

Abstract
Micropatterning is a powerful technique to control cell shape and position on a culture substrate. In this chapter, we describe the method to reproducibly create large numbers of micropatterned heterotypic cell pairs with defined size, shape, and length of cell–cell contact. These cell pairs can be utilized in patch clamp recordings to quantify electrical interactions between cardiomyocytes and non-cardiomyocytes.
Keywords: Micropatterning, Cardiac cell therapy, Electrical coupling, Stem cells, Patch clamp
1 Introduction
Systematic studies of structural and functional interactions between cardiomyocytes and other cells are critical for understanding cardiac development, function, and diseases (e.g., infarction, arrhythmias) as well as for improving the safety and efficacy of cardiac cell therapies. Specifically, a large variety of different donor cells including skeletal myoblasts [1], bone marrow-derived mesenchymal stem cells (MSCs) [2], heart-derived stem cells [3], pluripotent stem cell-derived cardiomyocytes [4], and genetically engineered somatic cells [5, 6] are currently being evaluated for their utility in cell-based cardiac repair. Direct studies of in situ interactions between these cells and host cardiomyocytes, although critical, are hampered by our inability to access and identify sites of host–donor cell contact and by high variability among different hearts and treatment outcomes. Traditional cell co-cultures have been used to aid in general understanding of in situ host–donor cell interactions; however, heterocellular studies at a single-cell level have been hampered by the inability to control individual cell size, shape, number, and type of interacting cells. To overcome these challenges, we have utilized micropatterning techniques to develop and optimize methods for the generation of large numbers of individual heterotypic cell pairs with controllable size, shape, and cell–cell contact length [7, 8]. Dual whole-cell voltage clamp recordings [9] are used to quantify the electrical coupling between the two cells in the pair, while whole-cell current clamp is employed to assess the resulting changes in electrical properties of cardiomyocytes [8].
2 Materials
2.1 Micropatterning Components
Reagents
Negative photoresist (SU8-2, Microchem Inc.).
Silane solution [(tridecafluoro-1,1,2,2-tetrahydro octyl)-1-trichlorosilane].
Propylene glycol methyl ether acetate (PGMEA): CAUTION: Wear protective goggles, gloves, and clothing, and ensure proper ventilation.
Isopropyl alcohol.
Poly-dimethylsiloxane (PDMS) silicone elastomer base and curing agent (Sylgard 184, Dow Corning).
Ethanol, 70 % in DI H2O.
Fibronectin (Human), powder.
Dulbecco’s phosphate-buffered saline, 1× (DPBS).
Triton X-100.
Paraformaldehyde, 16 % in DI H2O (PFA): CAUTION: Wear protective goggles, gloves, and clothing, and ensure proper ventilation.
Equipment
4″ Silicon wafer.
22 mm Glass cover slips.
Tissue culture 12-well plate.
Polystyrene tissue culture Petri dish, 100 mm diameter.
Crystallizing dishes, 100 mm diameter, 50 mm tall.
Programmable spin coater.
Digital hot plate.
Mask aligner with 350 W mercury arc lamp UV light source.
Vacuum desiccator and vacuum pump.
Sonicator bath.
UV decontamination system.
N2 tank and filtered N2 gun.
Wafer grip tweezers.
Fine forceps, straight.
Standard forceps, curved.
Syringe needle (21G 1½″) with tip bent 90° using needle-nose pliers.
Oven at 80 °C.
Incubator at 37 °C, 5 % CO2.
Water bath at 37 °C.
Metallurgical microscope and standard 10× objective.
Miscellaneous: Transfer pipettes, razor blades, Pasteur pipettes (vacuum linked).
Software
Metamorph image acquisition software package (Molecular Devices).
AutoCAD software package (Autodesk).
MATLAB software package (The Mathworks).
2.2 Cell Sources and Media
For a list of potential cell types, their respective culture media, and the source from which they can be obtained please see Table 1.
Table 1.
