

Abstract
In this report we describe a straightforward approach to synthesize polymers with end-groups that bind site-specifically to two different proteins. Telechelic biotin, maleimide poly(N-isopropylacrylamide) (pNIPAAm) was synthesized for the formation of streptavidin (SAv)-bovine serum albumin (BSA) polymer conjugates. Reversible addition-fragmentation chain transfer (RAFT) polymerization of NIPAAm was conducted in the presence of biotinylated chain transfer agents (CTAs) with either ester or amide linkages, and the resultant α-biotinylated pNIPAAm were formed with low polydispersity indices (PDI ≤ 1.09). UV-Vis analysis of the trithiocarbonate chain-ends indicated 88% or greater retention of the group. A maleimide was introduced to the ω chain-end via a radical cross-coupling reaction with a functionalized azo-initiator. The polymer structures were characterized by 1H NMR spectroscopy and gel permeation chromatography (GPC). The resultant biotin-maleimide heterotelechelic polymer was used to form a SAv-BSA heterodimer conjugate. Bioconjugate formation was confirmed by gel electrophoresis.
Introduction
Merging synthetic polymers with proteins allows the properties of the protein to be finely tuned for applications in medicine, biotechnology and nanotechnology.1, 2 Polymer bioconjugates are typically prepared by attaching pre-formed reactive polymers to defined locations on the protein surface. A popular class of bioconjugates is modified with polyethylene glycol (PEG). These protein conjugates have increased stabilities, decreased immunogenicities and improved pharmacokinetics compared to the non-PEGylated counterparts.3 Another class of bioconjugates is formed from “smart” polymers, such as poly(N-isopropylacrylamide) (pNIPAAm). These polymers respond to external stimuli including changes in temperature or pH, and can be used to modulate protein activity.4
Numerous cellular processes are the result of multivalent interactions; therefore, protein valency should have a major influence on the application of a bioconjugate.5, 6 Protein oligomerization plays many roles in cell signaling and has been induced using small molecule ligands.6-8 Indeed, multivalent polymers or dendrimers modified with several biomolecules have improved biomimetic features.1, 9-16 For example, polymers containing side-chains modified with ligands often exhibit enhanced binding affinities to their respective cell-surface receptors compared to the ligand itself.9 Thus, incorporating two proteins into a polymer conjugate should result in improved properties for biological applications. If the proteins are different this might provide additional benefits such as the ability to orient immobilized proteins on surfaces.17 Despite these advantages, the majority of polymer conjugates consist of a single protein modified with one or more polymers. This is likely due to a lack of available methods to easily prepare telechelic protein-reactive polymers. In this report we describe a straightforward method to synthesize heterotelechelic polymers with end-groups that enable the formation of heterodimeric protein-polymer conjugates.
Conjugates, where the polymer has a narrow molecular weight distribution and the site of attachment is defined, have superior properties to heterogeneous conjugates.18 Therefore, methods to prepare well-defined functional polymers that react with specific sites on proteins are important.19 Controlled radical polymerizations (CRPs) such as atom transfer radical polymerization (ATRP)20, 21 and reversible addition-fragmentation chain transfer (RAFT) polymerization22, 23 have emerged as powerful methods to prepare such polymers. Protein-reactive ATRP initiators have enabled the synthesis of low polydispersity α-functional polymers for modification of lysine side-chains,24, 25 chemospecific reactions with cysteines,26, 27 ketones,28 and association with the ligand binding sites of streptavidin (SAv).29-31 RAFT polymerization produces end-functional polymers by judicious choice of a chain transfer agent (CTA). In addition, the dithioester and trithiocarbonate chain ends can reduced to a thiol32, 33 and are susceptible to radical reactions.34 RAFT polymerization has been used to prepare biotin,35, 36 and pyridyl disulfide37 at one chain end for bio-conjugation.
Herein, we describe the synthesis of α biotin, ω maleimide polymers using RAFT polymerization. There has been one reported example of a polymer synthesized with a biotin at one end and a pyridyl disulfide at the other end.38 In that approach, an azide-functionalized CTA was employed, and the biotin was introduced after polymerization. A pyridyl disulfide group was linked to the ω chain end via a hydrolysable trithiocarbonate group. The authors reported that they obtained low protein conjugation efficiencies (∼10%) likely because the reaction had to be conducted at a low pH to prevent hydrolysis and elimination of the pyridyl disulfide group during the reaction. In this report, the described strategy is distinct (Scheme 1). The CTA already contained a biotin group and the maleimide was installed by heating the polymer in the presence of designed radical initiator to form a stable group at the ω chain-end. The resultant bis-functionalized polymers were used to form heterodimeric protein-polymer conjugates.
Scheme 1.

Polymer synthesis and formation of protein-heterodimer conjugates (protein structures from the PDB: 1SWA and 1P56).
Experimental Section
Materials
AlexaFluor 350 streptavidin was purchased from Invitrogen. Azobisisobutyronitrile (AIBN) (Sigma) was recrystallized twice from ethanol. 4,4,-Azobis(N,N,-cyanopentanoic acid) (V501) (Sigma) was dried under high vacuum at 23 °C for 12 h before using. N-Isopropylacrylamide (NIPAAm) (Sigma) was recrystallized twice from hexanes. Triethylamine (TEA) (Sigma) was stored over potassium hydroxide, dichloromethane (DCM) (Fisher) was distilled from calcium hydride and stored under argon, and tetrahydrofuran (THF) (Fisher) was distilled from sodium/benzophenone and stored under argon. 2-(Hydroxyethoxyethyl) biotin (1),30 4-(3-hydroxy-propyl)-10-oxa-4-aza-tricyclo[5,2,1,02,6] dec-8-ene-3,5-dione (3),39 and biotin ethylamine40 were synthesized according to literature procedures. All other chemicals were purchased from Sigma or Fisher Scientific and used as received.
