

Abstract
Whereas adipocytes have a unique capacity to store excess free fatty acids in the form of triglyceride in lipid droplets, non-adipose tissues, such as liver, have a limited capacity for storage of lipids. Saturated long-chain fatty acids, such as palmitate, are the major contributors to lipotoxicity. Silymarin is a mixture of flavonolignans, extracted from the milk thistle (Silibum marianum). Its hepatoprotective properties have been studied both in vitro and in vivo; however, its effect on palmitate-induced lipotoxicity has not been investigated. The objective of this study was to investigate (i) whether silymarin could protect HepG2 cells from palmitate-induced cell death in an in vitro model, and (ii) possible mechanisms involved in this hepatoprotective role of silymarin. HepG2 cells were treated with palmitate in the absence or presence of silymarin and supernatants or cell lysates were collected at varying time-points. Cell death was assayed by measuring DNA fragmentation, caspase-3 activity and lactate dehydrogenase release. Lipid peroxidation was assessed by measuring malondialdehyde and 4-hydroxyalkenals. Akt kinase activity was also measured. Incubation with palmitate caused significant death in HepG2 cells. Palmitate incubation did not cause significant changes in reactive oxygen species production or intracellular glutathione content, but markedly inhibited Akt kinase activity. Pre-treatment of HepG2 cells with silymarin prevented palmitate-induced inhibition of Akt kinase activity and attenuated cell death. Our results suggest that silymarin may be an effective agent in protecting hepatocytes from saturated fatty acids-induced cell death. These data also provide a further rationale for exploration of the use of silymarin in the treatment of non-alcoholic steatohepatitis.
Non-alcoholic steatohepatitis (NASH), a part of the spectrum of non-alcoholic fatty liver disease (NAFLD), is becoming an increasingly recognized problem in the USA and around the world because of the prevalence of obesity, diabetes and insulin resistance syndrome [1–5]. Histopathology on liver biopsy in NASH is almost identical to the damage induced by alcohol abuse. The presence of macrovesicular steatosis, inflammatory cell infiltration, hepatocyte ballooning and apoptosis/necrosis are the main histological features of NASH [6]. NASH is the most prevalent form of progressive liver disease in the USA. It is estimated that approximately 50% of NASH patients developed liver fibrosis, 15–30% developed cirrhosis and 3% may progress to liver failure [7]; thus, there is an increasing need to understand the aetiology and treatment of this liver disease.
The widely accepted ‘two-hit’ model of NASH is viewed as the first ‘hit’ being the development of fatty liver followed by the second ‘hit’ that leads to hepatocyte injury; however, the aetiology of NASH remains elusive [8]. Recent studies have reported that obesity and insulin resistance are associated with an increased frequency of NASH independent of the severity of steatosis. Elevated serum levels of free fatty acids due to obesity and/or insulin resistance cause the accumulation of excess free fatty acids in non-adipose tissues. Emerging evidence suggests that elevated serum free fatty acid levels play an aetiologic role in the pathogenesis of NASH and are postulated to be a critical link between obesity, insulin resistance and the risk of NASH [4].
Adipocytes have a unique capacity to store excess free fatty acids in the form of triglyceride in lipid droplets, but non-adipose tissues, such as liver, have a limited capacity for storage of lipids. Accumulation of excess lipid in non-adipose tissues may lead to cell dysfunction and/or cell death, a phenomenon known as lipotoxicity. However, saturated and unsaturated free fatty acids differ significantly in their contribution to lipotoxicity. Many studies have shown that saturated long-chain fatty acids are the major contributor to lipotoxicity. Palmitate, a long-chain (C16:0) saturated fatty acid, has been reported to induce apoptosis in many cell types, including cardiomyocytes [9], haematopoietic cells [10], pancreatic β-cells [11] and astrocytes [12]. Although the mechanism(s) by which palmitate induces cell damage and cell death in these cell types are not completely elucidated, it is generally accepted that reactive oxygen species (ROS) and cytochrome c are two major intracellular mediators of palmitate effects [13–17].
