degree with the Electrical Engineering Department, University of Wisconsin, Milwaukee, WI, USA. Her research focus in Biophotonics Laboratory is on optical studies of the metabolic status of different tissues and mostly on signal and image processing techniques and algorithms.

Said H. Audi is an Associate Professor of biomedical engineering with Marquette University. His research interest has been primary in mathematical modeling of physiologic systems, including in the areas of pulmonary mass transfer, pulmonary hemodynamics, and functional imaging. His research focuses on using indicator dilution techniques, optical fluorescence and single photon emission computed tomography imaging, as well as computational modeling to elucidate the underlying mechanisms of acute lung injury.

Kevin S. Staniszewski received the B.S. degree in electrical engineering from the University of Wisconsin Milwaukee, Milwaukee, WI, USA, in 2009, and the M.S. degree in electrical engineering with the same university. He began working in the industry with a Test Engineer Internship at Cooper Power Systems in 2009. After finishing his graduate studies, he continued to work as a Research Associate in the Biophotonics Laboratory, University of Wisconsin Milwaukee. He is currently a Software Engineer with Prairie Technologies, Inc., Middleton, WI, USA.

Steven T. Haworth is a Research Scientist of pulmonary and critical care medicine with the Medical College of Wisconsin and Research Engineer at the Zablocki VA Medical Center. His interests focus on using a range of medical imaging modes (microcomputed tomography, micro single-photon emission computed tomography) as tools for understanding the overall structural, functional and metabolic relationships of the pulmonary vascular system under various compromised diseased states.

Elizabeth R. Jacobs is a Professor of pulmonary and critical care medicine at the Medical College of Wisconsin and the Associate Chief of Staff at the Zablocki VA Medical Center. Her interest is in mechanisms underlying acute lung injury as well as novel diagnostics to better identify pulmonary injury.

Mahsa Ranji is an Assistant Professor with the Electrical Engineering Department, University of Wisconsin-Milwaukee, Milwaukee, WI, USA. She is the Director of the Biophotonics Laboratory with research focus on optical imaging, and image processing of tissue metabolism. Her group has implemented optical devices to study cardiopulmonary injuries.
Funding Statement
This work was supported in part by the University of Wisconsin Milwaukee RGI 7 Grant, the Clinical and Translational Science Institute KL2 under Grant NIH 8Kl2TR000056, Wisconsin Applied Research Grant (Wi-ARG), the NIH under Grant 8UL1TR000055 (CTSI), NIH LH116530, the VA Merit Review Award under Grant BX001681, and the Department of Veterans Affairs.
References
- [1].Saugstad O. D., “Optimal oxygenation at birth and in the neonatal period,” Neonatology, vol. 91, pp. 319–322, Jun. 2007. [DOI] [PubMed] [Google Scholar]
- [2].Dunne P. J., et al. , “Respiratory care year in review 2011: Long-term oxygen therapy, pulmonary rehabilitation, airway management, acute lung injury, education, and management,” Respir. Care, vol. 57, no. 4, pp. 590–606. [DOI] [PubMed] [Google Scholar]
- [3].Kallet R. H. and Matthay M. A., “Hyperoxic acute lung injury,” Respir. Care, vol. 58, no. 1, pp. 123–141, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Davis W. B., Rennard S. I., Bitterman P. B., and Crystal R. G., “Pulmonary oxygen toxicity: Early reversible changes in human alveolar structures induced by hyperoxia,” New England J. Med., vol. 309, pp. 878–883, Oct. 1983. [DOI] [PubMed] [Google Scholar]