Analytical Techniques
1H and 13C NMR spectra were acquired on a Bruker Avance 500 MHz or 600 MHz DRX and spectra were processed using Topspin 1.2 NMR software. UV-Vis spectra were obtained on a Biomate 5 Thermo Spectronic UV-Vis spectrometer using quartz cells. Matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometry was performed on an Applied Biosystems Voyager-DE STR. Electrospray ionization mass spectrometry (ESI-MS) was performed on an IonSpec Ultima 7T ICR, (Varian Inc.). Both mass spectrometers are located in the Molecular Instrumentation Center (MIC) at the University of California, Los Angeles (UCLA). Infrared absorption spectra were recorded using a PerkinElmer FT-IR equipped with an ATR accessory. TLC plates were pre-coated with silica gel 60 F254 and were developed in the indicated solvent systems. Merck 60 (230-400 Mesh) silica gel was used for column chromatography. GPC was conducted on a Shimadzu HPLC system equipped with a refractive index detector RID-10A, one Polymer Laboratories PLgel guard column, and two Polymer Laboratories PLgel 5 μm mixed D columns. LiBr (0.1 M) in DMF at 40 °C was used as the eluent (flow rate: 0.80 mL/min). Calibration was performed using near-monodisperse poly(methyl methacrylate) standards from Polymer Laboratories. Analytical and preparatory reverse phase HPLC were carried out on a Shimadzu HPLC system equipped with a UV detector using Alltech Prevail C18 columns (analytical: 5μm, 150 × 4.6 mm, flow rate 1 mL/min; preparatory: 5μm, 150 × 22 mm, flow rate 23 mL/min) with monitoring at λ = 280 nm and 220 nm. Chromatograms were processed using the EZStart 7.2 chromatography software. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was carried out using 4-15% tris(hydroxymethyl)aminomethane (TRIS)-glycine precast gradient gels (Invitrogen), and samples were dissolved in TRIS buffer containing SDS, bromophenol blue and glycerol.
Methods
Synthesis of 2-(Ethylsulfanylthiocarbonylsulfanyl)propionic acid (2)
This compound was synthesized using a literature procedure.40 δ 1H NMR (500 MHz, CDCl3): 4.86 (q, J = 7.5 Hz, 1H, CH3CH), 3.37 (q, J = 7.4 Hz, 2H, CH3CH2S), 1.63 (d, J = 7.5 Hz, 3H, CH3CH), 1.36 (t, J = 7.4 Hz, 3H, CH3CH2S); 13C NMR (500 MHz, CDCl3): 222.17, 175.49, 47.37, 31.91, 16.69, 13.06; IR (cm -1): 2980, 2933, 2872, 2653, 2580, 2496, 1691, 1449, 1419, 1405, 1378, 1366, 1293, 1254, 1224, 1213, 1196, 1089, 1066, 1049, 1037, 968, 922, 861, 824, 770, 748, 731; λmax (SC=SS) = 308 nm (MeOH).
Synthesis of biotinylated CTA 1
Biotinylated alcohol 1 (200 mg, 0.603 mmol) and trithiocarbonate acid 2 (140 mg, 0.666 mmol) were dissolved in anhydrous DMF (5.0 mL). N,N′-dicyclohexylcarbodiimide (DCC) (193 mg, 0.935 mmol) and 4-(dimethylamino)pyridine (DMAP) (9.9 mg, 0.081 mmol) were added in one portion and the contents were stirred at 22 °C for 10 h. The solvent was then removed in vacuo, and the crude product was purified by silica gel chromatography (9:1 DCM:MeOH), yielding CTA1 as yellow solid in 19% yield (60 mg). δ 1H NMR (600 MHz, CD3OD): 4.83 (q, J = 7.4 Hz, 1H, CH(CH3)S), 4.49 (dd, J = 4.4, 7.8 Hz, 1H, NHCHCH2), 4.30 (dd, J = 4.4, 7.9 Hz, 1H, NHCHCH), 4.29-4.27 (m, 2H, CH2C=O), 3.69-3.67 (m, 2H, CH2OCH2), 3.55 (t, J = 5.5 Hz, 2H, CH2OCH2), 3.40 (q, J = 7.4 Hz, 2H, SCH2CH3), 3.35 (t, J = 5.5 Hz, 2H, NHCH2CH2), 3.22-3.19 (m, 1H, NHCHCHS), 2.93 (dd, J = 5.0, 12.7, 1H, NHCH2S), 2.70 (d, J = 6.4 Hz, 1H, NHCH2S), 2.24-2.21 (m, 2H, CH2C=ONH), 1.76-1.59 (m, 4H, SCHCH2CH2H2), 1.56 (d, J = 3.1, 3H, O=CH(CH3)), 1.47-1.42 (m, 2H, SCHCH2CH2H2), 1.34 (t, J = 7.4 Hz, 3H, S=CSCH2CH3). δ 13C NMR (600 MHz, CD3OD): 223.79, 176.16, 172.58, 166.11, 70.62, 69.73, 66.09, 63.39, 61.64, 57.02, 41.06, 40.34, 36.77, 32.34, 29.77, 29.51, 26.85, 17.03, 16.92, 13.43. IR (cm -1): 3238, 2926, 2863, 2376, 1732, 1681, 1548, 1453, 1260, 1126, 1077, 866, 813. ESI-MS observed (predicted): [M+Na] 546.12 (546.12), [M+K] 562.09 (562.09). UV-Vis (MeOH): λmax (SC=SS) = 306 nm
Synthesis of biotinylated trithiocarbonate CTA2
Biotin ethylamine (64.8 mg, 0.226 mmol) and 2 (49.5 mg, 0.235 mmol) were weighed into a round bottom flask and dissolved in DMF (5 mL). N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) hydrochloride (45.6 mg, 0.238 mmol) and N-hydroxybenzotriazole (HOBT) (32.1 mg, 0.237 mmol) were added in one portion and the contents were stirred for 23 h at 23 °C. The DMF was removed in vacuo and the crude product was purified first by silica gel chromatography (9:1 DCM:MeOH) and then by octadecyl-modified silica gel (60/40 MeOH:H2O) reversed-phase chromatography. Fractions that were yellow in color were combined, and then lyophilized yielding CTA2 as a yellow solid in 27% yield (32.3 mg). δ 1H NMR (500 MHz, CD3OD): 4.74 (q, J = 7.3 Hz, 1H, CH(CH3)S), 4.52 (dd, J = 4.9, 7.8, 1H, NHCHCH2), 4.33 (dd, J = 4.3, 7.8 Hz, 1H, NHCHCH), 3.42 (q, J = 7.3 Hz, 1H, SCH2CH3), 3.26-3.22 (m, 1H, CHCHS) 2.96 (dd, J = 5.0, 12.8 Hz, 1H, CHCH2S), 2.73 (d, J = 12.8, 1H, CHCH2S), 2.24-2.22 (m, 2H, CH2C=ON), 1.77-1.62 (m, 4H, SCHCH2CH2), 1.58 (d, J = 3.4Hz, 3H, CH(CH3)2), 1.49-1.45 (m, 2H, SCHCH2), 1.36 (t, J = 7.4 Hz, 3H, SCH2CH3); 13C NMR (500 MHz, CD3OD): 224.37, 176.43, 173.43, 63.35, 61.64, 56.96, 50.74, 50.72, 41.07, 40.41, 39.85, 36.86, 32.25, 29.79, 29.47, 26.76, 17.85, 13.45; IR (cm -1): 2927, 2406, 1680, 1633, 1459, 1446, 1239, 1080, 1026, 821; MALDI-TOF observed (predicted): [M+Na] 501.1 (501.0), [M+K] 517.1 (517.0); λmax (SC=SS) = 307 nm.