Silymarin, a mixture of flavonolignans, is extracted from the milk thistle (Silibum marianum). It consists of several flavonolignans: silybin A, silybin B, isosilybin A, isosilybin B, silydianin and silychristin [18]. It has been reported to prevent liver injuries induced by various chemicals or toxins. In vivo studies have demonstrated that silymarin exerts a protective effect in hepatocytes exposed to a range of molecules, including tert-butyl hydroperoxide, phenylhydrazine, acetaminophen and carbon tetrachloride [19–22].
This study was conducted to investigate (i) whether palmitate could induce cell death in cultured HepG2 cells, (ii) whether silymarin supplementation has a protective role in palmitate-induced liver injury in an in vitro model, and (iii) possible mechanism(s) involved in both palmitate-induced hepatocyte death and protection by silymarin.
Materials and Methods
Cells and culture conditions
HepG2 cells, a human hepatoma cell line, were obtained from the American Type Culture Collection (Manassas, VA, USA) and were cultured in Dulbecco’s modified Eagle’s medium, containing 10% (v/v) foetal bovine serum, 2 mM glutamine, 5 U/ml penicillin and 50 μg/ml streptomycin at 37°C in a humidified O2/CO2 (19:1) atmosphere.
Preparation of palmitate-albumin complexes
Palmitate-albumin complex preparation was produced by a saponification technique according to Goldstein et al. [23] using palmitate (C16:0) for complexing to bovine serum albumin (BSA), which is essentially fatty-acid free. Briefly, palmitate was dissolved in ethanol and saponified with sodium hydroxide. The sodium salt was dried, resuspended in saline and heated for 5 min. at 70°C. While the solution was still warm, 25% (w/v) BSA was added and the mixture was stirred at room temperature for 4 hr to allow palmitate to bind to albumin. Palmitate-BSA complex (6.4 mmol/l fatty acid: 0.9 mmol/l BSA, molar ratio, 7:1) was then sterilized by filtering, aliquoted and frozen for future use. These conditions were chosen to mimic pathophysiologic states in which free fatty acid : bovine serum albumin molar ratio approaches 7:1[24] and yield unbound free fatty acid concentrations in the several hundred nM range.
Lactate dehydrogenase assay
Cell death was measured by the release of lactate dehydrogenase (LDH) into the culture medium. LDH activity was determined spectrophotometrically at 340 nm by following the rate of NAD+ reduction in the presence of L-lactate.
ELISA assay for DNA fragmentation
DNA fragmentation was measured 6 hr after treatments using the cell death ELISA kit manufactured by Roche (Indianapolis, IN, USA).
Caspase activities assay
Caspase-3 activity was measured using the assay kit from Calbiochem (San Diego, CA, USA) according to the manufacturer’s instructions.
HPLC assay for intracellular glutathione
Intracellular reduced glutathione (GSH) was quantified by HPLC with electrochemical detection according to the method of Richie and Lang [25], with slight modifications. In brief, 20-μl samples were injected onto a reverse-phase C18 column (Val-U-Pak HP, fully end-capped ODS, 5μm, 250 × 4.6 mm; ChromTech Inc., Apple Valley, MN, USA). The mobile phase, which consisted of a solution of 0.1 M mono-chloroacetic acid and 2 mM heptanesulfonic acid at pH 2.8 (98%) and acetonitrile (2%), was delivered at a flow rate of 1 ml/min. The compounds were detected in the eluent with a Bioanalytical Systems dual LC4B amperometric detector, using two Au/Hg electrodes in series with potentials of −1.2 V and +0.15 V for the upstream and downstream electrodes, respectively. Standard curves for the analyses were plotted as peak area versus concentration of the analyse.
