

Abstract
The National Institutes of Health Undiagnosed Diseases Program evaluates patients for whom no diagnosis has been discovered despite a comprehensive diagnostic workup. Failure to diagnose a condition may arise from the mutation of genes previously unassociated with disease. However, we hypothesized that this could also co-occur with multiple genetic disorders. As demonstration of a complex syndrome caused by multiple disorders, we report two siblings manifesting both similar and disparate signs and symptoms. They shared a history of episodes of hypoglycemia and lactic acidosis, but had differing exam findings and developmental courses. Clinical acumen and exome sequencing combined with biochemical and functional studies identified three genetic conditions. One sibling had Smith-Magenis Syndrome and a nonsense mutation in the RAI1 gene. The second sibling had a de novo mutation in GRIN2B, which resulted in markedly reduced glutamate potency of the encoded receptor. Both siblings had a protein-destabilizing homozygous mutation in PCK1, which encodes the cytosolic isoform of phosphoenolpyruvate carboxykinase (PEPCK-C). In summary, we present the first clinically-characterized mutation of PCK1 and demonstrate that complex medical disorders can represent the co-occurrence of multiple diseases.
Keywords: Hypoglycemia, lactic acidemia, dysmorphism, developmental delay, protein structure-function, multiple genetic disorders
1. INTRODUCTION
Genome-Scale or “Next Generation” Sequencing (NGS) has become widely available for clinical use. The field of rare and new disease discovery leads the way in the application of NGS, and the NIH Undiagnosed Diseases Program (UDP) typifies this effort. The UDP features intensive phenotyping, diagnostic tools agnostic to disease causation, newly developed NGS analytics, and collaborative research aimed at establishing the disease-causality of candidate mutations by biochemists, cell biologists, and physiologists.
A traditional search for the cause of a heritable disease seeks a single, highly penetrant, large-effect gene. In this model, unusually severe or treatment-refractory presentations represent novel versions of known disorders and are considered to reflect the influence of genetic and/or environmental modifiers. However, case reports support the epidemiologic expectation that multiple rare disorders will occasionally occur in the same individual [1, 2] [2, 3]. Experience in the UDP has confirmed that a substantial number of apparently unique, undiagnosed medical conditions will, in fact, be combinations of multiple genetic disorders in single individuals or families. Illustrating this, we present a non-consanguineous family with two affected children in which three different functionally verified mutations contribute to their phenotypes. For undiagnosed, unique and extensively investigated individuals such as those evaluated by the UDP, physicians should consider complex alternatives to a single, unifying genetic explanation.
The family reported here included two siblings who presented with variable developmental delay, hypotonia, and fasting hypoglycemia with lactic acidemia. One had prominent dysmorphic features and obesity; the other did not. Each had extensive metabolic and genetic testing by several specialists before the final diagnoses were confirmed by DNA sequencing, functional, and structural analyses.
2. METHODS
2.1 Patients
The family was enrolled in clinical protocol 76-HG-0238, “Diagnosis and Treatment of Patients with Inborn Errors of Metabolism and Other Genetic Disorders”. The parents gave written informed consent for their children, who were enrolled in the NIH UDP. This initiative evaluates individuals and families in whom previous diagnostic investigations have been unrevealing [3–5].
22.2 NGS Sequencing and SNP Analysis
SNP oligo array analysis for filtering of exome sequence data was performed using Illumina Bead Array Platform and Genome Studio software (Illumina Corp, San Diego, CA), as described previously [6, 7]. NGS data were generated using the Sureselect Human All Exon System (Agilent Technologies Inc, Santa Clara, CA) and a GAIIx sequencer (Illumina Inc, San Diego CA). Library construction, sequence generation, sequence alignment to the reference genome, variant calling and potential pathogenic variant identification were performed as described [8, 9].
Initial sequence-variant analysis was performed using an autosomal recessive genetic model. A second round of analysis employed a new-dominant model, identifying sequence variants that met the following criteria: (1) excellent coverage in all samples from prior UDP NGS projects; (2) variation from the human reference sequence (HG18) occurring in only the family being studied; and (3) appearance of the candidate mutation only in affected members of the family.
2.3 Sanger sequencing
The primer pairs RAI1-F-5′-CCAGGGCTGTTTGAAGACC -3′ and RAI1-R-5′-AAGTCGGCGGTGGAACAG -3′, PCK1-F-5′-CTCTGCAGAAATGCCTCCTC -3′ and PCK1-R-5′-CACCTTTCTCCACCAACTCC -3′ and GRIN2B-F-5′-AATCCCACAGCTCAAAAGCA -3′ and GRIN2B-R-5′-ATGTTGGCACTTGTTGGCTT -3′ were used to amplify respectively from genomic DNA the putative variants NM_030665:c.2273G>A, NM_000834.3:c.1238A>G, NM_002591.3:c.134T>C and 200 flanking nucleotides. PCR amplification was performed using Qiagen HotStarTaq master mix (Qiagen, Valencia, CA). The following conditions were used for amplification: 1 cycle of 95°C for 5 min, followed by 40 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 5 min. Unincorporated primers and nucleotides were removed using ExoSAP-IT reagent (USB, Cleveland, OH, USA).
Sanger sequencing of the PCR products was performed by Macrogen (Rockville, MD). The sequences were aligned and analyzed using Sequencher v.5.0.1 (Gene Codes, Ann Arbor, MI, USA). Mutation interpretation analysis was conducted using Alamut 2.0 (Interactive Biosoftware, San Diego, CA, USA).
2.4 RAI1 mRNA expression analysis
Total RNA was isolated from patients’ or control lymphoblastoid cells using the RNeasy Mini-Kit (Qiagen, Valencia, CA). RNA was subsequently treated with DNA-free DNase (Applied Biosystems). RNA concentration and purity were assessed on a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). First strand cDNA was synthesized using a high capacity RNA-to-cDNA kit (Applied Biosystems). qPCR was performed utilizing a RAI1 Assays-On-Demand Taqman primer-probe assay (Applied Biosystems), Hs00430773_m1 (located at the RAI1 exon 2–3 boundary) and a control assay for the beta-actin housekeeping gene (Hs99999903_m1). PCR amplifications were performed on 100ng of cDNA using TaqMan Gene Expression Master Mix reagent (Applied Biosystems) and were carried out on an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems). Results were analyzed with the comparative CT method as described [10, 11]. All assays were performed in triplicate. Displayed values in Figure 3 represent the relative quantification (RQ) normalized to the average of all control assays in all three control cell lines (arbitrarily set to 1).