- [5].Fisher A. B. and Beers M. F., “Hyperoxia and acute lung injury,” Amer. J. Physiol. Lung Cellular Molecular Physiol., vol. 295, no. 6, pp. L1066–L1066, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].O'Brodovich H. M. and Mellins R. B., “Bronchopulmonary dysplasia. Unresolved neonatal acute lung injury,” Amer. Rev. Respir. Disease, vol. 132, no. 3, pp. 694–709, 1985. [DOI] [PubMed] [Google Scholar]
- [7].Ratner V., Starkov A., Matsiukevich D., Polin R. A., and Ten V. S., “Mitochondrial dysfunction contributes to alveolar developmental arrest in hyperoxia-exposed mice,” Amer. J. Respir. Cell Molecular Biol., vol. 40, no. 5, pp. 511–518, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Crapo J. D., Barry B. E., Foscue H. A., and Shelburne J., “Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen,” Amer. Rev. Respir. Disease, vol. 122, no. 1, pp. 123–143, 1980. [DOI] [PubMed] [Google Scholar]
- [9].Crapo J. D., Peters-Golden M., Marsh-Salin J., and Shelburne J. S., “Pathologic changes in the lungs of oxygen-adapted rats: A morphometric analysis,” Lab Invest., J. Tech. Methods Pathol., vol. 39, no. 6, pp. 640–653, 1978. [PubMed] [Google Scholar]
- [10].Zmijewski J. W., et al. , “Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury,” Amer. J. Respir. Critical Care Med., vol. 178, no. 2, pp. 168–179, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Audi S. H., Merker M. P., Krenz G. S., Ahuja T., Roerig D. L., and Bongard R. D., “Coenzyme Q1 redox metabolism during passage through the rat pulmonary circulation and the effect of hyperoxia,” J. Appl. Physiol., vol. 105, no. 4, pp. 1114–1126, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gan Z., Roerig D. L., Clough A. V., and Audi S. H., “Differential responses of targeted lung redox enzymes to rat exposure to 60 or 85% oxygen,” J. Appl. Physiol., vol. 111, no. 1, pp. 95–107, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gan Z., Audi S. H., Bongard R. D., Gauthier K. M., and Merker M. P., “Quantifying mitochondrial and plasma membrane potentials in intact pulmonary arterial endothelial cells based on extracellular disposition of rhodamine dyes,” Amer. J. Physiol. Lung Cellular Molecular Physiol., vol. 300, no. 5, pp. L762–L772, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bassett D. J., Bowen-Kelly E., and Reichenbaugh S. S., “Rat lung glucose metabolism after 24 h of exposure to 100% oxygen,” J. Appl. Physiol., vol. 66, no. 2, pp. 989–996, 1989. [DOI] [PubMed] [Google Scholar]
- [15].Bassett D. J., Elbon C. L., and Reichenbaugh S. S., “Respiratory activity of lung mitochondria isolated from oxygen-exposed rats,” Amer. J. Physiol., vol. 263, no. 4, pp. L439–L445, 1992. [DOI] [PubMed] [Google Scholar]
-
[16].Fisher A. B., “Energy status of the rat lung after exposure to elevated
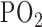 ,” J. Appl. Physiol., vol. 45, no. 1, pp. 56–59, 1978. [DOI] [PubMed] [Google Scholar]
,” J. Appl. Physiol., vol. 45, no. 1, pp. 56–59, 1978. [DOI] [PubMed] [Google Scholar] - [17].Audi S. H., et al. , “Effect of chronic hyperoxic exposure on duroquinone reduction in adult rat lungs,” Amer. J. Physiol. Lung Cellular Molecular Physiol., vol. 289, no. 5, pp. L788–L797, 2005. [DOI] [PubMed] [Google Scholar]
- [18].Staniszewski K., Audi S. H., Sepehr R., Jacobs E. R., and Ranji M., “Surface fluorescence studies of tissue mitochondrial redox state in isolated perfused rat lungs,” Ann. Biomed. Eng., vol. 41, no. 4, pp. 827–836, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fisher A. B., “Intermediary metabolism of the lung,” Environ. Health Perspect., vol. 55, pp. 149–158, Apr. 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ramanujam N., Richards-Kortum R., Thomsen S., Mahadevan-Jansen A., Follen M., and Chance B., “Low temperature fluorescence imaging of freeze-trapped human cervical tissues,” Opt. Exp., vol. 8, no. 6, pp. 335–343, 2001. [DOI] [PubMed] [Google Scholar]