Measurement of intracellular ROS production
2′,7′-Dichloro-hydrofluorescein diacetate (DCFH-DA; Molecular Probes, Eugene, OR, USA) is a cell-permeable non-fluorescent compound that is cleaved into fluorescent products in the presence of H2O2 and other ROS molecules and esterases. Thus, it allows for the measurement of ROS production within cells. Two hours before the end of the incubation with the indicated stimulus, the medium was removed and the cells were washed and placed in HEPES-DMEM without serum. DCFH-DA was added to each well of a 24-well plate at a final concentration of 10 μM. At the completion of the incubation, the cells were placed on ice, washed three times with ice-cold phosphate-buffered saline, and then lysed in 0.3 ml of cold Cell Signaling Lysis Buffer (Cell Signaling Technology, Beverly, MA, USA). The cells were scraped off the plate and sonicated on ice for 10 times, to rupture the cells. The lysates were then centrifuged at 18,300 ×g at 4°C for 5 min. The supernatants were transferred to a 96-well plate in which fluorescence was measured at an emission wavelength of 530 nm, using an excitation wavelength of 485 nm for the 2′,7′-dichloro-fluorescein fluorophore. All data are representative of at least three independent experiments and are expressed as means ± S.D.
Lipid peroxidation measurement
Lipid peroxides were measured using the assay kit from Calbiochem according to manufacturer’s instruction. Malondialdehyde and 4-hydroxyalkenals are end-products derived from peroxidation of polyunsaturated fatty acids and related esters. Measurement of such aldehydes provides a convenient index of lipid peroxidation.
Western blot analysis
Cytosolic and nuclear protein extracts were isolated according to the method of Dignam [26]. Protein extracts were analysed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis electrophoresis. The nitrocellulose membrane was incubated with appropriate polyclonal or monoclonal antibody against target proteins. The blot in each case was developed using an enhanced chemiluminescence (ECL system; Amersham Pharmacia Biotech, Piscataway, NJ, USA) according to the manufacturer’s instructions.
ELISA assay for Akt kinase activity
Akt kinase activity was quantified using ELISA kits (BioSource, Camarillo, CA, USA) in accordance with manufacturer’s instructions. All assays were run in triplicate.
Statistical analysis
All data are expressed as mean ± S.D. Statistical analysis was performed using one-way ANOVA and further analysed by Newman–Keuls test for statistical significance. Differences between treatments were considered to be statistically significant at P < 0.05.
Results
Silymarin protects HepG2 cells from palmitate-induced cell death
Cell death was measured by DNA fragmentation ELISA. As shown in fig. 1A, inclusion of palmitate in the medium caused a dose-dependent increase in cell death in HepG2 cells. Palmitate at 0.25 mM has already caused a significant increase in DNA fragmentation; therefore, this concentration of palmitate was used in all following experiments. Figure 1B shows time-course effects of palmitate on cell death in HepG2 cells. Two-hour pre-treatment of HepG2 cells with silymarin prevented palmitate-induced DNA fragmentation in a dose-dependent manner with 0.2 mM silymarin having an optimal protective effect (fig. 2A). Therefore, we used this concentration in all following mechanistic studies. Moreover, our results also show that incubation of HepG2 cells with palmitate caused a significant increase in necrosis demonstrated by elevated LDH release into the medium and silymarin pre-treatment attenuated palmitate-induced hepatocyte necrosis (fig. 2B).
Fig. 1.
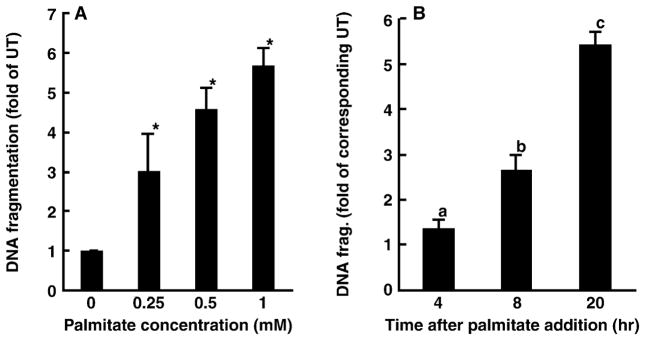
Palmitate induces cell death in HepG2 cells. Cell death was measured by DNA fragmentation ELISA assay and expressed as fold of untreated. (A) Dose response of cell death induced by palmitate. HepG2 cells were incubated with increased concentration of palmitate (0, 0.25, 0.5 and 1 mM) for 20 hr. At least three experiments were conducted for each measurement. *Value is significantly different from that seen in control cells, P < 0.05. (B) Time-course change of cell death induced by palmitate. HepG2 cells were treated with 0.25 mM palmitate for different period. Values in bars that do not share a letter differ significantly. At least three experiments were conducted for each measurement.