Fig. 3.

Structural and Functional Properties of the Mutant PEPCK-C. (A) The overall structure of crystallized recombinant I45T PEPCK-C indicating the location of the I45T mutation with respect to the overall enzyme structure and the active site (indicated by the molecules of βSP and GTP rendered as ball and stick models colored by atom type). The N- and C-termini are also labeled. (B) The comparison of WT (light blue, PDB 3DT7) and I45T PEPCK-C (grey, PDB 4OX2, this work) in the vicinity of I45 illustrates that the local structure of the hydrophobic pocket is not perturbed upon the threonine for isoleucine substitution. 2Fo-Fc density corresponding to the side-chain of T45 is illustrated as a blue mesh rendered at 1.2 σ. (C) Kinetic characterization of recombinant I45T PEPCK-C compared with the wild type (WT) enzyme, showing very similar kinetic parameters (KM for both OAA and GTP and kcat), and heat stability (t1/2) at two temperatures (*WT data is from [20]). (D) Western blots of PEPCK-C and PEPCK-M in frozen liver from patient 1 and two controls. Figures above show western blots specific for PEPCK-C and PEPCK-M and the reference proteins β-actin or GAPDH, with the amount of micrograms of protein homogenate beneath, for patient 1 and human controls A or B. Graphs below these respective blots comparing the normalized signal intensity (corrected for the ratio to their reference proteins) of ratios of PEPCK-C (blue) and PEPCK-M (red) in the patient vs controls A and B. (E) Half lives of mutant I45T and wt PEPCK-C in T-Rex 293 cultured stable inducible cell lines. Figure above shows Western blots at indicated time intervals before and after tetracycline induction of PEPCK-C I45T (upper row), PEPCK-C wt (lower row), and GAPDH as a reference protein. Graph below shows relative amounts of PEPCK-C I45T/GAPDH (solid circles) and PEPCK-C/GAPDH wt (solid squares) over 24 h after induction, to establish the mutant and wild type protein half-lives.
2.5 GRIN2B physiology and structural modeling
Site-directed mutagenesis was performed using the QuikChange protocol (Stratagene, La Jolla, CA); amino acid numbering refers to full length protein (initiating methionine is 1) [12]. cRNA was prepared from cDNA for mutant and wild type human NMDA receptor subunits in pCI-neo (GenBank: NP_015566 and NP_000825) and injected into Xenopus laevis oocytes as previously described [13, 14]. Two-electrode voltage-clamp recordings from oocytes were made 2–4 days post-injection (23°C). The recording solution contained (in mM) 90 NaCl, 1 KCl, 10 HEPES, 0.5 BaCl2, 0.01 EDTA (pH 7.4); recording electrodes were filled with 0.3–3 M KCl. Homology models of the human GluN2B ligand binding domain [15] were simulated with molecular dynamics bound L-glutamate and explicit solvent at 310K and 1 bar under periodic boundary conditions using the program NAMD [16].
2.5 PEPCK-C enzyme activity, immunoassay, and expression of the recombinant mutant form for structural and functional analysis
PEPCK total enzyme activity (including PEPCK-C and PEPCK-M) was assayed in disrupted cultured skin fibroblasts and in frozen liver homogenates by PEP (phosphoenolpyruvate)-dependant 14CO2 fixation [17, 18]. Immunoblot assays of PEPCK-C and PEPCK-M in frozen liver from one sibling and controls, using specific monoclonal antibodies that distinguish the two isoforms (Santa Cruz Biotechnology, Dallas, Texas), and the tissue reference proteins, β-actin and GAPDH [19]. Control fibroblasts and frozen liver samples were obtained from unused extra material retained in the Center for Inherited Disorders of Energy Metabolism laboratory (UHCMC).
Recombinant and mutant I45T PEPCK were expressed and purified as previously described [20], with the exception that the I45T-PEPCK was initially grown at 37°C to an OD of 1.2–1.5 and then the temperature was lowered to 30°C and the cells were induced with 0.05mM IPTG and grown for a further 15–20 hours. Crystals of I45T in complex with βSP and GTP were grown as previously described for the WT enzyme [20, 21]. Diffraction data on the cryocooled (100K) I45T-Mn2+-BSP-Mn2+GTP crystals were collected at the University of Waterloo on a Rigaku Micro-Max rotating anode X-ray generator equipped with an RaxisIV++ detector and Oxford cryostream. The 2.0 Å structure was solved by the molecular replacement method using MOLREP in the CCP4 package and the previously solved structure of the WT-Mn2+-BSP-Mn2+GTP complex [22–24]. The molecular replacement solution was refined using Refmac5 followed by manual model adjustment and rebuilding using COOT [25]. Ligand, metal, and water addition and validation were also performed in COOT. The crystallographic data and model statistics are presented in Table S-1. All structural data were validated using Molprobity (http://molprobity.biochem.duke.edu) [26, 27], and the statistics from this analysis are also presented in Table S-1.
For kinetic analyses, WT and I45T PEPCK-C were assayed as previously described [20, 21]. To determine of the half-life stability for the enzymes, WT and I45T PEPCK-C were incubated in 25 mM HEPES pH 7.5, 10mM DTT at either 37 or 50° C. At various time points a quantity of enzyme was removed from the incubation mix and assayed for activity using the standard PEPCK assay in the direction of OAA formation [20, 21]. The inactivation rate (kinact) was determined by plotting the natural log of the remaining activity vs time. This value was used to calculate the apparent ½ life for the enzymes using the equation t1/2 = ln2/kinact.
2.6 Expression of PEPCK-C in HEK293 cells and measurement of half-life
The PCK1 wild type cDNA (NM_002591.3) was cloned into the pENTR vector (Life Technologies, Grand Island). The mutation c.134T>C was introduced by site directed mutagenesis using QuikChange II kit (Agilent, Santa Clara). The wild type and mutant cDNAs were then recombined into the pDEST31 (Life Technologies, Grand Island) expression vector using the GateWay system (Life Technologies, Grand Island).
