

Abstract
Aims
Liver fibrosis occurs as a result of several chronic liver diseases and leads to portal hypertension, cirrhosis and liver failure, often requiring liver transplantation. Activated hepatic stellate cells (HSC) are known to contribute to liver fibrosis, but currently there are no effective therapies for the treatment of established liver fibrosis. Activation of the acidic sphingomyelinase (ASM) has been shown to be involved in HSC activation. In the present study we investigated whether treatment with the ASM inhibitor, amitriptyline (TCA), could prevent and/or reverse fibrosis induced in mice by carbon tetrachloride (CCl4).
Methods
Mice were treated with CCl4 for 8 weeks to induce fibrosis. Concurrently, mice received drinking water with or without 180 mg/L TCA.
Results
Mice receiving TCA in the water had decreased hepatic collagen deposition and reduced liver mRNA expression of the fibrogenic mediators, TGF-β1, TIMP-1, collagen, and TNFα. TCA treatment also reduced HSC activation determined by α-smooth muscle actin staining. In a separate set of experiments, mice were treated with CCl4 for 5 weeks prior to treatment with TCA, to test whether TCA had any effect on established fibrosis. Remarkably, in mice with established fibrosis, treatment with TCA significantly reduced collagen deposition, HSC activation, and prevented portal hypertension and improved hepatic architecture. Treatment of isolated HSC in vitro with TCA completely inhibited TGF-β1-induced collagen expression and PDGF-BB-induced proliferation.
Conclusions
The data suggest that ASM is a critical signaling component in HSC for the development of liver fibrosis and represents an important therapeutic target.
Keywords: liver fibrosis, non-alcoholic steatohepatitis, stellate cells
Introduction
Hepatic fibrosis may result from a variety of different etiologies including viral hepatitis, cholestatic hepatitis, alcoholic steatohepatitis and non-alcoholic steatohepatitis. In many patients, hepatic fibrosis will progress to cirrhosis and end stage liver disease, for which liver transplantation is the only life-saving therapy.1, 2 However, a significant gap exists between the number of liver transplantation candidates and the availability of organs3, 4 Therefore, developing strategies to limit the development and progression of hepatic fibrosis are of great clinical interest.
It is established that perisinusoidal hepatic stellate cells (HSCs) are the primary cell type responsible of liver fibrosis.5 The retinoid-storing HSCs are quiescent physiologically, but become progressively activated during liver injury. The activation process is characterized by their transformation into a proliferating, contractile and fibrogenic myofibroblast-like phenotype. Thus, targeting activated HSCs as a therapeutic approach to control or reverse hepatic fibrosis has been intensely investigated.6, 7 Unfortunately, despite these efforts, there are no effective therapies for the treatment of liver fibrosis at present.
Ceramide is a sphingolipid which has been previously implicated in the development and progression of liver disease. Ceramide is produced by hydrolysis of sphingomyelin catalyzed by acidic sphingomyelinase (ASM). ASM is a lysosomal glycoprotein, which translocates to the cell surface and plays an important role in cellular stress responses.8 High concentrations of ceramide in the cellular membranes serve to form ceramide-enriched domains, which regroup receptors and signaling molecules.9 This reorganization of the plasma membrane facilitates and amplifies various signaling processes related to cellular responses to injury, which include cell migration, survival, contraction, proliferation, gene expression and cell-to-cell interaction.10–13
ASM activation and resulting ceramide production have been implicated in a variety of human diseases including atherosclerosis, emphysematous lung disease, neurodegenerative disease, insulin resistance and diabetes mellitus, as well as non-alcoholic steatohepatitis.14–16 As such, the inhibition of ASM is appealing given the many potential therapeutic implications. One ASM inhibitor, amitriptyline, is a tricyclic antidepressant that causes detachment of ASM from the inner lysosomal membrane, resulting in subsequent inactivation by lysosomal proteases.8, 17 Amitriptyline treatment has been used to influence ceramide producing pathways in both murine in vitro and in vivo studies, as well as in phase II clinical trials for cystic fibrosis.18–20
In the present study, we sought to determine whether therapeutic inhibition of ASM with amitriptyline would reduce hepatic fibrosis induced by carbon tetrachloride (CCl4). Previous studies have demonstrated that ASM heterozygous mice are protected from developing hepatic fibrosis in cholestatic and CCl4 models of liver disease in association with reduced activation of HSCs 19, and other studies have shown that treatment with amitriptyline reduces liver pathology in a rat model of Wilson’s Disease.21 However, we tested the ability of amitriptyline to treat established liver fibrosis. Secondly, we investigated the mechanism by which ASM inhibition with amitriptyline in HSCs controls their fibrogenic activity.