Synthesis of V501-4-(3-hydroxy-propyl)-10-oxa-4-aza-tricyclo[5,2,1,02,6] dec-8-ene-3,5-dione -dimer
V501 (2.54 g, 9.1 mmol), thionyl chloride (10.0 mL, 137.1 mmol), DMF (0.05 mL) and ethyl acetate (20.0 mL) were added to a 100 mL round bottom flask under an argon atmosphere, and warmed to 60 °C. After the solution turned clear (approximately 10 min) the temperature was kept at 60 °C for another 20 min, then cooled to 23 °C and the solvent was removed in vacuo. The acid chloride derivative of V501 was obtained as a yellow solid, and was used in the next step without further purification.
4-(3-hydroxy-propyl)-10-oxa-4-aza-tricyclo[5,2,1,02,6] dec-8-ene-3,5-dione 4
Compound 3 (2.90 g, 13.0 mmol) and Et3N (1.41 g, 13.9 mmol) were dissolved in dry THF (50 mL) and stirred in an ice-water bath under an argon atmosphere for 20 min. Next, a solution of the acid chloride (1.59 g, 5.0 mmol) in THF (20 mL) was added drop wise over 20 min. The ice-water bath was removed and the system was kept at 23 °C for 4 h and then filtered to remove the solids. The THF was removed in vacuo and the crude product was purified by silica gel chromatography (3:7 hexane:EtOAc to 100% EtOAc) yielding 4 as a white powder in 58% overall yield (2.00 g). δ 1H NMR (600 MHz, CDCl3): 6.49 (s, 4H, exo, CH=CH), 5.23 (bs, 4H, exo, CHO), 4.05-4.02 (m, 4H, CH2CH2O), 3.55 (t, J = 6.8 Hz, 4H, NCH2), 2.82 (s, 4H, exo, CHC=O), 2.54-2.33 (m, 8H, O=CCH2CH2C), 1.93-1.89 (m, 4H, NCH2CH2), 1.73 (s, 3H, CCH3), 1.67 (s, 3H, CCH3); δ 13C NMR (500 MHz, CDCl3): 176.27 (exo), 171.39, 136.58, 117.72, 81.02 (exo), 72.00/71.90 (anti/syn), 61.87, 47.47 (exo), 35.63, 33.15, 29.18/29.10 (anti/syn), 26.53, 23.99/23.74 (anti/syn); IR (cm-1): 1775, 1728, 1693, 1449, 1404, 1177, 1022, 872; ESI-MS observed (predicted): [M+Na] 713.24 (713.25), [M+K] 729.22 (729.22).
Extinction coefficient determination of CTA1
A series of solutions containing CTA1 ranging from 4.21 × 10-6 M to 5.06 × 10-5 M were prepared in volumetric glassware in methanol. The absorbance of each sample was measured at 306 nm (λmax SC=SS). A line was fit to these data points though the origin, which gave a molar absorptivity (ε) of 15069 M-1 cm-1.
Trithiocarbonate end-group analysis by UV-Vis spectroscopy
In a typical experiment, polymer solutions ranging from 1.46 ×10-5 M to 5.85 × 10-5 M were prepared in methanol (molecular weights were calculated from 1H NMR). The absorbance at 306 nm was measured for each sample. The percentage of trithiocarbonate end-group was determined using the calculated molar extinction coefficient of 15069 M-1cm-1; the percentage is the average of five trials.
Typical RAFT polymerization of NIPAAm with CTA1 and CTA2
RAFT polymerization was conducted using molar ratios of [1]:[0.1]:[100] for CTA:AIBN:NIPAAm. CTA1 (26.8 mg, 0.051 mmol), AIBN (0.90 mg, 0.005 mmol), and NIPAAm (570 mg, 5.04 mmol) were loaded into a Schlenk tube, and the flask was evacuated and refilled with argon three times. DMF (1 mL) was added and the flask was subjected to three freeze-pump-thaw cycles. The polymerization was initiated by immersion of the Schlenk tube into a 70 °C oil bath. After 2 h (80% conversion) the reaction flask was removed from the oil bath and opened to the atmosphere. The DMF was removed in vacuo and the polymer was purified by dialysis against MeOH (MWCO 6-8,000 Da). Polymer conversions were calculated by 1H NMR in methanol using the following equation: 1-Mt/M0 where Mt is the average of the integral value of the vinylic protons of the monomer and M0 is the methine peak at 3.97 ppm from monomer and polymer overlap. P1: δ 1H NMR (500 MHz, CD3OD): 7.97-7.61 (NH, pNIPAAm), 4.51-4.84 (m), 4.32-4.30 (m), 4.24-4.22 (m), 3.97 (br, CH(CH3)2, pNIPAAm), 3.69-3.67 (m), 3.57-3.55 (m), 3.21-3.20 (m), 2.93 (dd, J = 13.0, 5.2 Hz), 2.71 (d, J = 12.5 Hz), 2.09 (bs, CH2CHC=O, pNIPAAm), 1.59 (bs, CH2CHC=O, pNIPAAm), 1.45-1.43 (m), 1.34-1.29 (m), 1.16 (bs CH(CH3)2, pNIPAAm). Mn g/mol: (theory) 11,200; (GPC) 17,300; PDI: 1.08; (1H NMR) 10,900; MALDI-TOF 10,400; UV-Vis (MeOH): λmax = 306 nm; trithiocarbonate end-group retention by UV-Vis analysis: 93%
Trithiocarbonate terminated biotinylated pNIPAAm prepared from CTA2
δ 1H NMR (500 MHz, CD3OD): 7.92-7.61 (NH, pNIPAAm), 4.51-4.49 (m), 4.32-4.30 (m), 3.97 (bs, CH(CH3)2, pNIPAAm), 3.26-3.22 (m), 2.93 (dd, J = 12.6, 4.9 Hz, 1H, CHCH2S), 2.72 (d, J = 13.3 Hz, 1H, CHCH2S), 2.09 (bs, CH2CHC=O, pNIPAAm), 1.60 (bs, CH2CHC=O, pNIPAAm), 1.43, 1.35-1.32 (m, SCH2CH3), 1.16 (bs, CH(CH3)2, pNIPAAm); Mn g/mol: (theory) 9,420; (GPC): 16,330; PDI: 1.07; (1H NMR): 10,100; MALDI-TOF 9,750; UV-Vis (MeOH): λ max = 307 nm (SC=SS). Trithiocarbonate end-group retention by UV-Vis: 88%.