The pDEST31-PCK1wt and pDEST-PCK1134T>C constructs were transfected into T-REx-293 cell line (Life Technologies, Grand Island) using Lipofectamine 2000 according to the manufacturers protocol (Life Technologies, Grand Island). Cells stably transfected with each construct were selected by subsequently growing the transfected cells in DMEM containing 10% FBS, 5 μg/ml blasticidin and 120 μg/ml G418. Expression of the transgene was induced in each stable cell line by treatment with 1 μg/ml tetracycline for 24 h. To measure protein decay, the cells were treated with 1 μg/ml tetracycline for 24 h and then washed with PBS and grown in DMEM containing blasticidin, G418 and 50 μg/ml cycloheximide. The cells were harvested at 0, 2, 4, 6, 12, 18 and 24 h by scraping and the cell pellets were stored at −80°C.
Cell pellets were lysed by addition of 1 volume of 2X Laemmli Sample Buffer (Biorad, Hercules) and boiling for 10 min. After a brief centrifugation, the supernatants were fractionated over a 12% PAGE SDS gel. The proteins were transferred to nitrocellulose membranes detected with anti-PEPCK-C (Sigma-Aldrich, St. Louis) and anti-GAPDH (Novus Biologicals). Primary antibodies bound to the membrane were detected with infrared fluorescence labeled secondary antibodies (LI-COR, Lincoln). The images were captured by Odyssey infrared imaging system (LI-COR, Lincoln). Analysis of band intensity was performed using Image J (http://imagej.nih.gov). The amount of PEPCK-C was normalized to GAPDH to adjust for loading differences. Decay was measured from the maximal PEPCK-C level detected.
2.7 PCK1 mRNA expression analysis
For extraction of total RNA, T-Rex -PCK1wt and -PCK1134T>C cells were treated with 1 μg/ml tetracycline for 24 h and then washed with PBS and grown in DMEM containing blasticidin, G418 and 50 μg/ml cycloheximide for 0 or 6 h. We purified RNA using the RNeasy mini kit (Qiagen, Valencia, CA). One μg total RNA was reverse transcribed with SuperScript VILO cDNA kit (Life Technologies, Grand Island) and used for PCK1 transgene PCR amplification. To specifically amplify the transgene, one the forward primer was placed within the amino-terminal 6xHis tag: PCK1_his_Fw CCATCACACCGGTGATATCCT and PCK1 Rv AGCCAACCAGCAGTTGTCAT. PCR amplification was performed using Qiagen HotStarTaq master mix (Qiagen, Valencia, CA) under the following conditions: 1 cycle of 95°C for 5 min, followed by 40 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 5 min. The PCR products were run on 1.2% agarose gels.
3. RESULTS
3.1 Case Reports
The propositae are two sisters born to non-consanguineous parents with Ashkenzi Jewish ancestry and a non-contributory family history.
3.1.1 Patient 1
The elder (Patient 1) was born at term following an uncomplicated pregnancy. Her birth weight, length and head circumference were 3118 g (25–50th %ile), 48.3 cm (25–50th %ile) and 33.75 cm (5.5 %ile), respectively. She did not have neonatal hypoglycemia. Her growth during infancy and early childhood was normal, although she did have problems with gastroesophageal reflux and esophagitis as well as allergies to soy and milk proteins. By mid-childhood she had become obese, and at 11 years, she had a BMI of 33.5 kg/m2 (>97th %ile).
Her early motor development was late; she sat and walked independently by age 8 and 16 months, respectively. Gradually, she manifested intellectual disability (talking after 3 years), gross and fine motor delays, easy fatigability, sleep disturbance, low muscle tone, and transient unexplained neuropathy with an abnormal EMG. She also exhibited coarse facial features, brachydactyly, fifth finger clinodactyly and widely spaced teeth (Fig. 1). At 11 years of age, she could walk and talk commensurate with her cognitive abilities, was not toilet trained, and required significant assistance with fine motor and executive tasks. She had disrupted sleep architecture and aggressive and obstinate behavior when tired. On examination, her weight, height and head circumference were 68.9 kg (>97th %ile), 143.4 cm (25–50th %ile) and 60 cm (>97th %ile), respectively. Other notable features included synophrys, brachycephaly hypotelorism, hypoplastic toenails and abdominal striae.
Fig. 1.
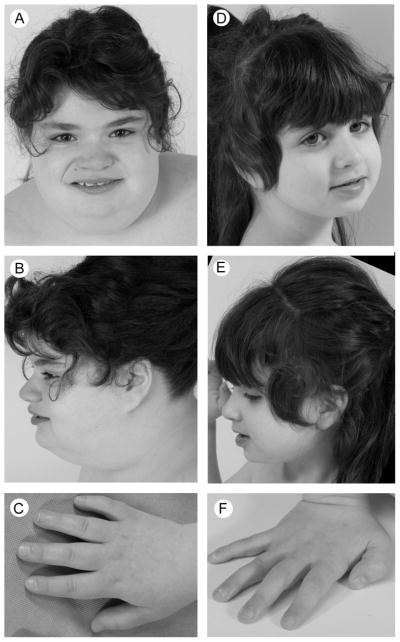
Photographs of propositae. Patient 1 (A–C) was obese and had a broad square-shaped face, midfacial hypoplasia, mildly upslanting palpebral fissures, a broad nasal bridge, a full-tipped nose, short tapering fingers and mild 5th finger clindodactyly. In contrast, patient 2 (D–F) was slight and had no dysmorphic features.
At 20 months, she presented with fever, lethargy, fasting hypoglycemia (blood glucose 29 mg/dl) and ketonuria. She had appropriately elevated growth hormone and cortisol levels, undetectable insulin and normal C-peptide levels. Over the subsequent 5 years, she had at least 12 similar episodes of illness or fasting associated hypoglycemia, on occasion associated with significant lactic acidemia (9.6 mM), leading to consideration of a defect of gluconeogenesis. Gluconeogenic enzyme assays included liver glucose-6-phosphatase, blood fructose-1,6-diphosphatase, skin fibroblast pyruvate carboxylase and total phosphoenolpyruvate carboxykinase (not excluding cytosolic PEPCK deficiency), which all were within their reference ranges.