Fig. 2.

Silymarin attenuates palmitate-induced cell death in HepG2 cells. (A) Silymarin protected HepG2 cells from palmitate-induced cell death in a dose-dependent manner. HepG2 cells were pre-treated with different dose of silymarin for 2 hr followed by 0.25 mM palmitate treatment for 20 hr. At least three experiments were conducted for each measurement. *Value is significantly different from that seen in control cells, P < 0.05. (B) Silymarin protects HepG2 cells from palmitate-induced lactate dehydrogenase (LDH) (U/l) release in a dose-dependent manner. HepG2 cells were pre-treated with a different dose of silymarin for 2 hr followed by 0.25 mM palmitate treatment for 20 hr. At least three experiments were conducted for each measurement. *Value is significantly different from that seen in control cells; †Value is statistically different from that seen in the absence of silymarin, P < 0.05.
Silymarin prevents palmitate-induced caspase-3 activation
As shown in fig. 3A, incubation of HepG2 cells with palmitate in the presence of Z-DQMD-FMK, a cell-permeable and irreversible inhibitor of caspase-3, significantly attenuated cell death, suggesting that apoptosis was the major form of cell death induced by palmitate in HepG2 cells. The time-course effects of silymarin on caspase-3 activity were next examined. As shown in fig. 3B, palmitate caused a 2-fold increase in caspase-3 activity in comparison with untreated cells at 8 hr and the increase was maintained at 16 hr after palmitate treatment. Silymarin pre-treatment markedly attenuated palmitate-induced increase of caspase-3 activity at both time-points. This result was also confirmed by Western blot as shown in fig. 3C.
Fig. 3.

Effects of silymarin on palmitate-induced caspase-3 activation in HepG2 cell. (A) Caspase-3 inhibitor inhibited palmitate-induced cell death in HepG2. Apoptosis was measured by DNA fragmentation ELISA assay and expressed as fold of untreated. At least three experiments were conducted for each measurement. Values in bars that do not share a letter differ significantly, P < 0.05. Both (B) enzymatic activity assay and (C) Western blot analysis indicated that silymarin pre-treatments prevented palmitate-induced caspase-3 activation. *Value is statistically different from that seen in the absence of silymarin, P < 0.05.
Silymarin protects against palmitate-induced cell death independently of its antioxidant properties
It has been widely accepted that one of major hepatoprotective activities of silymarin derives from its antioxidant property; therefore, we first investigated if palmitate caused lipid peroxidation in HepG2 cells by measuring intracellular malondialdehyde + 4-hydroxyalkenal content. As shown in fig. 4A, culturing HepG2 cells with 0.25 mM palmitate for 20 hr did not cause lipid peroxidation as shown by unchanged intracellular malondialdehyde + 4-hydroxyalkenal levels in comparison with control cells. Intracellular GSH plays a central role in cellular antioxidant defence, and its loss may render cells more prone to oxidative stress. We then examined the effect of palmitate on intracellular GSH levels with or without pre-treatment of either silymarin or N-acetylcysteine, a precursor of GSH. As shown in fig. 4B, intracellular GSH levels were unexpectedly increased by palmitate treatment after 4 hr with no difference detected at 8 hr. Both silymarin and N-acetylcysteine pre-treatment increased intracellular GSH levels significantly; however, silymarin protected palmitate-induced cell death while N-acetylcysteine did not (fig. 4C). To further verify our results, we directly measured ROS production by HepG2 cells using DCFH-DA and results were shown in fig. 4D. The membrane-permeable dye DCFH-DA enters cells and produces a fluorescent signal after intracellular oxidation by ROS such as hydrogen peroxide and hydroxyl radical. HepG2 cells were incubated with palmitate for 1, 4 and 20 hr, respectively. Incubation of HepG2 cells with palmitate did not lead to increased oxidation of DCFH-DA at all time-points, while incubation of HepG2 cells with 300 μM H2O2 for 1 hr resulted in a significant increase in the fluorescence in comparison with control cells, suggesting that protective effect of silymarin was independent of its antioxidant properties.