-
[21].Audi S. H., Roerig D. L., Haworth S. T., and Clough A. V., “Role of glutathione in lung retention of
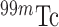 -hexamethylpropyleneamine oxime in two unique rat models of hyperoxic lung injury,” J. Appl. Physiol., vol. 113, no. 4, pp. 658–665, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
-hexamethylpropyleneamine oxime in two unique rat models of hyperoxic lung injury,” J. Appl. Physiol., vol. 113, no. 4, pp. 658–665, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar] - [22].Barrientos A., Fontanesi F., and Diaz F. “Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays,” in Current Protocols in Human Genetics, New York, NY, USA: Wiley, 2009, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
[23].Bassett D. J. and Reichenbaugh S. S., “Tricarboxylic acid cycle activity in perfused rat lungs after
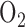 exposure,” Amer. J. Physiol., vol. 262, no. 4, pp. L495–L501, 1992. [DOI] [PubMed] [Google Scholar]
exposure,” Amer. J. Physiol., vol. 262, no. 4, pp. L495–L501, 1992. [DOI] [PubMed] [Google Scholar] - [24].Gardner P. R., Nguyen D. D., and White C. W., “Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs,” Proc. Nat. Acad. Sci. USA, vol. 91, no. 25, pp. 12248–12252, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Currie W. D., Pratt P. C., and Sanders A. P., “Hyperoxia and lung metabolism,” Chest, vol. 66, pp. 19S–21S, Jul. 1974. [DOI] [PubMed] [Google Scholar]
- [26].Bhandari V., “Molecular mechanisms of hyperoxia-induced acute lung injury,” Front Biosci., vol. 13, pp. 6653–6661, 2008. [DOI] [PubMed] [Google Scholar]
-
[27].Brueckl C., et al. , “Hyperoxia-induced reactive oxygen species formation in pulmonary capillary endothelial cells
 ,” Amer. J. Respir. Cell Molecular Biol., vol. 33, no. 4, pp. 455–462, 2006, doi: 10.1165/rcmb.2005-0223OC. [DOI] [PubMed] [Google Scholar]
,” Amer. J. Respir. Cell Molecular Biol., vol. 33, no. 4, pp. 455–462, 2006, doi: 10.1165/rcmb.2005-0223OC. [DOI] [PubMed] [Google Scholar] - [28].Fisher A. B., Forman H. J., and Glass M., “Mechanisms of pulmonary oxygen toxicity,” Lung, vol. 162, no. 1, pp. 255–259, 1984. [DOI] [PubMed] [Google Scholar]
- [29].Zhao H. W., Ali S. S., and Haddad G. G., “Does hyperoxia selection cause adaptive alterations of mitochondrial electron transport chain activity leading to a reduction of superoxide production?,” Antioxid. Redox Signal., vol. 16, no. 10, pp. 1071–1076, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Campian J. L., Qian M., Gao X., and Eaton J. W., “Oxygen tolerance and coupling of mitochondrial electron transport,” J. Biol. Chem., vol. 279, no. 45, pp. 46580–46587, 2004. [DOI] [PubMed] [Google Scholar]
- [31].Li J., Gao X., Qian M., and Eaton J. W., “Mitochondrial metabolism underlies hyperoxic cell damage,” Free Radical Biol. Med., vol. 36, no. 11, pp. 1460–1470, 2004. [DOI] [PubMed] [Google Scholar]
- [32].Turrens J. F., “Mitochondrial formation of reactive oxygen species,” J. Physiol., vol. 552, pp. 335–344, Oct. 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sanders S. P., et al. , “Hyperoxic sheep pulmonary microvascular endothelial cells generate free radicals via mitochondrial electron transport,” J. Clinical Invest., vol. 91, no. 1, pp. 46–52, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ruchko M., Gorodnya O., LeDoux S. P., Alexeyev M. F., Al-Mehdi A. B., and Gillespie M. N., “Mitochondrial DNA damage triggers mitochondrial dysfunction and apoptosis in oxidant-challenged lung endothelial cells,” Amer. J. Physiol. Lung Cellular Molecular Physiol., vol. 288, no. 3, pp. L530–L535, 2005. [DOI] [PubMed] [Google Scholar]