In the context of illness related hypoglycemia with lactic acidosis and steatosis, she was also evaluated for glycogen storage disease (GSD) and fatty acid oxidation. Analysis of a muscle biopsy performed at 2 years however showed normal muscle histopathology, glycogen content, glucose-1-phosphate/glucose ratio, debranching enzyme activity, pyruvate dehydrogenase and total phosphorylase activity. Subsequently, histopathological studies of a liver biopsy at age 4 years showed no iron deposits, and no abnormal lipid or glycogen, although there was mild-moderate portal inflammation, mild portal fibrosis, and patchy, moderate, macrovesicular steatosis. Furthermore, assessment of the biopsied liver tissue did not detect deficiency of debranching enzyme or phosphorylase, phosphorylase kinase, and testing of peripheral blood DNA did not identify pathogenic mutations of G6PC, SLC37A4, ALDOA, or GSK3B deficiency. Consistent with these findings, she lacked the GSD features of hepatomegaly, growth failure, hypertriglyeridemia, or hyperuricemia.
To exclude mitochondrial disorders, she had additional studies and another muscle biopsy at 4 years of age. Analysis of this muscle biopsy showed normal histopathology and electron transport chain enzymology. She also had no detectable mutations of the muscle mitochondrial DNA or of POLG, TK2, COX10, ACAD9 and DGUOK in her blood DNA.
She had a normal cardiac ECHO, renal ultrasound, brain MRI, electroretinogram, karyotype, TSH and free T4 levels, plasma amino acid and acylcarnitine profiles, urine organic acid profile, ammonia and lactate levels (when she was well), complete blood counts, and liver function tests. Other molecular testing found normal SNRPN methylation and detected no mutations of BBS1 or FMR1. Abnormal non-diagnostic test results included array comparative genomic hybridization (CGH) Xq13.3 (hg18:74,015,588bp–74,683,357bp) x3 paternal origin.
3.1.2 Patient 2
The younger sister of Patient 1 (Patient 2) was born at term following a pregnancy complicated by premature labor. Her birth weight, length and head circumference were 3515 g (50th–75th %ile), 48.3 cm (25–50th %ile) and 33.25 cm (2.4 %ile), respectively. She did well in the newborn period, without hypoglycemia.
In contrast to her sister, she had earlier delayed development and hypotonia. She rolled at 6 months, sat independently at 19 months, cruised at 3.5 years and walked independently at 4 years. At 5 years she was not toilet trained and was unable to dress or undress herself or feed herself with a spoon. She spoke only one comprehensible word “mom” and was unable to communicate by pointing or signing. Her neuropsychiatric evaluation at age 5 years assessed her gross motor, visual reception, fine motor, receptive language and expressive language age equivalents as 15 months, 5 months, 12 months, 10 months and 4 months, respectively; she had no daily living skills. No autism spectrum features could be clearly differentiated from her general profound intellectual disability.
At approximately 8 months, she also presented with illness-associated lethargy, fasting hypoglycemia (blood glucose 31 mg/dl) elevated urine lactate, and ketonuria. At 10 months, she had a similar episode, with blood glucose 20 mg/dl and lactate 11.4 mM. Like her sister, she has had several repeated episodes of illness or fasting-associated hypoglycemia, sometimes associated with acidosis.
On examination at 5 years, her weight, height and head circumference were 17.8 kg (25th–50th %ile), 106.1 cm (25th–50th %ile) and 50 cm (25th–50th %ile), respectively. Aside from the absence of dysmorphism, her notable features included an unsteady gait, hypotonia, stereotypic hand movements, absent pincher grasp, sensitivity to texture and pes planus. She did not have microcephaly or a webbed neck.
She had extensive testing that did not identify the cause her hypoglycemia or intellectual disability. The normal blood test results included plasma amino acid and acylcarnitine profiles, urine organic acid and acylglycine profiles, blood ammonia and lactate (when she was well), complete blood counts, and liver function tests. Similarly, testing of CSF neurotransmitter, lactate and glucose levels did not identify any abnormalities. Muscle biopsies performed at ages 4 and 6 years showed normal muscle electron transport and histopathology. She had a normal karyotype and no detectable mutations of muscle mitochondrial DNA, ETFDH, ETFA, ETFB, ALDOB, COX10, G6PC, SLC37A4, FMR1 and ACADVL. Patient 2 also had a normal cardiac ECHO, abdominal ultrasound (showing upper normal liver size), and brain MRI. Her EEG was within the broad range of normal limits for age in the awake and drowsy state and without focal, paroxysmal, or epileptiform abnormalities. By array CGH she also had inherited the paternal duplication of Xq13.3 (hg18:74,015,588bp–74,683,357bp).
3.2 Diagnosis #1: Smith-Magenis syndrome
Patient 1 had signs and symptoms consistent with Smith-Magenis Syndrome (SMS) including obesity, brachydactyly, developmental delay, cognitive impairment, behavioral outbursts, sleep disruption and facial appearance (Fig. 1). Since the common deletion causing SMS had been excluded by the CGH array, we tested for mutations in RAI1. Sanger sequencing identified NM_030665:c.2273G>A (NP_109590.3:p.W758X), a mutation previously reported in SMS [28]. This mutation was detected only in patient 1, not in her parents or sister (Fig. 2B). The RAI1 mRNA in patient lymphoblastoid cells was decrease to 62%±2% of the level in control cells (Fig. 2C), likely due to RNA decay of the nonsense allele.
Fig. 2.

Identification and characterization of RAI1 and PCK1 mutations. (A) Family pedigree. (B) Sanger sequence traces showing a heterozygous RAI1 mutation (NM_030665:c.2273G>A, NP_109590.3:p.W758X) only in patient 1 (II-1). (C) Quantification of RAI1 steady state mRNA levels in cultured skin fibroblasts of patient 1 and an unaffected control. The nearly 50% decrease in RAI1 steady state mRNA levels in patient lymphoblastoid cells compared to unaffected control lymphoblastoid cells suggests that the p.W758X causes nonsense mediated mRNA decay. β-actin mRNA levels were used for normalization. (D) Sanger sequence traces showing that both patients (II-1 and II-2) had a homozygous PCK1 mutation (NM_002591.3:c.134T>C, NP_002582.3:p.I45T), whereas the parents (I-1 and I-2) were heterozygous for the mutation. (E) NP_002582.3:p.I45T alters an isoleucine (arrowhead) conserved across species to Drosophila melanogaster (see also Figure S-1 for more extensive species comparison, indicating this hydrophobic residue is conserved in the GTP utilizing enzymes all the way through Ascaris suum).