Typical radical cross-coupling of trithiocarbonate end-functional polymers with protected maleimide azo-initiator
P1 (34 mg, 3.30 μmol) and 4 (49.0 mg, 70.9 μmol) were loaded into a Schlenk tube, and then evacuated and argon-refilled three times. A 1/1 v/v solution of DMF/dioxane (660 μL) was added and the reaction flask was subjected to three freeze-pump-thaw cycles. The reaction flask was immersed into a 70 °C oil bath. After four hours the reaction flask was removed from the oil bath and opened to the atmosphere. The solvent was removed in vacuo and the polymer was purified by dialysis against 1:1 EtOAc:MeOH (MWCO 6-8,000 Da) to afford the protected maleimide adduct. P2: δ 1H NMR (500 MHz, CD3OD): 7.93-7.57 (NH, pNIPAAm), 6.55, 5.16, 4.69, 4.50, 4.31, 4.23, 3.97 (bs, CH(CH3)2, pNIPAAm), 3.67, 3.56-3.55 (m), 3.10-3.06 (m), 2.93, 2.72 (d, J = 13.0 Hz), 2.09 (bs, CH2CHC=O, pNIPAAm), 1.59 (bs, CH2CHC=O, pNIPAAm), 1.42, 1.29, 1.16 (bs, CH(CH3)2). Mn g/mol: (GPC) 18,430; PDI: 1.09
Protected maleimide pNIPAAm intermediate from CTA2 polymerization
δ 1H NMR (500 MHz, CD3OD): 7.98-7.62 (NH, pNIPAAm), 6.55 (s), 5.16 (s), 4.51-4.49 (m), 4.32-4.30 (m), 3.97 (bs, CH(CH3)2, pNIPAAm), 3.64-, 3.56, 2.92 (s), 2.72 (d, J = 13.0 Hz), 2.57-2.44 (m), 2.10 (bs, CH2CHC=O, pNIPAAm), 1.60 (bs, CH2CHC=O, pNIPAAm), 1.43, 1.16 (bs, CH(CH3)2, pNIPAAm); Mn g/mol (GPC): 19,100; PDI: 1.08.
Regeneration of maleimide functional group to form biotin-maleimide pNIPAAm (P3)
P2 (1.0 mg, 0.085 μmol) was loaded into a Schlenk tube and then dissolved in MeOH (100 μL). The solvent was removed in vacuo, and while under vacuum it was placed in a 120 °C oil bath for 2 h to afford biotin-maleimide P3. 1H NMR (500 MHz, CD3OD) δ: 7.94-7.57 (br, NH, pNIPAAm), 6.84, 4.69, 4.55-4.50 (m), 4.31, 4.23, 3.96 (bs, CH(CH3)2, pNIPAAm), 3.67, 3.55 (m), 3.09-3.06 (m), 3.00, 2.95-2.93 (m), 2.72 (d, J = 12.4 Hz), 2.61, 2.09 (bs, CH2CHC=O, pNIPAAm), 1.58 (bs, CH2CHC=O, pNIPAAm), 1.43, 1.29, 1.16 (bs, CH(CH3)2,, pNIPAAm). Mn g/mol: (GPC) 18,570; PDI: 1.10
Biotin-maleimide pNIPAAm (P4)
δ 1H NMR (500 MHz, CD3OD): 7.97-7.62 (NH, pNIPAAm), 6.83 (d, J = 6.84 Hz), 4.69, 4.51-4.49(m), 4.32 (m), 3.97 (bs, CH(CH3)2, pNIPAAm), 3.64-3.56 (m), 2.95-2.92 (m), 2.72 (d, J = 12.1 Hz), 2.62, 2.10 (bs, CH2CHC=O, pNIPAAm), 1.59 (bs, CH2CHC=O, pNIPAAm), 1.43, 1.16 (bs, CH(CH3)2, pNIPAAm); Mn g/mol: (GPC) 19,560; PDI: 1.06.
Formation of biotin-pNIPAAm-BSA (conjugate 1)
In typical experiment, biotin-pNIPAAm-maleimide (P3, 1.94 mg) was dissolved in 200 μL of phosphate buffer (0.05M, 25 mM EDTA, 5 mM TCEP pH 7.0) (buffer1) and 200 μL of a BSA stock solution (1 mg/mL in buffer1) was added to the polymer solution. The sample was incubated at 4 °C for 12 h. The conjugate was purified by gel filtration (Sephadex G100) and eluted with dH2O. The absorbance of each fraction was measured at 280 nm and fractions containing protein were lyophilized and then used for SDS-PAGE.
Formation of SAv-pNIPAAm-BSA heterodimer (conjugate 2)
One fraction of the purified and lyophilized conjugate 1 was re-dissolved in 75 μL of dH2O. Then, 5 μL of SAv-AlexaFluor 350 (1 mg/mL in dH2O) was added directly to the solution and then left to incubate at 4 °C for 12 h. The conjugate was purified by gel filtration (Sephadex G100) and eluted with dH2O. Fractions containing an absorbance at 280 nm were combined and lyophilized. The isolated conjugate was analyzed by SDS-PAGE. The gel was first visualized under UV light and then stained with coomassie Blue.