Methods
Model of Liver Fibrosis
Male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were used in all experiments. The University of Cincinnati Animal Care and Use Committee approved all animal care and procedures performed. To induce fibrosis, mice were treated twice weekly with intraperitoneal injections of 1 mL/kg CCl4 (Sigma-Aldrich, St. Louis, MO) diluted in three volumes of mineral oil.22 Age and sex matched control animals were treated twice weekly with similar volumes of mineral oil injected intraperitoneally. To pharmacologically inhibit ASM, mice were provided 180 mg/L of amitriptyline (Sigma) in their drinking water, which was changed every 3 days. This regimen has been shown to effectively inhibit ASM in vivo.18, 23, 24 In one set of experiments, mice were given amitriptyline in the water two weeks prior to treatment with CCl4 to ensure that ASM was inhibited prior to induction of fibrosis. Mice were sacrificed 2, 4, 6 and 8 weeks after treatment with CCl4 with or without amitriptyline. In a second set of experiments, mice received amitriptyline in their water beginning 5 weeks after initiation of CCl4 treatment to determine whether ASM inhibition could arrest and reverse established fibrosis. These mice were sacrificed 9, 12 and 15 weeks after initiation of CCl4 treatment.
Blood and Tissue Analysis
Blood was obtained via cardiac puncture for analysis. Liver tissues were fixed in 10% neutral-buffered formalin (Richard Allen Scientific, Kalamazoo, MI), processed and then embedded in paraffin for light microscopy. Serial sections were stained with hematoxylin and eosin for histological examination and sirius red for determination of collagen deposition. Activated HSCs were identified by immunohistochemical staining of alpha-smooth muscle actin (α-SMA, Abcam, Cambridge, MA). Sections were examined by a blinded hepatopathologist and hepatic fibrosis was assessed using the criteria described by Kleiner et al.25
Isolation and Culture of Hepatic Stellate Cells
Murine HSCs were isolated from male C57BL/6 mice, as previously described.26, 27 Livers were perfused and digested in situ with Hank’s balanced salt solution containing collagenase and protease. The liver was excised, minced and strained through a steel mesh sieve. The resulting cell suspension was centrifuged 3 to 4 times at 65 g for 2 minutes to remove hepatocytes and cellular debris. The supernatant was then centrifuged at 650 g for 10 minutes and hepatic stellate cells were clarified from the resulting pellet via Nycodenz gradient centrifugation. HSCs were suspended in Dulbecco’s Modified Eagle Medium containing 10% FBS, 10% horse serum, 100 mg/mL penicillin and 100 U/mL streptomycin (Invitrogen, Carlsbad, CA). Cells were seeded at a density of 1.75 × 106 cells/mL. Media was renewed on the following day and on alternating days thereafter. Cells were passaged after becoming approximately 70% confluent and were used during their third and fourth passage. Passaged cells were treated with or without 25 μM of amitriptyline in reduced serum media (2% FBS) for 48 hours to inhibit ASM.19 Cells where then treated with 5 nM of transforming growth factor-beta (TGF-β1, R&D Systems, Minneapolis, MN) or 50 nM platelet derived growth factor-beta, beta (PDGF-BB, Peprotech, Rocky Hill, NJ) with or without the presence of amitriptyline for 24 hours. Genetic expression of collagen was assessed by real-time polymerase chain reaction (RT-PCR) and cellular proliferation was assessed by bromodeoxyuridine incorporation.
RT-PCR Analysis
Total RNA was extracted from liver tissue and HSCs using Trizol (Invitrogen, Carlsbad, CA). cDNA was generated through the reverse-transcription of 2 μg of RNA using random primers and Superscript RNase H-reverse transcriptase (Invitrogen). Samples were incubated at 20°C for 10 minutes, 42°C for 30 minutes, 99°C for 5 minutes to inactive the reverse transcriptase and then cooled at 5°C for 5 minutes. The amount of mRNA was quantified by real-time reverse transcriptase polymerase chain reaction per the manufacturer’s specifications (Eppendorf, Mastercycler Real-Time PCR). Four microliters of diluted cDNA samples (1:10 dilution) were used for quantitative two-step PCR using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). Each sample was analyzed in duplicate. Beta-actin (β-actin) was used as a housekeeping control gene. Threshold cycles were automatically calculated by the iCycler iQ Real-Time Detection System. Threshold cycle values were normalized to the housekeeping control (β-actin) to give relative genomic equivalence. Primers utilized are listed in the Table 1.
Table 1.