3.3 Diagnosis #2: Cytosolic PEPCK deficiency
The episodic fasting hypoglycemia and lactic acidemia of both siblings remained unexplained, even after investigating possible gluconeogenic defects (including assaying total PEPCK in cultured skin fibroblasts). This prompted exome sequencing. Assuming a recessive genetic model, variant filtering yielded 19 homozygous and 9 compound heterozygous variants. A homozygous variant in cytosolic PCK1 (PEPCK-C, NM_002591.3:c.134T>C) was found in both children (Fig. 2D); it changed the highly conserved isoleucine 45, which resides in a β-pleated sheet in the N-terminal domain, to a threonine (Fig. 2E and Fig. S1). NM_002591.3:c.134T>C (rs202197769) occurs with a frequency of 0.2% in Europeans and has not been detected in other populations or previously reported to occur in homozygosity. Consequently, this homozygous variant was of immediate interest due to the critical role of PEPCK-C in gluconeogenesis [29, 30].
We determined that recombinant I45T PEPCK-C was predominantly expressed in E. coli as an insoluble protein located largely in inclusion bodies, compared with the normal expression and distribution of the WT enzyme (1mM IPTG, 37°C, 4 h) (data not shown). Only upon lowering the induction temperature to 30°C and inducing with 0.05mM IPTG for a total of 15–20 hours was a sufficient amount of the enzyme present in a soluble, active form. Even with these altered conditions, a majority of the enzyme was present in the insoluble fraction.
The 2.0Å crystal structure of I45T PEPCK-C was determined in complex with beta-sulfopyruvate (βSP, a substrate analog of OAA) and GTP; this state approximates the OAA-GTP Michaelis complex. The enzyme-complex crystallized under identical conditions to the same complex with the WT enzyme [23], and the overall structure is essentially identical to that of the WT enzyme with an overall Cα RMSD of 0.2Å (Fig. 3A and Supplementary Table 1). Consistent with the similarity in overall structure, the local structure around the I45T, most notably the hydrophobic pocket formed in part by I45 (Fig. 3B), is identical to that in the WT enzyme. However, the participation of I45 in formation of this hydrophobic pocket, when viewed in conjunction with other data, is consistent with this hydrophobic cluster being important in the folding and stability of PEPCK-C. These findings are consistent with the mutation destabilizing the folded state and suggest that the N-terminal domain possibly acts as an intra-molecular chaperone for correct folding.
In addition, the kinetic parameters (KM of both substrates and kcat) of the recombinant mutant and WT enzymes are virtually identical within error of determination (Fig. 3C). Heat stability of the mutant PEPCK-C (t1/2) was 59% of that for the WT enzyme at 37°C, and 24% at 50°C (also Fig. 3C).
These findings are consistent with the low level of immunoreactive PEPCK-C in frozen liver from patient 1 (Fig. 3D). Western blotting of frozen liver from patient 1 showed greatly reduced amounts (≤ 10%) of immunoreactive PEPCK-C relative to human controls, indicating that the I45T mutation caused PEPCK-C instability; surprisingly, the amount of PEPCK-M was increased (Fig. 3D). Total enzymatic activity of PEPCK was increased in frozen liver (217% of control mean) and normal in cultured skin fibroblasts (94% of control mean). Attempts to assay PEPCK-C and PEPCK-M activity separately in frozen liver or cultured fibroblasts by specific immunoprecipitation or inhibition were unsuccessful. The relatively increased level of PEPCK-M in liver from patient 1 accounts for the increased activity of total PEPCK in frozen liver.
Induced expression of both WT and mutant PEPCK-C in stable cultured human 293 cells, using the T-Rex system, further confirmed these observations. The half-life of immunoreactive WT PEPCK under these conditions was more than 24 h, whereas the half-life of the I45T mutant form of PEPCK-C was no more than 2 h (Figure 3E), and based on RT_PCR, this was not attributable to differences in stability of the WT and mutant PCK1 mRNAs (data not shown).
3.4 Diagnosis #3: Mutation of GRIN2B
The severe neurological and developmental problems of patient 2 still required explanation, so we re-analyzed the exome sequence data using a model to identify new-dominant variants. For patient 2 but not in Patient 1 or either parent, this procedure identified a heterozygous NM_000834.3:c.1238A>G (NP_000825.2:p.E413G) mutation in GRIN2B, the gene encoding GluN2B, an NMDA receptor subunit (Fig. 4A). The p.E413G mutation resides close to residues that correspond to those shown to be in direct atomic contact with the co-agonist L-glutamate in structures of GluN2A and GluN2D glutamate binding domains (Fig. 4B) [31, 32]. We hypothesized that substitution of a glycine at this position would reshape the glutamate binding pocket (Fig. 4B). Two electrode voltage clamp recordings following co-expression with the GluN1 subunit in Xenopus laevis oocytes showed that the p.E413G mutation reduced the glutamate potency >50-fold compared to the wild type receptor (Fig. 4C). This mutation had no effect on the potency of glycine, which binds to the GluN1 subunit (Fig. 4D). Moreover, molecular dynamic simulations for GluN2B (E413G) suggested that H486 in E413G has fewer atomic interactions with neighboring residues, making it more mobile compared to the wild-type receptor (Wells, Yuan, Synder, and Traynelis, unpublished results). These data suggest that the E413G mutation reduced the sensitivity of GluN2B-containing NMDA receptors to synaptic and non-synaptic glutamate through a decrease in EC50 value and thus agonist potency.
Fig. 4.

Identification and characterization of a de novo GRIN2B (GluN2B) mutation in patient 2. (A) Sanger sequence traces showing a heterozygous GRIN2B mutation (NM_000834.3:c.1238A>G, NP_000825.2:p.E413G) only in patient 2 (II-2). (B) Predicted quaternary structure of tetrameric human GluN1/GluN2B, based on the GluA2 AMPA receptor structure (left; 3KG2) [52]. The bi-lobed ligand binding domain (yellow) of GluN2B and related glutamate receptors adopts a clamshell-like structure, which binds glutamate (blue) within the cleft. Ligand-protein interactions between L-glutamate (spheres) and wild-type GluN2B (middle) or GluN2B-E413G (right) ligand binding domains, are modeled from GluN1/GluN2A crystallographic data (2A5T) [15]. Nearby residues and crystallographically conserved waters are shown (sticks). Hydrogen bonds are depicted by the black dashed lines. (C) The composite concentration-response curves and fitted EC50 values are shown for human GluN1/GluN2B (wild type) or GluN1/GluN2B-E413G (E413G) current responses (100 μM glycine in all solutions, VHOLD −40 mV). (D) The composite glycine concentration-response curves and fitted EC50 values at human GluN1/GluN2B (wild type) or GluN1/GluN2B-E413G (E413G) receptors (1 mM glutamate present in all solutions). EC50 values were obtained by fitting the curves in (C) and (D) with Response (%) = 100 / (1 + (EC50 / [agonist])N). N is the Hill slope, which ranged between 1.1 – 1.4; n = 11–20 oocytes).