Fig. 4.

Protective effect of silymarin on palmitate-induced cell death is independent of its antioxidant property. (A) Changes of intracellular malondialdehyde (MDA) + 4-hydroxyalkenals (HAE) content (μM). HepG2 cells were incubated with 0.25 mM palmitate for 20 hr with or without silymarin or N-acetylcysteine (NAC) pre-treatment. (B) Changes of intracellular glutathione (GSH) content (nmol/million cells). HepG2 cells were incubated with 0.25 mM palmitate for 4 and 8 hr, respectively, with or without silymarin or N-acetylcysteine (NAC) pre-treatment. *Value is significantly different from that seen in the presence of palmitate alone, P < 0.05. (C) NAC did not protect palmitate-induced cell death in HepG2 cells. Apoptosis was measured by DNA fragmentation ELISA assay and expressed as fold of untreated. Values in bars that do not share a letter differ significantly. (D) Changes of 2′,7′-dichlorohydrofluorescein diacetate fluorescent products. HepG2 cells were incubated with 300 μM H2O2 for 1 hr or with palmitate for 1, 4 and 20 hr, respectively. Data are expressed as Arbitrary Fluorescence Units. *Value is significantly different from that seen in the control cells, P < 0.05. At least three experiments were conducted for each measurement.
Silymarin prevents palmitate-induced decrease of Akt kinase activity
Akt kinase is a critical survival factor in hepatocytes and plays an important role in preventing hepatocytes from injury induced by a variety of pro-apoptotic stimuli. To examine whether the alteration of Akt kinase activity was involved in palmitate-induced cell death in HepG2 cells and whether silymarin pre-treatment maintained Akt kinase activity, we measured Akt kinase activiy in HepG2 cells treated with silymarin. As shown in fig. 5A, palmitate at 0.25 mM significantly lowered intracellular Akt kinase activity, and pre-treatment with silymarin counteracted the effect of palmitate. Moreover, inhibition of Akt kinase activity by Akt inhibitor-IV (Calbiochem), a specific Akt kinase inhibitor, further increased palmitate-induced cell death, indicating that inhibition of Akt kinase activity plays a critical role in palmitate-induced cell death in HepG2 cells (fig. 5B).
Fig. 5.

Maintenance of Akt kinase activation involved in the protective effect of silymarin on palmitate-induced cell death. (A) Silymarin prevented palmitate-induced inhibition on Akt kinase activation in HepG2 cells. Akt kinase activity (U/mg protein) was measured when incubation of HepG2 cells with 0.25 mM palmitate with or without silymarin pre-treatment. At least three experiments were conducted for each measurement. Values in bars that do not share a letter differ significantly, P < 0.05. (B) Inhibition of Akt kinase induced cell death in HepG2. At least three experiments were conducted for each measurement. *Value is significantly different from that seen in the control cells; †Value is significantly different from that seen in the presence of palmitate alone, P < 0.05.
Discussion
Lipotoxicity has been implicated in the pathogenesis of NAFLD, and free fatty acids appear to be important contributors of lipotoxicity [27]. In humans with NAFLD, circulating free fatty acids are elevated and their levels correlate with disease severity [28]. Therefore, agents with the ability to prevent or attenuate free fatty acids induced hepatocyte death represent a promising therapeutic choice for NAFLD. In this study, we examined the potential protective effects of silymarin on palmitate (C16:0)-induced lipotoxicity in cultured HepG2 cells. Our results demonstrate that inclusion of palmitate in the medium induced cell death in HepG2 cells, which was associated with its inhibitory effects on Akt kinase. Silymarin has a property of preventing inhibition of Akt kinase induced by palmitate exposure and protecting hepatocytes from palmitate-induced cell death.