A listing of the potential cell types that can be used in cell-pair patterning experiments, their respective culture media, and the sources for each cell type
| Cell type | Media | Cell sources |
|---|---|---|
| Neonatal rat ventricular myocytes (NRVMs) |
Seeding media: DMEM/F-12 media supplemented with 10 % calf serum and 10 % horse serum [7] Maintenance media: M199 media supplemented with 2 % horse serum, HEPES (10 mM), MEM nonessential amino acids, GlutaMAX (2 mM, Invitrogen), vitamin B 12 (2 μg/ml), and penicillin-G (20 U/ml) [8] |
Dissociated from the ventricles of 2-day-old Sprague-Dawley rats by trypsin and collagenase and then enriched using two 1-h differential preplating steps to remove faster-adhering nonmyocytes [10] |
| Neonatal rat ventricular fibroblasts (NRVFs) | Fibroblast growth media: M199 medium supplemented with 10 % FBS and HEPES (10 mM), nonessential amino acid solution (×1), L-glutamine (2 mM), dextrose (3.5 mg/ml), vitamin B12 (4 μg/ml), and penicillin G (100 U/ml) [11] | Recovered from NRVM isolation during the preplating steps and, upon reaching confluence, passaged at 1:2 ratio and used at passage 1 [7] |
| Adult rat mesenchymal stem cells (MSCs) | MSC growth media: α-MEM medium supplemented with 20 % fetal bovine serum (FBS), 50 U/ml penicillin, and 50 μg/ml streptomycin [12] | Isolated from the bone marrow of 3-month-old male Sprague-Dawley rats using previously published methods [13] |
| Adult rat skeletal myoblasts (SKMs) | SKM media: DMEM supplemented with 20 % FBS and 50 μg/ml gentamicin [7] | Isolated from the hindlimb soleus muscle of female Sprague-Dawley rats as previously described [14] |
| Mouse C2C12 myoblasts | Myoblast growth media: DMEM supplemented with 10 % FBS, 10 % calf serum, 2.8 ml chick embryo extract, and 50 μg/ml gentamicin [11] | American Type Culture Collection (ATCC): Cells need to be kept below 70 % confluence to prevent myotube formation |
| Mouse embryonic stem cell-derived cardiomyocytes (mESC-CMs) or cardiovascular progenitor cells (mESC-CVPs) | Maintenance media: N2B27 media [15] with 0 or 2 % FBS | Derived from D3 mESCs (ATCC CRL-11632) using previously published differentiation protocols [16, 15] |
| Wild-type or genetically engineered human embryonic kidney 293 cells (HEK293) | HEK media: DMEM low glucose supplemented with 10 % FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin [11] | Wild-type HEK293 cells were purchased from ATCC (CRL-1573). Cells were stably transfected and selected for monoclonal lines with Cx43 [11], Kir2.1+Cx43 [8], or Na v1.5+Kir2.1+Cx43 [17] |
2.3 Patch Clamp Components
Custom-made Faraday cage for electromagnetic interference reduction.
Anti-vibration N2 air table.
Inverted fluorescence microscope.
Multiclamp 700B amplifier (Axon Instruments, Inc.).
NIDAQ-MX computer interface (National Instruments).
WINWCP software package (John Dempster, University of Strathclyde) or any other patch clamp analysis software.
Patch glass pipette 1.5 mm O.D. × 1.16 mm I.D.
Micropipette puller.
MF-200 microforge (World Precision Instruments, Inc.) and stereomicroscope (with 40× objective) for fire-polishing glass pipette.
Patch pipette is filled with pipette solution containing (in mM) 140 KCl, 10 NaCl, 1 CaCl2, 10 EGTA, 10 HEPES, 2 MgCl2, and 5 MgATP (adjusted to pH 7.2 with KOH and 270–280 mOsm with glucose) [8].
MicroFil syringe needle for filling micropipettes 34 gauge/67 mm long (World Precision Instruments, Inc.). The needle is attached to a 10 mm syringe via a 22 μm syringe filter.
Perfusion chamber: Cell culture dish 35 mm outer diameter with glass bottom (23.5 mm in diameter). Perfusion chamber is filled with Tyrode’s solution containing (in mM) 135 NaCl, 5.4 KCl, 1.8 CaCl2, 5 HEPES, 5 glucose, 1 MgCl2, and 0.33 sodium phosphate [8].
Temperature controller with heating stage.
Micromanipulator with controller.
Straight microelectrode holder with Ag/AgCl wire, 1 mm pin, and 2 mm pressure port, for 1.5 mm O.D. glass. Frequent chloriding of the electrode is done by immersion in household bleach for about 15 min.