4. DISCUSSION
This family’s unaffected parents had two daughters, each of whom had PEPCK-C deficiency (associated with proven instability of the mutant protein), one of whom had SMS (documented by RAI1 mutation), and the other of whom had a GRIN2B mutation (shown by functional assay of the p.E413G mutation’s effect on GluN2B). The SMS explains the dysmorphisms and behavioral phenotype. Mutations in the GRIN2B gene have been associated with a wide range of neurological disorders, including intellectual disability (MRD6, OMIM #613970), autism spectrum disorders, attention deficit hyperactivity disorder, schizophrenia, epilepsy, developmental and other cognitive phenotypes [33–42]. Most reported individuals with GRIN2B mutations have developmental delay, mental retardation, or intellectual disability, often accompanying other neurological problems. The GluN2B alteration (E413G) could lead to hypo-function of GluN2B-containing NMDA receptors, which are expressed early in development and play an important role in development of the brain [43–49]. The PEPCK-C deficiency explains the previously unexplained repeated events of fasting hypoglycemia with lactic acidemia. This report of a mutation in the PCK1 gene and its protein consequences is the first molecularly characterized case of PEPCK-C deficiency in humans and the second functionally well-characterized case [30].
The respective functional roles of PEPCK-C and PEPCK-M, their tissue distribution and interaction in different species are not currently fully explained [29, 50]. It is clear the PEPCK-C is critical for hepatic gluconeogenesis in mammals, and expression of that gene (PCK1) is responsive to physiological and hormonal regulation. The functional role of PEPCK-M is less clear, except that it appears to be important for birds in flight. In mammals, it constitutes approximately 50% of total hepatic PEPCK; PEPCK-M appears to play an important role glycerogenesis, amino acid catabolism, insulin secretion, and thermogenesis [29, 50]. No human case of primary PEPCK-M deficiency has yet been confirmed. Although PEPCK-C deficiency was suspected prior to molecular diagnosis in these siblings, the assays of total PEPCK in cells and frozen liver from patient 1 were not informative because of the usual predominance of PEPCK-M in fibroblasts and the unanticipated increase in levels of PEPCK-M in liver (which apparently failed to replace the gluconeogenic role of PEPCK-C). Possible separation of mitochondria and cytosol from fresh liver did not materialize, but was performed in the one previous, well-documented case of functional diagnosis of PEPCK-C deficiency [30]. In retrospect, exome sequencing or sequencing of a panel of gluconeogenic genes may be the initial diagnostic method of choice in cases of suspected defects of gluconeogenesis; it could be more efficient, comprehensive, and more economical than a fresh liver biopsy, which would require on-site separation of mitochondria and cytosol and comprehensive assays of all the gluconeogenic enzymes. Most medical centers would find this difficult to execute, as evident in the lack of reported cases of PEPCK-C deficiency for nearly 30 years. Of course, the diagnostic benefits of exome sequencing in these rather different siblings extend beyond the elucidation of PEPCK-C deficiency.
In summary, the occurrence of three particularly rare disorders in one family is an unlikely event. This case illustrates the larger issue of multiple overlapping heritable diseases combining to produce a constellation of findings that are difficult to diagnose. As an example, we can crudely estimate the frequency of finding a patient with two autosomal recessive diseases. Assume that about one in 200 individuals will have any one of the many known recessive disorders. Then, within the US population of 316 million individuals, 1.6 million will have a single recessive disorder and, by coincidence, 8000 of those will have a second recessive disorder and 40 a third disorder. This estimate does not account for genetic variants that produce mild effects when present by themselves, but have a profound synergy when combined with a second, or third, heritable disease [51]. It is not surprising then, that a program like the UDP, which stringently selects for complicated disorders, would uncover a family with multiple genetic aberrations.
5. CONCLUSION
In the current era, in which multiple, sophisticated methodologies are employed for molecular diagnostics, more and more families with multiple genetic abnormalities are likely to be ascertained. This challenges the tenet of Occam’s razor, i.e., that one explanation should account for all of a patient’s symptoms. In particular, our family illustrates the need to consider additional diagnoses for cases that present in an atypical fashion or are not fully explained by mutation(s) in a single gene.
Supplementary Material
Highlights.
Diseases can be the co-occurrence of multiple disorders in individuals and siblings
Deficiency of PECK-C gives rise to infantile and childhood hypoglycemia
Acknowledgments
This work was supported by the Intramural Research Program of the National Human Genome Research Institute, by grant NS036654 (S.F.T.); by the NIH Undiagnosed Diseases Program (HSN268201300162P, H.Y.); the Center for Inherited Disorders of Energy Metabolism (D.S.K); the Allison Foundation, grant 116491 (R.W.H.); the National Center for Research Resources Grant P20 RR17708 and the Natural Sciences and Engineering Research Council of Canada (T.H.).