Primers used for RT-PCR.
| Gene | Sequence | |
|---|---|---|
| Collagen 1 α1 | Sense Antisense |
GAGCGGAGAGTACTGGATCG GCTTCTTTTCCTTGGGGTTC |
| α-SMA | Sense Antisense |
GTCCCAGACATCAGGGAGTAA TCGGATACTTCAGCGTCAGGA |
| TIMP-1 | Sense Antisense |
CCTTGCAAACTGGAGAGTGACA AAGCAAAGTGACGGCTCTGGT |
| TGF-β1 | Sense Antisense |
TTGCCCTCTACAACCACAA GGCTTGCGACCCACGTAGTA |
| TNFα | Sense Antisense |
TCGTAGCAAACCACCAAGTG AGATAGCAAATCGGCTGACG |
| GAPDH | Sense Antisense |
TGTTGAAGTCACAGGAGACAACCT AACCTGCCAAGTATGATGACATCA |
Cellular Proliferation
DNA synthesis was estimated using bromodeoxyuridine (BrdU) incorporation and enzyme-linked immunosorbent assay kit (Abcam). Passaged cells were incubated with BrdU for 4 hours. BrdU uptake was then measured according to standard enzyme-linked immunosorbent assay protocol per the manufactures’ instructions.
ASM Activity
ASM activity was measured by incubating immunoprecipitated ASM with [14C]sphingomyelin and quantifying the release of [14C]choline as described.28 The liver was removed, snap-frozen in liquid nitrogen, and lysed in 250 mM sodium acetate (pH 5.0), 1% NP40, and 1.3 mM EDTA for 15 min. The tissues were then homogenized with a tip sonicator. Aliquots of the lysates were diluted to 250 mM sodium acetate (pH 5.0), 0.1% NP40, and 1.3 mM EDTA and incubated with 50 nCi per sample [14C]sphingomyelin for 30 min at 37 °C. The substrate was dried prior to the assay, resuspended in 250 mM sodium acetate (pH 5.0), 0.1% NP40, and 1.3 mM EDTA and bath-sonicated for 10 min to obtain micelles. The enzyme reaction was terminated by the addition of 800 μL chloroform/methanol (2:1, v/v), phases were separated by centrifugation and radioactivity of the aqueous phase was measured by using liquid scintillation counting to determine the release of [14C]phosphorylcholine from [14C]sphingomyelin as a measure of ASM activity.
Statistical Analysis
All data are expressed as mean ± standard error of the mean (SEM). Data were analyzed with a one-way analysis of variance with subsequent Student Newman Keuls test. Differences were considered significant when p < 0.05.
Results
Inhibition of ASM reduces hepatic fibrogenesis by suppressing activation of HSCs
Using a murine model of liver fibrosis induced by CCl4, animals received amitriptyline in their drinking water to inhibit ASM throughout an 8-week course of CCl4 administration. Hepatic ASM activity was determined after 8 weeks of treatment. While vehicle- and CCl4-treated animals had similar levels of liver ASM activity, amitriptyline treatment significantly decreased ASM activity compared to both vehicle and CCl4-treated animals (Figure 1). Over the course of 8 weeks of treatment with CCl4, mice developed increasing hepatic fibrosis, as determined by collagen deposition using sirius red staining as well as pathological scoring (Figure 2). However, inhibition of ASM with amitriptyline significantly reduced the degree of fibrosis (Figure 2). To investigate if inhibition of ASM with amitriptyline decreased HSC activation as a mechanism for the reduced hepatic fibrosis observed, we performed RT-PCR on liver samples prior to induction of fulminant hepatic fibrosis. Liver samples were obtained after two weeks of treatment. As shown in Figure 3A, inhibition of ASM significantly decreased CCl4-induced expression of TGF-β1, TIMP-1, collagen, and TNFα, all markers associated with HSC activation and liver fibrosis (Figure 3A). This effect persisted throughout the duration of treatment (data not shown). Supporting these findings, immunohistochemical staining for α-SMA revealed decreased activation of HSCs in animals treated with amitriptyline at all time points relative to animals treated with CCl4 alone (Figure 3B).
Figure 1.
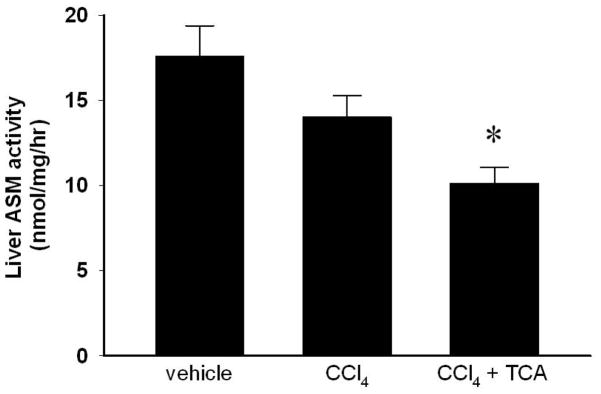
Amitriptyline (TCA) treatment decreases hepatic acidic sphingomyelinase (ASM) activity. Hepatic ASM activity was determined after 8 weeks of treatment. TCA treatment decreased ASM activity compared to vehicle and CCl4 treatment. n=4–6 per group. Data are expressed as mean and SEM. *p<0.05 compared to all other groups.