Pressure system: Pressure port on microelectrode holder is connected to a custom-made U-tube manometer via silicone tubing. A three-way stopcock is used to switch between a constant positive pressure of 10 cmH2O and manual suction by mouth or syringe.
Bath electrode is made of Ag/AgCl pellet. Frequent chloriding of the electrode is done by immersion in household bleach for about 15 min.
3 Methods
3.1 Cell Pair Micropatterning
3.1.1 Photomask Design
Draw micropatterns using custom AutoLISP software in AutoCAD (Autodesk, San Rafael, CA), and then print high-resolution photo-masks (chrome on soda lime) [18]. The patterns consist of pairs of rectangular islands, with one island held constant at 30 × 30 μm2 while the other varied from 20 × 10 μm2 to 20 × 36 μm2 in 2 μm increments (Fig. 1). The contact length between the islands is held constant at 20 μm. The patterns should be arranged such that approximately 5,000 island pairs fit within a single 22 mm diameter cover slip [8]. Other custom patterns can be generated using AutoLISP and AutoCAD software.
Fig. 1.

Photomask design of the micropatterned cell pairs
3.1.2 High-Resolution Photolithography
All photolithography steps should be performed in a chemical fume hood inside a clean room, wearing proper lab coats, shoe covers, bonnets, gloves, and UV goggles.
Use wafer tweezers to transfer a silicon wafer to a 200 °C hot plate for 5 min to dehydrate the surface. Transfer wafer to a Petri dish and let cool to room temperature.
Clean dust from the wafer with the N2 gun and UV-decontaminate for 8 min.
Using wafer tweezers, transfer and center the wafer onto the
Transfer approximately 5 mL of Su8-2 photoresist onto the wafer with a transfer pipette.
To obtain 5 μm tall features, spin-coat at 1,000 rpm for 1 min.
Release the vacuum, and, wearing sterile gloves, transfer the wafer by hand to a 65 °C hot plate and cover with a crystallizing dish, avoiding contact with the photoresist-coated surface of the wafer (see Note 1).
Heat at 65 °C for 1 min and then at 95 °C for 3 min.
Turn off hot plate and let cool to room temperature. Using wafer tweezers, return wafer to Petri dish. PAUSE POINT: Once baked, photoresist-coated wafers can be stored in dark, ambient conditions.
Position the wafer and glass photomask properly in the mask aligner, and apply chamber vacuum for “hard contact.”
Expose to 350 W collimated UV light for 20 s.
Using wafer tweezers, transfer wafer back to the hot plate (now at room temperature). Ramp up to 65 °C, bake for 1 min, ramp up to 95 °C, and bake for an additional 3 min. Finally, ramp down to 25 °C at 8 °C/h (see Note 2).
Using wafer tweezers, transfer wafer to a crystallizing dish containing PGMEA for 2 min, then remove, and rinse with iso-propyl alcohol over an empty crystallizing dish (see Note 3).
Dry wafer with N2 gun, and observe under the metallurgic microscope to ensure uniform patterned lines and line spacings, the absence of dust or other impurities, and no feature detachment. The finished wafer should be void of any defects.
Leave the template wafer overnight (12 h) at room temperature inside a vacuum desiccator containing a micro slide with one drop of silane solution. The evaporating silane solution will coat the wafer, creating a “non-stick” surface. Store silanized wafer in a Petri dish at room temperature. PAUSE POINT.
PDMS stamp molding
Mix approximately 20 g PDMS base and 2.0 g curing agent (10:1 by mass) thoroughly in a weigh boat for 1 min using a transfer pipette. Degas in vacuum desiccator for 20 min or until all air bubbles are extracted.
Gently pour the PDMS solution onto the template wafer in the 100 mm diameter Petri dish, filling the dish to a height of approximately 5 mm.
Cure the PDMS-covered wafer in an oven at 80 °C for 2 h. Remove and let cool to room temperature.
Tap Petri dish containing PDMS-covered wafer upside down until PDMS mold (and attached wafer) release from dish (see Note 4). Gently peel PDMS mold from wafer (avoid cracking the wafer), and place PDMS mold, feature side up, on a hard surface. Cut around the square border of the desired pattern with a clean razor blade. PAUSE POINT.