Abbreviations
- BSP
3-sulfopyruvate
- CGH
comparative genomic hybridization
- COOT
Crystallographic Object-Oriented Toolkit
- DMEM
Dulbecco’s Modified Eagle Medium
- DTT
dithiothreitol
- ECHO
echocardiogram
- EC50
half maximal effective concentration
- EEG
electroencephalogram
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GTP
guanosine-5′-triphosphate
- GRIN2B/GluN2B
N-methyl-D-aspartic (NMDA) receptor subunit 2B gene/protein
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- KCl
potassium chloride
- KM
the Michaelis constant of an enzyme, the concentration of substrate at half-maximum activity
- μg/dl
micrograms/deciliter
- μM
micromolar
- mM
millimolar
- NaCl
sodium chloride
- NAMD
Nanoscale Molecular Dynamics, a freeware molecular dynamics simulation package
- NMDA
N-methyl D-aspartate
- OAA
Oxaloacetic acid
- OD
optical density
- PAGE SDS
polyacrylamide gel electrophoresis in sodium dodecyl sulfate
- PCK1
phosphoenolpyruvate carboxykinase 1
- PDB
protein data bank
- PEPCK-C
phosphoenolpyruvate carboxykinase cytosolic isoform
- PEPCK-M
mitochondrial isoform
- RAI1
retinoic acid induced 1
- RMSD
root-mean-square-deviation
- SMS
Smith-Magenis Syndrome
- SNP
single nucleotide polymorphism
- WT
wild type
- kcat
rate constant of an enzyme, maximum catalytic activity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guy EL, Peckham DG, Brownlee KG, Conway SP, Lee TW. Cystic fibrosis coexisting with trisomy 21. J Cyst Fibros. 2010;9:330–331. doi: 10.1016/j.jcf.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Marin S, Casano-Sancho P, Villarreal-Pena N, Sebastiani G, Pinillos S, Perez-Duenas B, Hwa V, Rosenfeld RG, Ibanez L. Triple A syndrome in a patient with genetic growth hormone insensitivity: phenotypic effects of two genetic disorders. Horm Res Paediatr. 2012;77:63–68. doi: 10.1159/000335235. [DOI] [PubMed] [Google Scholar]
- 3.Gahl WA, Boerkoel CF, Boehm M. The NIH Undiagnosed Diseases Program: bonding scientists and clinicians. Dis Model Mech. 2012;5:3–5. doi: 10.1242/dmm.009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gahl WA, Markello TC, Toro C, Fajardo KF, Sincan M, Gill F, Carlson-Donohoe H, Gropman A, Pierson TM, Golas G, Wolfe L, Groden C, Godfrey R, Nehrebecky M, Wahl C, Landis DM, Yang S, Madeo A, Mullikin JC, Boerkoel CF, Tifft CJ, Adams D. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med. 2012;14:51–59. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gahl WA, Tifft CJ. The NIH Undiagnosed Diseases Program: lessons learned. JAMA. 2011;305:1904–1905. doi: 10.1001/jama.2011.613. [DOI] [PubMed] [Google Scholar]
- 6.Carr IM, Markham SA, Pena SD. Estimating the degree of identity by descent in consanguineous couples. Hum Mutat. 2011;32:1350–1358. doi: 10.1002/humu.21584. [DOI] [PubMed] [Google Scholar]
- 7.Markello TC, Han T, Carlson-Donohoe H, Ahaghotu C, Harper U, Jones M, Chandrasekharappa S, Anikster Y, Adams DR, Gahl WA, Boerkoel CF NISC Comparative Sequencing Program. Recombination mapping using Boolean logic and high-density SNP genotyping for exome sequence filtering. Mol Genet Metab. 2012;105:382–389. doi: 10.1016/j.ymgme.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams DR, Sincan M, Fuentes Fajardo K, Mullikin JC, Pierson TM, Toro C, Boerkoel CF, Tifft CJ, Gahl WA, Markello TC. Analysis of DNA sequence variants detected by high-throughput sequencing. Hum Mutat. 2012;33:599–608. doi: 10.1002/humu.22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teer JK, Bonnycastle LL, Chines PS, Hansen NF, Aoyama N, Swift AJ, Abaan HO, Albert TJ, Margulies EH, Green ED, Collins FS, Mullikin JC, Biesecker LG NISC Comparative Sequencing Program. Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 2010;20:1420–1431. doi: 10.1101/gr.106716.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Livak K. Comparative Ct method. Applied Biosystems, Inc; Grand Island, NY: 1997. [Google Scholar]
- 11.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 12.Low CM, Zheng F, Lyuboslavsky P, Traynelis SF. Molecular determinants of coordinated proton and zinc inhibition of N-methyl-D-aspartate NR1/NR2A receptors. Proc Natl Acad Sci U S A. 2000;97:11062–11067. doi: 10.1073/pnas.180307497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hedegaard M, Hansen KB, Andersen KT, Brauner-Osborne H, Traynelis SF. Molecular pharmacology of human NMDA receptors. Neurochem Int. 2012;61:601–609. doi: 10.1016/j.neuint.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burger PB, Yuan H, Karakas E, Geballe M, Furukawa H, Liotta DC, Snyder JP, Traynelis SF. Mapping the binding of GluN2B-selective N-methyl-D-aspartate receptor negative allosteric modulators. Mol Pharmacol. 2012;82:344–359. doi: 10.1124/mol.112.078568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atkin BM, Utter MF, Weinberg MB. Pyruvate carboxylase and phosphoenolpyruvate carboxykinase activity in leukocytes and fibroblasts from a patient with pyruvate carboxylase deficiency. Pediatr Res. 1979;13:38–43. doi: 10.1203/00006450-197901000-00009. [DOI] [PubMed] [Google Scholar]
- 18.Kerr DS, Ho L, Berlin CM, Lanoue KF, Towfighi J, Hoppel CL, Lusk MM, Gondek CM, Patel MS. Systemic deficiency of the first component of the pyruvate dehydrogenase complex. Pediatr Res. 1987;22:312–318. doi: 10.1203/00006450-198709000-00015. [DOI] [PubMed] [Google Scholar]