Results and Discussion
Polymer synthesis
In order to obtain well-defined bioconjugates, we chose end-groups that target ligand binding sites and free cysteines. Specifically, a biotin-maleimide heterotelechelic polymer was prepared. Biotin is a high affinity ligand for the protein streptavidin (SAv). Maleimides are excellent Michael acceptors of both thiols and amines, with the former addition proceeding 1000 times faster at pH 7.41 Maleimides react with free cysteine residues that occur infrequently on protein surfaces. Therefore, both groups target specific, yet different sites on proteins.
We explored RAFT polymerization as a convenient method to prepare α, ω end-functional protein-reactive polymers. RAFT is a controlled radical polymerization that results in polymers with narrow molecular weight distributions.42 The reaction proceeds in the presence of a CTA that is typically a dithioester or trithiocarbonate. This species is then present at the ω-end of the polymer and is available for further elaboration. We chose to install the requisite biotin via the CTA and the maleimide by a radical cross-coupling reaction between the trithiocarbonate chain-end and a protected-maleimide azo-initiator. Therefore, biotin was judiciously placed on the R-group of the functional CTA so that a stable α-biotinylated polymer would result. The biotinylated trithiocarbonate functionalized CTA1 was synthesized using DCC-mediated coupling with biotin alcohol 1 and trithiocarbonate acid 2, forming CTA1 in 19% yield (Scheme 2). We found that by using EDC as the coupling agent improved the yield up to 67%.
Scheme 2.

Synthesis of biotinylated chain transfer agent (CTA1).
Polymerization of NIPAAm was pursued next. PolyNIPAAm (pNIPAAm) is a thermoresponsive polymer used extensively for the creation of protein switches.4 It has a lower critical solution temperature (LCST) of approximately 32 °C and is therefore also attractive for drug delivery applications or for immobilization of proteins or cells on surfaces.43 As outlined in Scheme 3, AIBN initiated RAFT polymerization was conducted using molar ratios of 1:100:0.1 of CTA1:NIPAAm:AIBN in DMF at 70 °C. The polydispersity index (PDI) of the polymer (P1), determined from gel permeation chromatography (GPC), was low at 1.08 (Figure 2), demonstrating that the conditions provided narrow molecular weight polymers. The number average molecular weight (Mn) by GPC was 17,300 Da against non-authentic standards, while the Mn by 1H NMR (10,900 Da) and MALDI-TOF (10,400 Da) were close to the theoretical Mn (11,200 Da). Retention of the trithiocarbonate end-group was essential for further elaboration of the polymer and quantification by UV-Vis spectroscopy indicated that 93 % of the polymer chains contained the moiety. This is typical of RAFT polymerization because a small percentage of chains are initiated by AIBN, and thus not all chains are expected to contain the functional end-groups.23, 42
Scheme 3.

Synthesis of biotin-maleimide heterotelechelic pNIPAAm.
Figure 2.

GPC chromatograms of P1 (bottom), P2 (middle), and P3 (top). GPC was conducted in DMF containing 0.1 M LiBr at 0.80 mL/min.
A radical cross-coupling reaction between the trithiocarbonate chain-end and a protected maleimide azo-initiator was used to install the maleimide at the ω chain-end. Maleimide is an excellent monomer for radical polymerization,44 and therefore a protected maleimide azo-initiator was used during the radical cross-coupling reaction. Specifically, a furan-protected maleimide39 derivative of V501 was prepared. Thionyl chloride activation of V501 followed by esterification with the protected maleimide alcohol 3 provided the azo-derivative, 4, in 58% overall yield (Scheme 4).
Scheme 4.

Synthesis of protected maleimide V501 derivative.
Radical cross-coupling between 4 and P1 was performed at 70 °C in DMF:dioxane for 4 hours (Scheme 3). The reaction time was kept to a minimum in order to avoid deprotection of the maleimide. 1H NMR analysis of the resultant biotin-protected maleimide P2 confirmed incorporation of the protected maleimide moiety as evident by the appearance of peaks at 6.55, 5.16 and 2.93 ppm (Figure 1b). The GPC chromatogram (Figure 2) indicated that the Mn increased from 17,270 to 18,430. The resulting GPC trace was symmetric and did not contain a high molecular weight shoulder. The polydispersity remained narrow (PDI = 1.09) indicating retention of a well-defined polymer. The shift in Mn was attributed to a change in the hydrodynamic volume due to the incorporation of the protected-maleimide end-group.
Figure 1.
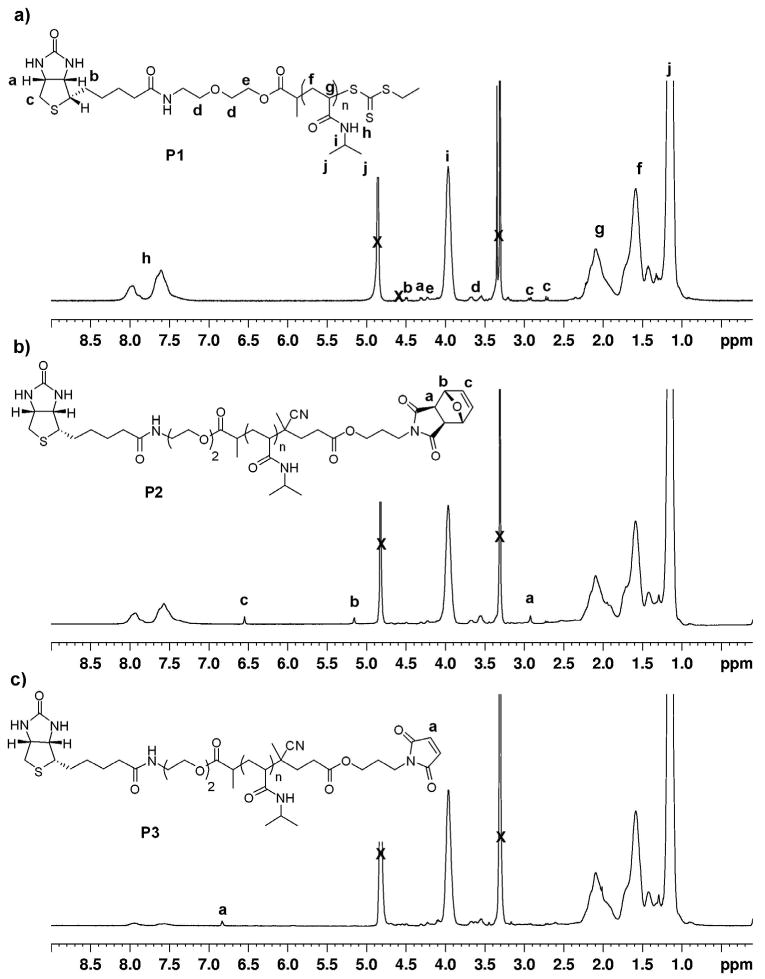
1H NMR spectra (CD3OD) of a) biotinylated pNIPAAm, b) biotin-protected maleimide pNIPAAm, and c) biotin-maleimide heterotelechelic pNIPAAm.