PDMS coating of glass cover slips
Mix PDMS base and curing agent (10:1 by mass) thoroughly in a weigh boat for 1 min using a transfer pipette. Degas in vacuum desiccator for 20 min or until all air bubbles are extracted.
Center a 22 mm diameter glass cover slip on the spin coater chuck, and apply vacuum. Using a transfer pipette, transfer approximately 250 μL of PDMS onto the cover slip, and spin- coat at 4,000 rpm for 20 s. Transfer cover slip by hand to a covered Petri dish, avoiding contact with the PDMS-coated surface (see Note 5).
Cure the PDMS-coated cover slips in an oven at 80 °C for 2 h. Remove and let cool to room temperature. Store cover slips covered at room temperature for microcontact printing.
3.1.3 Microcontact Printing
Sonicate PDMS stamp in 70 % ethanol for 45 min.
Remove stamp with standard curved forceps, dry with N2 gun in a sterile hood, and place, pattern side up, onto a Petri dish.
Coat stamp with 200 μL of 15 μg/ml fibronectin solution, spreading with the side of a sterile pipette tip, if necessary, to ensure even distribution. Coat at room temperature for at least 1 h.
Near the end of the fibronectin coating of the PDMS stamp, UV-decontaminate the surface of the PDMS-coated cover slip in a Petri dish for 8 min with lid off.
Immediately after UV decontamination of the PDMS-coated cover slips, use standard curved forceps to remove stamp from the Petri dish, then dry stamp again with N2 gun in a sterile hood, and return to the Petri dish, pattern side up.
Using fine forceps, gently place each cover slip PDMS side down onto a dried, fibronectin-coated stamp.
Very lightly, trace one tip of the forceps on the back of the cover slip along the features of the underlying stamp to ensure contact. Other than the weight of the forceps, no applied force should be necessary. Wait for 30 min to allow protein transfer (see Note 6).
Gently slide one tip of the fine forceps between the edge of the cover slip and the stamp. Slowly pry the cover slip from the stamp by running the tip of the forceps along the border of the stamp (see Note 7).
Transfer the cover slip into the well of a 12-well plate and coat with Pluronic F-127 (0.2 %, Invitrogen) for 10 min to block cell adhesion in unwanted areas.
Wash cover slip once with PBS and leave in 2 ml PBS in tissue culture hood at room temperature until ready for cell seeding.
Cell Seeding
Aspirate PBS. Seed NRVMs at a density of 104 cells/cm2 in cardiac seeding media. Incubate at 37 °C 5 % CO2 incubator overnight (day 0).
The morning after seeding, aspirate cardiac seeding media and wash with PBS. Gently agitate the plate to remove dead and unattached cells before aspirating PBS and replacing with seeding media (day 1).
About 48 h after seeding, aspirate seeding media and replace with cardiac maintenance media (day 2). Replace with fresh maintenance media on day 4.
On day 5, aspirate maintenance media before seeding non- NRVM cells to form heterotypic cell pairs. Seed non-NRVM cells at a density of 2 × 103 cells/cm2 (see Note 8). Incubate cells at 37 °C 5 % CO2 incubator.
On day 6, cell pairs are ready for patch clamp recordings.
3.2 Patch Clamp
In this section, we describe the patch clamp procedures using our setup and in accordance with previously described patch clamp methodology [19]. When different equipment is used, it is advisable for the user to follow the manufacturer’s instructions.
Software and equipment setup
Open the N2 tank connected to the anti-vibration table.
Open the perfusion system to fill the perfusion chamber with Tyrode’s solution (bath solution). After the chamber is filled to about 3 mm, adjust the perfusion output such that the solution level is maintained at a steady level. Make sure that the bath (ground) electrode, temperature probe, perfusion line, and suction line are all immersed in the bath solution.
Turn on the temperature controller, and set the bath temperature to 37 °C.
Turn on the micromanipulator controller.
Turn on Multiclamp 700B amplifier. Open Multiclamp software, and make sure that voltage clamp mode (VC) is selected.
Open WinWCP software. Create a new data file. Open the “Pipette seal test/Signal monitor” window, and apply a constant test pulse of 5 mV.
Patch pipette preparation
Install the glass pipette inside the pipette puller, and run the program. Program parameters depend on the type and size of pipette, position and shape of the heating filament, and intended use of the pipette. Follow the manufacturer’s instructions to optimize the program for the desired pipette tip size and shape.