Saturated and unsaturated free fatty acids differ significantly in their contribution to lipotoxicity. Much evidence has shown that saturated long-chain fatty acids are the major contributor to lipotoxicity [9]. Palmitate is one of the most common fatty acids encountered in mammals and induces apoptotic cell death in various cell types [9–17,24,25]. Although the mechanisms are still unclear, the generation of ROS has been postulated to be one of the major intracellular pathways. For instance, Yamagishi et al. [13] reported in a recent study that intracellular formation of ROS increased approximately 1.4-fold after treatment with 0.1 mM palmitate for 4 hr in cultured microvascular endothelial cells from human skin microvessels. Moreover, palmitate-induced inhibition of DNA synthesis and cell death were completely prevented by N-acetylcysteine, a GSH precursor. Similar results were found in cultured bovine retinal pericytes [14]. However, contrary to the above reports, Hickon-Bick et al. [24] has shown that palmitate-induced apoptosis in different cell types, including neonatal cardiomyocytes, rat fibroblasts and Chinese hamster ovary cells, was not dependent on the generation of ROS. Therefore, it is likely that molecular mechanisms involved in palmitate-induced cell death are cell-type dependent. Although palmitate-induced cell death in HepG2 cells has been reported, the mechanisms are still unclear [29,30]. In our experiments, we first examined the involvement of oxidative stress in palmitate-induced cell death and the protective role of antioxidants. We concluded that oxidative stress was not the principal mechanism underlying palmitate-induced death in HepG2 cells. This conclusion is based on the following results: (i) incubation of HepG2 cells with 0.25 mM palmitate for 20 hr did not cause lipid peroxidation as demonstrated by unchanged intracellular malondialdehyde + 4-hydroxyalkenal levels in comparison with control cells, (ii) palmitate incubation had no effects on intracellular GSH content, (iii) N-acetylcysteine, a precursor of GSH, did not provide protection against palmitate-induced cell death in our system, and (iv) most importantly, there was no increased H2O2 production in palmitate-treated cells.
Akt kinase is an important survival factor in a variety of cells. The anti-apoptotic property of phosphatidylinositol 3-kinase (PI3-K)/Akt pathway has been well documented in many studies involving different cell types including hepatocytes. Activation of PI3K/Akt pathway by repeated short-term, low-dose exposure to ethanol protected isolated neonatal rat cardiomyocytes from palmitate-induced apoptosis [31]. Moreover, via activating PI3-kinase/Akt kinase pathway, glucagon-like peptide-1 prevented apoptosis in human islets and INS832/13 cells induced by palmitate [32]. Furthermore, PI3K/Akt pathway plays a critical role in the tumour necrosis factor-induced hepatocyte apoptosis. Suppression of PI3K/Akt resulted in increased activation of caspase-3 and massive hepatocyte apoptosis in mice after tumour necrosis factor administration [33]. The observations from the current study demonstrate that apoptosis represents the major form of cell death induced by palmitate in HepG2 cells. Moreover, we examined the effect of palmitate on Akt kinase activity in HepG2 cells and demonstrated that palmitate caused significant inhibition of Akt kinase activation. Furthermore, our results show that Akt kinase inhibitor induced significant apoptosis, indicating that Akt kinase was a critical survival protein in HepG2 cells to counteract palmitate-induced cell death. To further investigate if activation of Akt kinase was involved in the protective effects of silymarin on palmitate-induced cell death, we examined the effect of silymarin supplementation on the activity of Akt kinase. Our results demonstrate that silymarin pretreatment attenuated the Akt kinase inhibition induced by palmitate. Conversely, Akt kinase inhibitor enhanced palmitate-induced cell death, indicating that prevention of palmitate-induced Akt kinase inhibition represented one major mechanism in silymarin’s hepato-protective function; however, the mechanism behind is still elusive.
In summary, this study shows that silymarin protected HepG2 cell against palmitate-induced cell death by preventing palmitate-induced inhibition of Akt kinase activity. The hepatoprotective activity of silymarin was independent of its antioxidant property. Our present study demonstrates that silymarin may be an effective agent in protecting hepatocytes against palmitate-induced lipotoxicity and suggest that silymarin may have beneficial effects in the treatment of fatty liver diseases such as non-alcoholic steatohepatitis.
Acknowledgments
This research was supported by the National Institutes of Health grants (Z. Song, C. J. McClain and D. Lee) and the Department of Veterans Affairs (C. J. McClain).