To expose the maleimide moiety, P2 was heated to 120 °C under vacuum (100 mTorr) for 4 hours. Under these conditions the retro Diels-Alder reaction produced the maleimide, while the furan byproduct was conveniently removed in situ. 1H NMR analysis of the biotin-maleimide telechelic P3 indicated the presence of the maleimide by appearance of a peak at 6.84 ppm, while the peaks at 6.55 and 5.16 and 2.93 ppm completely disappeared (Figure 1c). Further, the polydispersity remained narrow (PDI = 1.10) and no shift in retention time was observed in the GPC chromatogram (Figure 2).
Comparison of the integrations in the 1H NMR spectra of the P3 and P1 indicated a loss of the biotin end-group of ∼25 %. We hypothesized that this loss was due to hydrolysis of the ester bond between biotin and the polymer during the end group modification steps. Therefore, as bioconjugate formation was pursued with P3 as described below, the development of a polymer containing no hydrolytically susceptible esters was simultaneously explored.
The CTA was redesigned to contain only amide groups. The biotinylated trithiocarbonate functionalized CTA2 was synthesized using carbodiimide-mediated coupling with EDC and HOBT in 27% yield. Reversed-phase C18 chromatography was employed in addition to regular silica gel chromatography to rigorously remove any side products and residual starting materials. The low yield of the CTA was attributed to this additional purification step.
Outlined in Scheme 5, polymerization of NIPAAm was explored in the same fashion as for CTA1; the resulting polymer had a molecular weight of 10,100 Da by 1H NMR and 9,750 Da by MALDI-TOF compared to a theoretical molecular weight of 9,420 Da and a PDI of 1.07 by GPC (Fig. S4). UV-Vis analysis of the polymer indicated 88% retention of the trithiocarbonate end-group. Subsequent radical cross-coupling with 4, followed by a retro Diels-Alder to expose the maleimide group provided P4. 1H NMR analysis indicated complete retention of the biotin end-group during the post-polymerization reactions (Figure 3). This confirmed our theory that the loss in biotin end-group retention was the result of hydrolysis of the ester bond between biotin and the polymer. It might also be possible to prevent hydrolysis of the ester bonds by rigorous drying of the solvents and polymer prior to the end group modification steps.
Scheme 5.

Synthesis of biotin-maleimide pNIPAAm containing amide-linked biotin.
Figure 3.
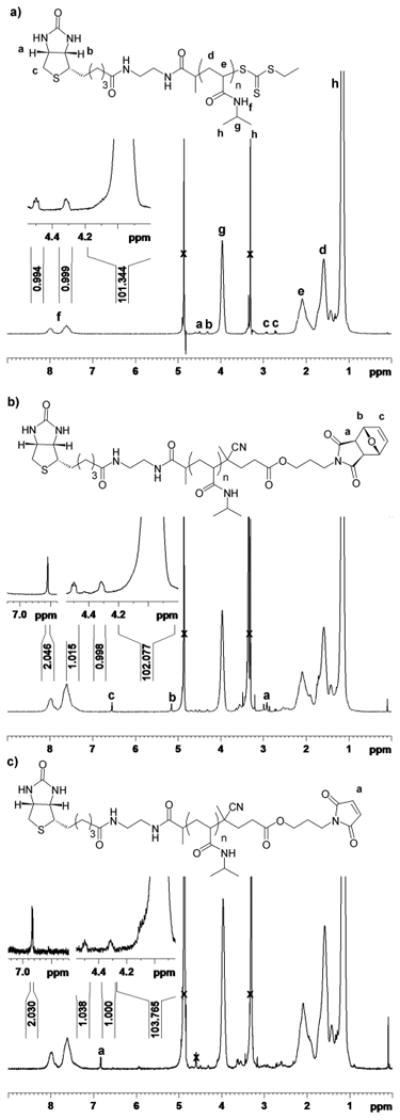
1H NMR spectra (CD3OD) of a) biotinylated pNIPAAm, b) biotin-protected maleimide pNIPAAm, and c) biotin-maleimide heterotelechelic pNIPAAm (4).
Formation of BSA-SAv heterodimer
One of the applications of these polymers is the formation of heterotelechelic protein dimers in solution. To demonstrate this, preparation of a SAv-bovine serum albumin (BSA)-heterodimer was investigated using the biotin-maleimide telechelic P3. BSA was employed because it contains one free surface accessible cysteine (Cys-34). In order to avoid hydrolysis of the maleimide, thioether formation was pursued first. BSA was incubated with a solution of the telechelic pNIPAAm P3 in phosphate buffer (Buffer 1) at 23 °C. The resultant biotin-pNIPAAm-BSA (conjugate 1) was purified by gel filtration to remove un-reacted polymer. SDS-PAGE confirmed conjugate formation by the shift to higher molecular weight (Fig. 4a). Residual BSA was also observed.
Figure 4.