Use a Bunsen burner to heat and bend the pipette in the middle at about a 35° angle. Depending on the relative position of the micromanipulator to the horizontal plane, this angle should be adjusted such that after clamped to the manipulator, the pipette is oriented as vertically as possible.
Using the microforge setup, fire-polish the pipette tip to about 1–2 μm in inner diameter.
Backfill the pipette with pipette solution (see Note 9).
Mount the pipette onto the electrode holder. Make sure that the pipette solution makes contact with the chlorided part of the silver electrode.
Whole-cell patch clamp
The current response to a 5 mV test pulse (applied with WinWCP software) is useful to help recognize the different steps of the patch clamp procedure (Fig. 2).
Fig. 2.
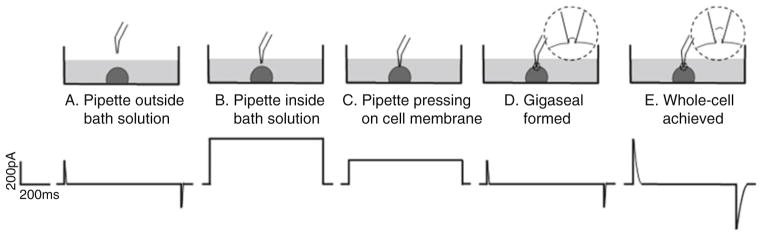
Current response to a 5 mV test pulse during each step of the whole-cell patch clamp procedure
With the manipulator at its coarsest mode, drive the pipette tip to the objective view, and then lower it down to the bath solution (see Note 10). After the pipette reaches the bath solution, the current response shown in the WINWCP software will change to a square shape (Fig. 2b). The displayed pipette resistance should be 1–2 MΩ (see Note 11).
Continue adjusting the position of the pipette tip so that it locates right above the cell to be patched (see Note 12).
Lower the pipette slowly and gradually until initial contact is made with the cell, which is shown as a slight increase in the pipette resistance (Fig. 2c). Keep pressing down on the cell until the resistance is increased by about 1.5–2 times.
Close the positive pressure applied on the pipette solution. The removal of the positive pressure should induce some initial suction on the cell membrane, shown as a further increase in pipette resistance.
Apply a slight, constant negative pressure (preferably by mouth suction) on the cell membrane until gigaseal is formed, indicated by an increase in pipette resistance to at least 1 GΩ (Fig. 2d). A good seal is usually formed instantly, within less than 5 s of suction.
Apply further negative pressure to rupture the membrane patch between the pipette tip and the cell to achieve whole-cell patch clamp configuration (Fig. 2e). This is indicated by a relatively broad capacitive transient in the current response on the WINWCP window (see Note 13).
Action potential recordings
After whole-cell configuration is achieved, switch to current clamp mode with zero applied current (I=0 mode in Multiclamp 700B window). Record the displayed membrane potential. If the cardiomyocyte is at rest and not contracting spontaneously, this is its resting membrane potential.
To record action potentials, switch to current clamp mode (IC in Multiclamp 700B) and use the “Record” function of WinWCP. In cells without spontaneous pacemaking activity, action potentials can be elicited by applying a 10-ms current pulse at 1.1× threshold amplitude (Fig. 3a). In spontaneously contracting cells, no applied current is necessary (Fig. 3b).
Fig. 3.
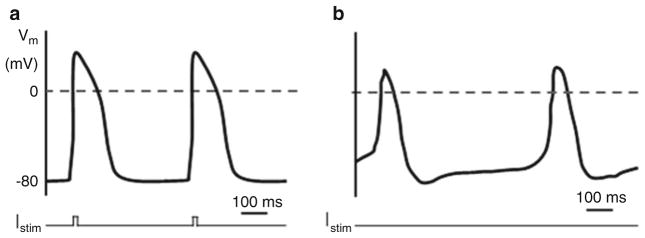
Action potential traces in NRVMs triggered by a stimulus current (Istim) (a) or in a spontaneously contracting cell (b)
Dual whole-cell voltage clamp recordings
With the cardiomyocyte kept in the stable whole-cell configuration, switch the manipulator to control the left-side manipulator.