References
- 1.Harrison SA, Neuschwander-Tetri BA. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Clin Liver Dis. 2004;8:861–79. doi: 10.1016/j.cld.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 2.Dixon JB, Bhathal PS, O’Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121:91–100. doi: 10.1053/gast.2001.25540. [DOI] [PubMed] [Google Scholar]
- 3.Marchesini G, Brizi M, Morselli-Labate AM, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999;107:450–5. doi: 10.1016/s0002-9343(99)00271-5. [DOI] [PubMed] [Google Scholar]
- 4.Choudhury J, Sanyal AJ. Insulin resistance in NASH. Front Biosci. 2005;10:1520–33. doi: 10.2741/1636. [DOI] [PubMed] [Google Scholar]
- 5.Choudhury J, Sanyal AJ. Insulin resistance and the pathogenesis of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:575–94. doi: 10.1016/j.cld.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 6.Brunt EM, Tiniakos DG. Pathological features of NASH. Front Biosci. 2005;10:1475–84. doi: 10.2741/1632. [DOI] [PubMed] [Google Scholar]
- 7.Bugianesi E, Leone N, Vanni E, et al. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123:134–40. doi: 10.1053/gast.2002.34168. [DOI] [PubMed] [Google Scholar]
- 8.Day CP, James OFW. Steatohepatitis: a tale of two ‘hits’? Gastroenterology. 1998;114:842–5. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 9.DeVries JE, Vork M, Roemen THM, et al. Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J Lipid Res. 1997;38:1384–94. [PubMed] [Google Scholar]
- 10.Paumen MB, Ishida Y, Muramatsu M, Yamamoto M, Honjo T. Inhibition of carnitine palmitoyltransferase I augments sphingolipid synthesis and palmitate-induced apoptosis. J Biol Chem. 1997;272:3324–9. doi: 10.1074/jbc.272.6.3324. [DOI] [PubMed] [Google Scholar]
- 11.Shimabukuro M, Zhou Y-T, Levi M, Unger RH. Fatty acid-induced β-cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA. 1998;95:2498–502. doi: 10.1073/pnas.95.5.2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blazquez C, Galve-Roperh I, Guzman M. De novo synthesized ceramide signals apoptosis in astrocytes via extracellular signal-regulated kinase. FASEB J. 2000;14:2315–22. doi: 10.1096/fj.00-0122com. [DOI] [PubMed] [Google Scholar]
- 13.Yamagishi S, Okamoto T, Amano S, et al. Palmitate-induced apoptosis of microvascular endothelial cells and pericytes. Mol Med. 2002;8:179–84. [PMC free article] [PubMed] [Google Scholar]
- 14.Cacicedo JM, Benjachareowong S, Chou E, Ruderman NB, Ido Y. Palmitate-induced apoptosis in cultured bovine retinal pericytes: roles of NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes. 2005;54:1838–45. doi: 10.2337/diabetes.54.6.1838. [DOI] [PubMed] [Google Scholar]
- 15.Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. 2001;276:14890–5. doi: 10.1074/jbc.M010286200. [DOI] [PubMed] [Google Scholar]
- 16.Sparagna GC, Hickson-Bick DL, Buja LM, McMillin JB. A metabolic role for mitochondria in palmitate-induced cardiac myo-cyte apoptosis. Am J Physiol Heart Circ Physiol. 2000;279:H2124–32. doi: 10.1152/ajpheart.2000.279.5.H2124. [DOI] [PubMed] [Google Scholar]
- 17.Sparagna GC, Hickson-Bick DL, Buja LM, McMillin JB. Fatty acid-induced apoptosis in neonatal cardiomyocytes: redox signaling. Antioxid Redox Signal. 2001;3:71–9. doi: 10.1089/152308601750100524. [DOI] [PubMed] [Google Scholar]
- 18.Lee YW, Liu YZ. Molecular structure and stereochemistry of silybin A, silybin B, and isosilybin A, isosilybin B, isolated from Silybum mariaanum. J Nat Prod. 2003;66:1171–4. doi: 10.1021/np030163b. [DOI] [PubMed] [Google Scholar]