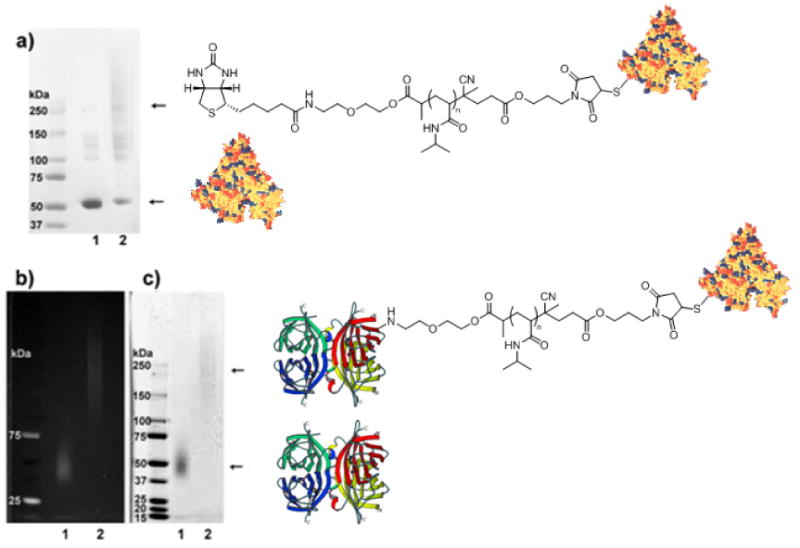
SDS-PAGE of purified pNIPAAm-BSA and SAv-BSA heterodimer. a) BSA-pNIPAAm conjugate 1; lane 1: BSA; lane 2: BSA-pNIPAAm conjugate 1; b) SAv-BSA heterodimer conjugate 2, visualized under UV light; c) SAv-BSA heterodimer visualized with Coomassie Blue stain. b), c) lane 1: SAv-AlexaFluor-350; lane 2: SAv-AlexaFluor350-pNIPAAm-BSA heterodimer conjugate 2. Figure 4. SDS-PAGE of purified pNIPAAm-BSA and SAv-BSA heterodimer. a) BSA-pNIPAAm conjugate 1; lane 1: BSA; lane 2: BSA-pNIPAAm conjugate 1; b) SAv-BSA heterodimer conjugate 2, visualized under UV light; c) SAv-BSA heterodimer visualized with coomassie Blue stain. b), c) lane 1: SAv-AlexaFluor-350; lane 2: SAv-AlexaFluor350-pNIPAAm-BSA heterodimer conjugate 2. Protein structures from PDB number 1P56 and reference 45.
To form a SAv-BSA heterodimer, purified conjugate 1 was mixed with SAv-AlexaFluor 350 in deionized water. Blue fluorescent SAv was used in order to have a method to visualize two proteins by UV light and coomassie blue stain. SDS-PAGE of the SAv-BSA heterodimer (conjugate 2) was visualized first under UV light (Fig. 4b) and then with coomassie stain (Fig. 4c). Using both visualization techniques it was clear that the SAv was present in the conjugate, and that the SAv-BSA heterodimer was formed. This demonstrated that heterotelechelic conjugate formation in solution was possible. Due to the number of binding sites on the SAv (3.2 binding sites by 2-(4-hydroxyphenylazo)benzoic acid (HABA) assay) it was likely that multiple BSAs were conjugated to the SAv thus forming a star protein-polymer conjugate. Such conjugates are also interesting for a variety of reactions. However, is required, a SAv-polymer-BSA heterodimer could be easily formed by using SAv that only has one biotin-binding site.
Conclusion
This report describes a straightforward approach to create protein-reactive heterotelechelic polymers using a functionalized CTA and radical cross-coupling of a trithiocarbonate with a functionalized azo-initiator. A maleimide was readily introduced onto the ω chain-end of an α-biotinylated polymer with this methodology. Using the biotin-maleimide telechelic polymer, SAv and BSA were readily conjugated to the same polymer chain. We illustrated incorporation of two types of protein-reactive groups (biotin and maleimide). However, other protein-reactive groups both at the α and ω ends should be easy to prepare by synthesizing different CTAs and azo initiators, respectively. This approach should also be amenable to conjugating peptides, fluorophores and other proteins besides SAv and BSA. We targeted pNIPAAm because it is a thermoresponsive polymer utilized in diverse applications including reversible cell adhesion and enzyme switches. Yet, likely a wide variety of polymers can be synthesized. Thus, we envision that an array of heterodimer protein-polymer conjugates can be prepared using this technique, and applied as therapeutics or for surface modifications. We are currently pursuing both of these applications.
Acknowledgments
This research was funded by the National Science Foundation Grant Number (CAREER CHE-0645793). KLH and GG thank the Christopher S. Foote Graduate Fellowship in Organic Chemistry. HDM thanks Alfred P. Sloan Foundation and Amgen (New Faculty Award) for additional funding.