- 19.Hakimi P, Yang J, Casadesus G, Massillon D, Tolentino-Silva F, Nye CK, Cabrera ME, Hagen DR, Utter CB, Baghdy Y, Johnson DH, Wilson DL, Kirwan JP, Kalhan SC, Hanson RW. Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse. J Biol Chem. 2007;282:32844–32855. doi: 10.1074/jbc.M706127200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson TA, Holyoak T. The Omega-loop lid domain of phosphoenolpyruvate carboxykinase is essential for catalytic function. Biochemistry. 2012;51:9547–9559. doi: 10.1021/bi301278t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson TA, Holyoak T. Increasing the conformational entropy of the Omega-loop lid domain in phosphoenolpyruvate carboxykinase impairs catalysis and decreases catalytic fidelity. Biochemistry. 2010;49:5176–5187. doi: 10.1021/bi100399e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bailey S. The Ccp4 Suite - Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 23.Sullivan SM, Holyoak T. Enzymes with lid-gated active sites must operate by an induced fit mechanism instead of conformational selection. Proc Natl Acad Sci U S A. 2008;105:13829–13834. doi: 10.1073/pnas.0805364105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vilboux T, Ciccone C, Blancato JK, Cox GF, Deshpande C, Introne WJ, Gahl WA, Smith AC, Huizing M. Molecular analysis of the Retinoic Acid Induced 1 gene (RAI1) in patients with suspected Smith-Magenis syndrome without the 17p11.2 deletion. PLoS One. 2011;6:e22861. doi: 10.1371/journal.pone.0022861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanson RW. Thematic minireview series: a perspective on the biology of phosphoenolpyruvate carboxykinase 55 years after its discovery. J Biol Chem. 2009;284:27021–27023. doi: 10.1074/jbc.R109.040519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vidnes J, Sovik O. Gluconeogenesis in infancy and childhood. III. Deficiency of the extramitochondrial form of hepatic phosphoenolpyruvate carboxykinase in a case of persistent neonatal hypoglycaemia. Acta Paediatr Scand. 1976;65:307–312. doi: 10.1111/j.1651-2227.1976.tb04890.x. [DOI] [PubMed] [Google Scholar]
- 31.Furukawa H, Singh SK, Mancusso R, Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- 32.Vance KM, Simorowski N, Traynelis SF, Furukawa H. Ligand-specific deactivation time course of GluN1/GluN2D NMDA receptors. Nat Commun. 2011;2:294. doi: 10.1038/ncomms1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tarabeux J, Kebir O, Gauthier J, Hamdan FF, Xiong L, Piton A, Spiegelman D, Henrion E, Millet B, team SD, Fathalli F, Joober R, Rapoport JL, DeLisi LE, Fombonne E, Mottron L, Forget-Dubois N, Boivin M, Michaud JL, Drapeau P, Lafreniere RG, Rouleau GA, Krebs MO. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry. 2011;1:e55. doi: 10.1038/tp.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, del Rosario M, Hoischen A, Scheffer H, de Vries BB, Brunner HG, Veltman JA, Vissers LE. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 35.Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, Milh M, Kortum F, Fritsch A, Pientka FK, Hellenbroich Y, Kalscheuer VM, Kohlhase J, Moog U, Rappold G, Rauch A, Ropers HH, von Spiczak S, Tonnies H, Villeneuve N, Villard L, Zabel B, Zenker M, Laube B, Reis A, Wieczorek D, Van Maldergem L, Kutsche K. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010;42:1021–1026. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- 36.Epi KC, Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu YF, Madou MR, Marson AG, Mefford HC, Esmaeeli Nieh S, O’Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin Thio L, Venkat A, Vining EP, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR P. Epilepsy Phenome/Genome. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freunscht I, Popp B, Blank R, Endele S, Moog U, Petri H, Prott EC, Reis A, Rubo J, Zabel B, Zenker M, Hebebrand J, Wieczorek D. Behavioral phenotype in five individuals with de novo mutations within the GRIN2B gene. Behav Brain Funct. 2013;9:20. doi: 10.1186/1744-9081-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kenny EM, Cormican P, Furlong S, Heron E, Kenny G, Fahey C, Kelleher E, Ennis S, Tropea D, Anney R, Corvin AP, Donohoe G, Gallagher L, Gill M, Morris DW. Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol Psychiatry. 2013 doi: 10.1038/mp.2013.127. [DOI] [PubMed] [Google Scholar]
- 39.Lemke JR, Hendrickx R, Geider K, Laube B, Schwake M, Harvey RJ, James VM, Pepler A, Steiner I, Hortnagel K, Neidhardt J, Ruf S, Wolff M, Bartholdi D, Caraballo R, Platzer K, Suls A, De Jonghe P, Biskup S, Weckhuysen S. GRIN2B mutations in west syndrome and intellectual disability with focal epilepsy. Ann Neurol. 2014;75:147–154. doi: 10.1002/ana.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myers RA, Casals F, Gauthier J, Hamdan FF, Keebler J, Boyko AR, Bustamante CD, Piton AM, Spiegelman D, Henrion E, Zilversmit M, Hussin J, Quinlan J, Yang Y, Lafreniere RG, Griffing AR, Stone EA, Rouleau GA, Awadalla P. A population genetic approach to mapping neurological disorder genes using deep resequencing. PLoS Genet. 2011;7:e1001318. doi: 10.1371/journal.pgen.1001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, Ernst C, Hanscom C, Rossin E, Lindgren AM, Pereira S, Ruderfer D, Kirby A, Ripke S, Harris DJ, Lee JH, Ha K, Kim HG, Solomon BD, Gropman AL, Lucente D, Sims K, Ohsumi TK, Borowsky ML, Loranger S, Quade B, Lage K, Miles J, Wu BL, Shen Y, Neale B, Shaffer LG, Daly MJ, Morton CC, Gusella JF. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Constantine-Paton M. Effects of NMDA receptor antagonists on the developing brain. Psychopharmacol Bull. 1994;30:561–565. [PubMed] [Google Scholar]
- 44.Feldmeyer D, Cull-Candy S. Functional consequences of changes in NMDA receptor subunit expression during development. J Neurocytol. 1996;25:857–867. doi: 10.1007/BF02284847. [DOI] [PubMed] [Google Scholar]
- 45.Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron. 1996;16:333–344. doi: 10.1016/s0896-6273(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 46.Loftis JM, Janowsky A. The N-methyl-D-aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol Ther. 2003;97:55–85. doi: 10.1016/s0163-7258(02)00302-9. [DOI] [PubMed] [Google Scholar]
- 47.Ozawa S, Kamiya H, Tsuzuki K. Glutamate receptors in the mammalian central nervous system. Progress in Neurobiology. 1998;54:581–618. doi: 10.1016/s0301-0082(97)00085-3. [DOI] [PubMed] [Google Scholar]
- 48.Vallano ML. Developmental aspects of NMDA receptor function. Crit Rev Neurobiol. 1998;12:177–204. doi: 10.1615/critrevneurobiol.v12.i3.20. [DOI] [PubMed] [Google Scholar]
- 49.Williams K, Russell SL, Shen YM, Molinoff PB. Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron. 1993;10:267–278. doi: 10.1016/0896-6273(93)90317-k. [DOI] [PubMed] [Google Scholar]
- 50.Yang J, Kalhan SC, Hanson RW. What is the metabolic role of phosphoenolpyruvate carboxykinase? J Biol Chem. 2009;284:27025–27029. doi: 10.1074/jbc.R109.040543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol. 2012;87:795–803. doi: 10.1002/ajh.23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.