Repeat the same procedure to get the second pipette to form gigaseal with the non-cardiac cell in the cell pair, and then break into whole-cell configuration.
In VC modes, hold the NRVM at 0 mV while applying a 4-s voltage pulse of various potentials from −40 to 40 mV on the non-cardiac cell.
For each voltage pulse, determine the gap-junctional conductance between the two cells by dividing the measured junctional current by the applied voltage difference after correcting for series resistance and membrane resistance as previously described [20].
Acknowledgments
We thank Dr. Robert Kirkton for reading of the manuscript. This work was supported by the National Heart, Lung, and BloodInstituteoftheNIHunderAwardNumbersR01-HL-104326 and R21-HL-106203. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Dust can adhere to wet photoresist, ultimately disrupting the micropattern. Make sure that gloves and surrounding region are clean. Transfer and cover wafer quickly.
Rapid cooling introduces stress to the photoresist–silicon bond, resulting in feature fragility. Gradual, ramped cooling is essential.
If white residue is seen during isopropyl alcohol rinse, return wafer to PGMEA for an addition minute and re-rinse with iso-propyl alcohol. Repeat as necessary.
If the wafer and attached PDMS mold fail to detach from the polystyrene Petri dish, use clippers to make several cuts along the circumference of the Petri dish wall and carefully break off wall segments until the contents of the dish can easily be removed.
Repeat for each cover slip. Dust can adhere to the wet PDMS and interfere with micropatterned protein transfer. Make sure that gloves and surrounding region are clean. Transfer and cover cover slips quickly.
The pattern should gradually appear as seal is formed between the cover slip and the PDMS stamp. Applied force other than the weight of the forceps could result in loss of the pattern due to sagging, which is also visible. In such case, reduce protein transfer time (no less than 15 min) to prevent further sagging.
Excessive force could result in breaking the cover slip or forcing undesired stamp contact and protein transfer. An alternative way is to bend the PDMS stamp gently to “pop off” the cover slip.
Use a syringe to hold the NRVM-stamped cover slip in place.
Make sure that the needle is inserted in the pipette as close to the pipette tip as possible before injecting the solution with the syringe to prevent large bubbles. To get rid of bubbles at the tip, gently flick the pipette with your finger near the tip end until the bubbles float up.
A slight positive pressure of around 10 cm of H2O should be applied on the pipette solution before lowering the pipette to the bath solution. This will help protect the pipette tip from debris at the air–solution interface.
If the resistance is too large (>5 MΩ), it usually means that either the pipette tip is too small or there is an air bubble at the pipette tip. Replace with new pipette in such case.
To locate the pipette in the field of view, it is easier to switch to a 10× objective. Move the pipette around in the x–y plane until its slightly dark, moving shadow can be recognized. Once the rough position of the pipette is identified, gradually lower the pipette while move up the focal plane of the microscope until the pipette tip comes into focus as a small circle in a dark background. Then, lower the focal plane and the pipette iteratively, making sure that the focal plane always lies between the cover slip and the pipette tip to prevent the pipette from accidentally hitting the cover slip. As the pipette tip gets close enough to the cell such that both are recognizable (but not in focus) in the same field of view, switch to a 40× objective, and switch micromanipulator to a finer mode.
There are various methods that can be used to break the membrane patch to get to whole-cell mode. However, for NRVMs, applying gradual negative pressure with a 3 mm syringe seems to give the highest success rate.