- [35].Swalwell H., et al. , “Respiratory chain complex I deficiency caused by mitochondrial DNA mutations,” Eur. J. Human Genet., vol. 19, pp. 769–775, Mar. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Paradies G., Petrosillo G., Pistolese M., and Ruggiero F. M., “Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage,” Gene, vol. 286, no. 1, pp. 135–141, 2002. [DOI] [PubMed] [Google Scholar]
- [37].Chicco A. J. and Sparagna G. C., “Role of cardiolipin alterations in mitochondrial dysfunction and disease,” Amer. J. Physiol. Cell Physiol., vol. 292, no. 1, pp. C33–C44, 2007. [DOI] [PubMed] [Google Scholar]
- [38].Huang L., Tang D., Yappert M. C., and Borchman D., “Oxidation-induced changes in human lens epithelial cells 2. Mitochondria and the generation of reactive oxygen species,” Free Radical Biol. Med., vol. 41, no. 6, pp. 926–936, 2006. [DOI] [PubMed] [Google Scholar]
- [39].Ide T., et al. , “Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction,” Circ Res, vol. 88, no. 5, pp. 529–535, 2001. [DOI] [PubMed] [Google Scholar]
- [40].Barrientos A. and Moraes C. T., “Titrating the effects of mitochondrial complex I impairment in the cell physiology,” J. Biol. Chem., vol. 274, no. 23, pp. 16188–16197, 1999. [DOI] [PubMed] [Google Scholar]
- [41].Fisher A. B., Furia L., and Chance B., “Evaluation of redox state of isolated perfused rat lung,” Amer. J. Physiol., vol. 230, no. 5, pp. 1198–1204, 1976. [DOI] [PubMed] [Google Scholar]
- [42].Suranadi I. W., Demaison L., Chate V., Peltier S., Richardson M., and Leverve X., “An increase in the redox state during reperfusion contributes to the cardioprotective effect of GIK solution,” J. Appl. Physiol., vol. 113, no. 5, pp. 775–784, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nuutinen E. M., “Subcellular origin of the surface fluorescence of reduced nicotinamide nucleotides in the isolated perfused rat heart,” Basic Res. Cardiol., vol. 79, no. 1, pp. 49–58, 1984. [DOI] [PubMed] [Google Scholar]
- [44].Aldakkak M., Stowe D. F., Lesnefsky E. J., Heisner J. S., Chen Q., and Camara A. K., “Modulation of mitochondrial bioenergetics in the isolated Guinea pig beating heart by potassium and lidocaine cardioplegia: Implications for cardioprotection,” J. Cardiovascular Pharmacol., vol. 54, no. 4, pp. 298–309, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kunz W. S. and Kunz W., “Contribution of different enzymes to flavoprotein fluorescence of isolated rat liver mitochondria,” Biochim. Biophys. Acta, vol. 841, no. 3, pp. 237–246, 1985. [DOI] [PubMed] [Google Scholar]
- [46].Kunz W. S. and Gellerich F. N., “Quantification of the content of fluorescent flavoproteins in mitochondria from liver, kidney cortex, skeletal muscle, and brain,” Biochem. Med. Metabolic Biol., vol. 50, no. 1, pp. 103–110, 1993. [DOI] [PubMed] [Google Scholar]
-
[47].Frank L., Iqbal J., Hass M., and Massaro D., “New ‘rest period’ protocol for inducing tolerance to high
 exposure in adult rats,” Amer. J. Physiol., vol. 257, no. 4, pp. L226–L231, 1989. [DOI] [PubMed] [Google Scholar]
exposure in adult rats,” Amer. J. Physiol., vol. 257, no. 4, pp. L226–L231, 1989. [DOI] [PubMed] [Google Scholar] - [48].Block E. R. and Fisher A. B., “Depression of serotonin clearance by rat lungs during oxygen exposure,” J. Appl. Physiol., vol. 42, no. 1, pp. 33–38, 1977. [DOI] [PubMed] [Google Scholar]
-
[49].Klein L. S., Fisher A. B., Soltoff S., and Coburn R. F., “Effect of
 exposure on pulmonary metabolism of prostaglandin E2,” Amer. Rev. Respir. Disease, vol. 118, no. 3, pp. 622–625, 1978. [DOI] [PubMed] [Google Scholar]
exposure on pulmonary metabolism of prostaglandin E2,” Amer. Rev. Respir. Disease, vol. 118, no. 3, pp. 622–625, 1978. [DOI] [PubMed] [Google Scholar] - [50].Chance B., Legallais V., Sorge J., and Graham N., “A versatile time-sharing multichannel spectrophotometer, reflectometer, and fluorometer,” Anal. Biochem., vol. 66, no. 2, pp. 498–514, 1975. [DOI] [PubMed] [Google Scholar]