- 19.Valenzuela A, Guerra R. Protective effect of the flavonoid silybin dihemisuccinate on the toxicity of phenylhydrazine on rat liver. FEBS Lett. 1985;181:291–4. doi: 10.1016/0014-5793(85)80278-7. [DOI] [PubMed] [Google Scholar]
- 20.Campos R, Garrido A, Guerra R, Valenzuela A. Silybin dihemisuccinate protects against glutathione depletion and lipid peroxidation induced by acetaminophen on rat liver. Planta Med. 1989;55:417–9. doi: 10.1055/s-2006-962055. [DOI] [PubMed] [Google Scholar]
- 21.Halim AB, el-Ahmady O, Hassab-Allah S, Abdel-Galil F, Hafez Y, Darwish A. Biochemical effect of antioxidants on lipids and liver function in experimentally-induced liver damage. Ann Clin Biochem. 1997;34 (pt 6):656–63. doi: 10.1177/000456329703400610. [DOI] [PubMed] [Google Scholar]
- 22.Letteron P, Labbe G, Degott C, et al. Mechanism for the protective effects of silymarin against carbon tetrachloride-induced lipid peroxidation and hepatotoxicity in mice: evidence that silymarin acts both as an inhibitor of metabolic activation and as a chain-breaking antioxidant. Biochem Pharmacol. 1990;39:2027–34. doi: 10.1016/0006-2952(90)90625-u. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein JL, Basu SK, Brown MS. Receptor-mediated endocytosis of low-density lipoproteins in cultured cells. Meth Enzymol. 1983;98:241–60. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 24.Hickson-Bick DL, Sparagna GC, Buja LM, McMillin JB. Palmitate-induced apoptosis in neonatal cardiomyocytes is not dependent on the generation of ROS. Am J Physiol Heart Circ Physiol. 2002;282:H656–64. doi: 10.1152/ajpheart.00726.2001. [DOI] [PubMed] [Google Scholar]
- 25.Richie JP, Jr, Lang CA. The determination of glutathione, cyst(e)ine, and other thiols and disulfides in biological samples using high-performance liquid chromatography with dual electrochemical detection. Anal Biochem. 1987;163:9–15. doi: 10.1016/0003-2697(87)90085-6. [DOI] [PubMed] [Google Scholar]
- 26.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feldstein AE, Werneburg NW, Canbay A, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185–94. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 28.Nehra V, Angulo P, Buchman A, Lindor KD. Nutritional and metabolic considerations in the etiology of nonalcoholic steatohepatitis. Dig Dis Sci. 2001;46:2347–52. doi: 10.1023/a:1012338828418. [DOI] [PubMed] [Google Scholar]
- 29.Aydin HH, Celik HA, Deveci R, et al. Induction of apoptosis by fatty acid ethyl esters in HepG2 cells. Food Chem Toxicol. 2005;43:139–45. doi: 10.1016/j.fct.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Hasaba A, Cluette-Brown JE, Laposata M. Stearic acid stimulates FA ethyl ester synthesis in HepG2 cells exposed to ethanol. Lipids. 2003;38:1051–5. doi: 10.1007/s11745-006-1160-3. [DOI] [PubMed] [Google Scholar]
- 31.Sparagna GC, Jones CE, Hickson-Bick DL. Attenuation of fatty acid-induced apoptosis by low-dose alcohol in neonatal rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2004;287:H2209–15. doi: 10.1152/ajpheart.00247.2004. [DOI] [PubMed] [Google Scholar]
- 32.Buteau J, El-Assaad W, Rhodes CJ, Rosenberg L, Joly E, Prentki M. Glucagon-like peptide-1 prevents beta cell glucolipo-toxicity. Diabetologia. 2004;47:806–15. doi: 10.1007/s00125-004-1379-6. [DOI] [PubMed] [Google Scholar]
- 33.Hatano E, Brenner DA. Akt protects mouse hepatocytes from TNF-alpha-and Fas-mediated apoptosis through NK-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1357–68. doi: 10.1152/ajpgi.2001.281.6.G1357. [DOI] [PubMed] [Google Scholar]