Footnotes
Supporting Information: Supporting Information Available: 1H NMR spectra of CTAs, initiator, UV-VIS study, GPC not in text, MALDI-TOF. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Duncan R. Nat Rev Drug Discovery. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman AS. Clinical Chem. 2000;46:1478–1486. [PubMed] [Google Scholar]
- 3.Caliceti P, Veronese FM. Adv Drug Deliv Rev. 2003;55:1261–1277. doi: 10.1016/s0169-409x(03)00108-x. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman AS, Stayton PS. Macromol Symp. 2004;207:139–151. [Google Scholar]
- 5.Mammen M, Choi SK, Whitesides GM. Angew Chem Int Ed. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 6.Veselovsky AV, Ivanov YD, Ivanov AS, Archakov AI, Lewi P, Janssen P. J Molec Recogn. 2002;15:405–422. doi: 10.1002/jmr.597. [DOI] [PubMed] [Google Scholar]
- 7.Bilgicer B, Moustakas DT, Whitesides GM. J Am Chem Soc. 2007;129:3722–3728. doi: 10.1021/ja067159h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 9.Choi SK, Mammen M, Whitesides GM. J Am Chem Soc. 1997;119:4103–4111. [Google Scholar]
- 10.Fichter FM, Zhang L, Kiick KL, Reineke TM. Bioconjugate Chem. 2008;19:76–88. doi: 10.1021/bc0701141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffith BR, Allen BL, Rapraeger AC, Kiessling LL. J Am Chem Soc. 2004;126:1608–1609. doi: 10.1021/ja037646m. [DOI] [PubMed] [Google Scholar]
- 12.Heath F, Haria P, Alexander C. Aaps Journal. 2007;9:E235–E240. doi: 10.1208/aapsj0902026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maynard HD, Okada SY, Grubbs RH. J Am Chem Soc. 2001;123:1275–1279. doi: 10.1021/ja003305m. [DOI] [PubMed] [Google Scholar]
- 14.Puffer EB, Pontrello JK, Hollenbeck JJ, Kink JA, Kiessling LL. ACS Chem Bio. 2007;2:252–262. doi: 10.1021/cb600489g. [DOI] [PubMed] [Google Scholar]
- 15.Strong LE, Kiessling LL. J Am Chem Soc. 1999;121:6193–6196. [Google Scholar]
- 16.Thomas TP, Shukla R, Kotlyar A, Liang B, Ye JY, Norris TB, Baker JR. Biomacromolecules. 2008;9:603–609. doi: 10.1021/bm701185p. [DOI] [PubMed] [Google Scholar]
- 17.Hatzakis NS, Engelkamp H, Velonia K, Hofkens J, Christianen PCM, Svendsen A, Patkar SA, Vind J, Maan JC, Rowan AE, Nolte RJM. Chem Commun. 2006:2012–2014. doi: 10.1039/b516551b. [DOI] [PubMed] [Google Scholar]
- 18.Kochendoerfer, et al. Science. 2003;299:884–887. doi: 10.1126/science.1079085. [DOI] [PubMed] [Google Scholar]
- 19.Heredia KL, Maynard HD. Org Biomol Chem. 2007;5:45–53. doi: 10.1039/b612355d. [DOI] [PubMed] [Google Scholar]
- 20.Kamigaito M, Ando T, Sawamoto M. Chem Rev. 2001;101:3689–3745. doi: 10.1021/cr9901182. [DOI] [PubMed] [Google Scholar]
- 21.Matyjaszewski K, Xia JH. Chem Rev. 2001;101:2921–2990. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 22.Lowe AB, McCormick CL. Prog Polym Sci. 2007;32:283–357. [Google Scholar]
- 23.Moad G. Aust J Chem. 2006;59:661–662. [Google Scholar]
- 24.Ladmiral V, Monaghan L, Mantovani G, Haddleton DM. Polymer. 2005;46:8536–8545. [Google Scholar]
- 25.Tao L, Mantovani G, Lecolley F, Haddleton DM. J Am Chem Soc. 2004;126:13220–13221. doi: 10.1021/ja0456454. [DOI] [PubMed] [Google Scholar]
- 26.Bontempo D, Heredia KL, Fish BA, Maynard HD. J Am Chem Soc. 2004;126:15372–15373. doi: 10.1021/ja045063m. [DOI] [PubMed] [Google Scholar]
- 27.Mantovani G, Lecolley F, Tao L, Haddleton DM, Clerx J, Cornelissen JJ, Velonia K. J Am Chem Soc. 2005;127:2966–2973. doi: 10.1021/ja0430999. [DOI] [PubMed] [Google Scholar]
- 28.Heredia KL, Tolstyka ZP, Maynard HD. Macromolecules. 2007;40:4772–4779. [Google Scholar]
- 29.Bontempo D, Li RC, Ly T, Brubaker CE, Maynard HD. Chem Commun. 2005:4702–4704. doi: 10.1039/b507912h. [DOI] [PubMed] [Google Scholar]
- 30.Qi K, Ma QG, Remsen EE, Clark CG, Wooley KL. J Am Chem Soc. 2004;126:6599–6607. doi: 10.1021/ja039647k. [DOI] [PubMed] [Google Scholar]
- 31.Vazquez-Dorbatt V, Maynard HD. Biomacromolecules. 2006;7:2297–2302. doi: 10.1021/bm060105f. [DOI] [PubMed] [Google Scholar]
- 32.Sumerlin BS, Lowe AB, Stroud PA, Zhang P, Urban MW, McCormick CL. Langmuir. 2003;19:5559–5562. [Google Scholar]
- 33.You YZ, Manickam DS, Zhou QH, Oupicky D. Biomacromolecules. 2007;8:2038–2044. doi: 10.1021/bm0702049. [DOI] [PubMed] [Google Scholar]
- 34.Perrier S, Takolpuckdee P, Mars CA. Macromolecules. 2005;38:2033–2036. [Google Scholar]
- 35.Bathfield M, D'Agosto F, Spitz R, Charreyre MT, Delair T. J Am Chem Soc. 2006;128:2546–2547. doi: 10.1021/ja057481c. [DOI] [PubMed] [Google Scholar]
- 36.Hong CY, Pan CY. Macromolecules. 2006;39:3517–3524. [Google Scholar]
- 37.Liu J, Bulmus V, Barner-Kowollik C, Stenzel MH, Davis TP. Macromol Rapid Commun. 2007;28:305–314. [Google Scholar]
- 38.Boyer C, Liu J, Bulmus V, Davis TP, Barner-Kowollik C, Stenzel MH. Macromolecules. 2008;41:5641–5650. [Google Scholar]
- 39.Neubert BJ, Snider BB. Org Lett. 2003;5:765–768. doi: 10.1021/ol034042e. [DOI] [PubMed] [Google Scholar]
- 40.Tao L, Geng J, Chen GJ, Xu YJ, Ladmiral V, Mantovani G, Haddleton DM. Chem Commun. 2007;(33):3441–3443. doi: 10.1039/b709080c. [DOI] [PubMed] [Google Scholar]
- 41.Hermanson GT. Bioconjugate Techniques. Academic Press; New York: 1996. [Google Scholar]
- 42.Moad G, Rizzardo E, Thang SH. Aust J Chem. 2006;59:669–692. [Google Scholar]
- 43.Hentschel J, Bleek K, Ernst O, Lutz J, Borner HG. Macromolecules. 2008;41:1073–1075. [Google Scholar]
- 44.Brandrup J, Immergut EH, Grulke EA. Polymer Handbook. 4th. Wiley; New York: 1999. [Google Scholar]
- 45.Carter DC, Ho JX. Adv Protein Chem. 1994;45:153–204. doi: 10.1016/s0065-3233(08)60640-3. [DOI] [PubMed] [Google Scholar]