- [51].Matsubara M., et al. , “In vivo fluorometric assessment of cyclosporine on mitochondrial function during myocardial ischemia and reperfusion,” Ann. Thoracic Surgery, vol. 89, no. 5, pp. 1532–1537, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mayevsky A. and Rogatsky G. G., “Mitochondrial function in vivo evaluated by NADH fluorescence: From animal models to human studies,” Amer. J. Physiol. Cell Physiol., vol. 292, pp. C615–C640, Feb. 2007. [DOI] [PubMed] [Google Scholar]
- [53].Wunsch H., Linde-Zwirble W. T., Angus D. C., Hartman M. E., Milbrandt E. B., and Kahn J. M., “The epidemiology of mechanical ventilation use in the United States,” Critical Care Med., vol. 38, no. 10, pp. 1947–1953, 2010. [DOI] [PubMed] [Google Scholar]
- [54].Kumar G., et al. , “Outcomes of morbidly obese patients receiving invasive mechanical ventilation: A nationwide analysis,” Chest, vol. 144, no. 1, pp. 48–54, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gajic O., et al. , “Ventilator-associated lung injury in patients without acute lung injury at the onset of mechanical ventilation,” Critical Care Med., vol. 32, no. 9, pp. 1817–1824, 2004. [DOI] [PubMed] [Google Scholar]
- [56].Trillo-Alvarez C., et al. , “Acute lung injury prediction score: Derivation and validation in a population-based sample,” Eur. Respir. J., vol. 37, no. 3, pp. 604–609, 2011. [DOI] [PubMed] [Google Scholar]
- [57].Rubenfeld G. D., et al. , “Incidence and outcomes of acute lung injury,” New England J. Med., vol. 353, pp. 1685–1693, Oct. 2005. [DOI] [PubMed] [Google Scholar]
-
[58].Nagase T., et al. , “Acute lung injury by sepsis and acid aspiration: A key role for cytosolic phospholipase
 ,” Nature Immunol., vol. 1, no. 1, pp. 42–46, 2000. [DOI] [PubMed] [Google Scholar]
,” Nature Immunol., vol. 1, no. 1, pp. 42–46, 2000. [DOI] [PubMed] [Google Scholar] - [59].Elie-Turenne M. C., et al. , “Lung injury prediction score for the emergency department: First step towards prevention in patients at risk,” Int. J. Emerg. Med., vol. 5, no. 1, pp. 5/33_1–5/33_11, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Phua J., et al. , “Has mortality from acute respiratory distress syndrome decreased over time? A systematic review,” Amer. J. Respir. Critical Care Med., vol. 179, no. 3, pp. 220–227, 2009. [DOI] [PubMed] [Google Scholar]
- [61].Panngom K., Baik K. Y., Nam M. K., Han J. H., Rhim H., and Choi E. H., “Preferential killing of human lung cancer cell lines with mitochondrial dysfunction by nonthermal dielectric barrier discharge plasma,” Cell Death Disease, vol. 4, pp. e642-1–e642-8, May 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sommer S. P., et al. , “Ischemia-reperfusion injury-induced pulmonary mitochondrial damage,” J. Heart Lung Transplant., vol. 30, no. 7, pp. 811–818, 2011. [DOI] [PubMed] [Google Scholar]
- [63].Matute-Bello G., Frevert C. W., and Martin T. R., “Animal models of acute lung injury,” Amer. J. Physiol. Lung Cellular Molecular Physiol, vol. 295, no. 3, pp. L379–L399, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Krisciukaitis A., et al. , “Heart tissue viability monitoring in vivo by using combined fluorescence, thermography and electrical activity measurements,” Biomed. Tech., vol. 50, no. 12, pp. 419–425, 2005. [DOI] [PubMed] [Google Scholar]
- [65].Ranji M., Jarggard D., Apreleva S., Vinogradov S., and Chance B., “Heart tissue viability monitoring in vivo by using combined fluorescence, thermography and electrical activity measurements,” Opt. Lett., vol. 31, pp. 2995–2997, Jan. 2006.17001378 [Google Scholar]
- [66].Ranji M., “Fluorescent images of mitochondrial redox states of in situ mouse hypoxic ischemic intestines,” J. Innovative Opt. Health Sci., vol. 2, pp. 365–374, Oct. 2009. [Google Scholar]