References
- 1.Menasche P, Hagege AA, Vilquin JT, et al. Autologous skeletal myoblast transplantation for severe postinfarction left ventricular dysfunction. J Am Coll Cardiol. 2003;41(7):1078–1083. doi: 10.1016/s0735-1097(03)00092-5. [DOI] [PubMed] [Google Scholar]
- 2.Assmus B, Schachinger V, Teupe C, et al. Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction (TOPCARE-AMI) Circulation. 2002;106(24):3009–3017. doi: 10.1161/01.cir.0000043246.74879.cd. [DOI] [PubMed] [Google Scholar]
- 3.Chugh AR, Beache GM, Loughran JH, et al. Administration of cardiac stem cells in patients with ischemic cardiomyopathy: the SCIPIO trial: surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation. 2012;126(11 Suppl 1):S54–S64. doi: 10.1161/circulationaha.112.092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laflamme MA, Chen KY, Naumova AV, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25(9):1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 5.Cho HC, Marban E. Biological therapies for cardiac arrhythmias: can genes and cells replace drugs and devices? Circ Res. 2010;106(4):674–685. doi: 10.1161/CIRCRESAHA.109.212936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gepstein L. Cell and gene therapy strategies for the treatment of postmyocardial infarction ventricular arrhythmias. Ann N Y Acad Sci. 2010;1188:32–38. doi: 10.1111/j.1749-6632.2009.05080.x. [DOI] [PubMed] [Google Scholar]
- 7.Pedrotty DM, Klinger RY, Badie N, et al. Structural coupling of cardiomyocytes and noncardiomyocytes: quantitative comparisons using a novel micropatterned cell pair assay. Am J Physiol Heart Circ Physiol. 2008;295(1):H390–H400. doi: 10.1152/ajpheart.91531.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McSpadden LC, Nguyen H, Bursac N. Size and ionic currents of unexcitable cells coupled to cardiomyocytes distinctly modulate cardiac action potential shape and pacemaking activity in micropatterned cell pairs. Circ Arrhythm Electrophysiol. 2012;5(4):821–830. doi: 10.1161/CIRCEP.111.969329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.del Corsso C, Srinivas M, Urban-Maldonado M, et al. Transfection of mammalian cells with connexins and measurement of voltage sensitivity of their gap junctions. Nat Protoc. 2006;1(4):1799–1809. doi: 10.1038/nprot.2006.266. [DOI] [PubMed] [Google Scholar]
- 10.Bursac N, Parker KK, Iravanian S, et al. Cardiomyocyte cultures with controlled macroscopic anisotropy: a model for functional electrophysiological studies of cardiac muscle. Circ Res. 2002;91(12):e45–e54. doi: 10.1161/01.res.0000047530.88338.eb. [DOI] [PubMed] [Google Scholar]
- 11.Klinger R, Bursac N. Cardiac cell therapy in vitro: reproducible assays for comparing the efficacy of different donor cells. IEEE Eng Med Biol Mag. 2008;27(1):72–80. doi: 10.1109/MEMB.2007.913849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pedrotty DM, Klinger RY, Kirkton RD, et al. Cardiac fibroblast paracrine factors alter impulse conduction and ion channel expression of neonatal rat cardiomyocytes. Cardiovasc Res. 2009;83(4):688–697. doi: 10.1093/cvr/cvp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mangi AA, Noiseux N, Kong D, et al. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat Med. 2003;9(9):1195–1201. doi: 10.1038/nm912. [DOI] [PubMed] [Google Scholar]
- 14.Taylor DA, Silvestry SC, Bishop SP, et al. Delivery of primary autologous skeletal myoblasts into rabbit heart by coronary infusion: a potential approach to myocardial repair. Proc Assoc Am Physicians. 1997;109(3):245–253. [PubMed] [Google Scholar]
- 15.Liau B, Christoforou N, Leong KW, et al. Pluripotent stem cell-derived cardiac tissue patch with advanced structure and function. Biomaterials. 2011;32(35):9180–9187. doi: 10.1016/j.biomaterials.2011.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christoforou N, Miller RA, Hill CM, et al. Mouse ES cell-derived cardiac precursor cells are multipotent and facilitate identification of novel cardiac genes. J Clin Invest. 2008;118(3):894–903. doi: 10.1172/JCI33942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirkton RD, Bursac N. Engineering biosynthetic excitable tissues from unexcitable cells for electrophysiological and cell therapy studies. Nat Commun. 2011;2:300. doi: 10.1038/ncomms1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Badie N, Satterwhite L, Bursac N. A method to replicate the microstructure of heart tissue in vitro using DTMRI-based cell micropatterning. Ann Biomed Eng. 2009;37 (12):2510–2521. doi: 10.1007/s10439-009-9815-x. [DOI] [PubMed] [Google Scholar]
- 19.Hamill OP, Marty A, Neher E, et al. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391(2):85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 20.Veenstra RD, Brink PR. Patch-clamp analysis of gap junctional currents. In: Stevenson B, Paul DL, Gallin W, editors. Cell-cell interactions: a practical approach. Oxford University Press; Oxford: 1992. [Google Scholar]