Figure 2.

Inhibition of ASM activity limits CCl4-induced hepatic fibrosis. (A) Sirius red staining of liver sections (magnification 50X) reveals increased collagen deposition over time in animals treated with CCl4 compared to animals treated with CCl4 and amitriptyline (TCA).
(B) Quantitation of hepatic fibrosis using pathological scoring. Decreased fibrosis in animals treated with CCl4 and TCA vs. CCl4 alone was confirmed via quantification of liver sections by a blinded pathologist. n=4–8 per group. Data are expressed as mean and SEM. *p<0.05 compared to all other groups. #p<0.05 compared to CCl4 group.
Figure 3.

Inhibition of ASM activity reduces hepatic stellate cell activity in vivo. (A) RT-PCR analysis of whole liver tissue after two weeks of treatment with CCl4 demonstrated that treatment with amitriptyline (TCA) decreased levels of transforming growth factor beta one (TGF-β1), tissue inhibitor of metalloprotease one (TIMP-1), collagen and tumor necrosis factor alpha (TNF-α) mRNA compared to CCl4 alone. n=6–8 per group. Data are expressed as mean and SEM. *p<0.05 compared to all other groups. (B) Hepatic stellate cell staining of α-SMA. Immunohistochemical staining of liver tissues for α-SMA reveals decreased activated HSC burden at each time point in animals treated with CCl4 and TCA relative to both vehicle and CCl4 treatment alone (magnification x 50). Data are representative of 4 separate histological analyses per group/time point. Original magnification was 50X.
Inhibition of ASM reduces established hepatic fibrosis and portal hypertension
In order to determine if inhibition of ASM could serve as a potential therapeutic modality for those with established hepatic fibrosis, mice were subjected to 5 weeks of CCl4 treatment to induce hepatic fibrosis, with some mice received amitriptyline in their drinking water along with continued CCl4 treatment until week 15. Mice treated with CCl4 alone developed increasing amounts of bridging fibrosis throughout the time course of treatment (Figure 4). However, mice treated with amitriptyline showed resolution of fibrosis at weeks 9 and 12 despite the continued profibrogenic stimulus. Interestingly, by 15 weeks, mice treated with amitriptyline did develop bridging fibrosis (Figure 4). However, liver architecture in these mice was preserved compared to those mice receiving only CCl4, which showed increased central fibrosis and hepatocellular loss (Figure 4). Immunohistochemical staining for α-SMA revealed decreased HSC activation in the mice treated with amitriptyline (Figure 5A). Furthermore, by week 15, CCl4-induced liver fibrosis was associated with splenomegaly, a putative marker of portal hypertension in the context of liver fibrosis 29, 30 (Figure 5B). Treatment with amitriptyline, abrogated this pathology (Figure 5B).
Figure 4.

Delayed treatment with amitriptyline (TCA) reduces CCl4-induced hepatic fibrosis. Sirius red staining of liver sections reveals progressive bridging fibrosis in animals treated with CCl4. In contrast, animals treated with CCl4 + TCA after establishment of fibrosis (TCA started after 5 weeks of CCl4 treatment) had significantly reduced hepatic fibrosis and slowed progression. (original magnification 50X) Quantitation of hepatic fibrosis using pathological scoring. Decreased fibrosis in animals treated with CCl4 and TCA in a delayed fashion vs. CCl4 alone was confirmed via quantification of liver sections by a blinded hepatopathologist. n=4–6 per group. Data are expressed as mean and SEM. *p<0.05 compared to all other groups, #p<0.05 compared to CCl4 alone group.
Figure 5.

Delayed treatment with amitriptyline reduces activated hepatic stellate cells and reduces portal hypertension after established hepatic fibrosis. (A) Representative samples of α-SMA immunohistochemical staining (magnification 50X) reveals decreased activated HSC burden by week 15 in animals that treated with amitriptyline (TCA) in a delayed fashion (after 5 weeks of treatment with CCl4) vs. animals treated with CCl4 alone. (B) Animals treated with CCl4 alone had more significant splenomegaly, reflective of their progressive liver disease, relative to the animals treated with CCl4 and TCA in a delayed fashion. n=4–6 per group. Data are expressed as mean and SEM. *p<0.05 compared to CCl4 alone group.
Inhibition of ASM prevents PDGF-BB and TGF-β1 stimulation of hepatic stellate cells
Because our in vivo data suggest that the beneficial effects of ASM inhibition are related to decreased HSC activation, we next evaluated the direct effects of amitriptyline on activated (passaged) HSCs in vitro. Isolated HSCs were passaged 3–4 times to establish a fully activated phenotype and then were stimulated in vitro with PDGF-BB or TGF-β1. Treatment with amitriptyline (25 μM) decreased collagen and alpha-smooth muscle actin (α-SMA) expression nearly 4-fold (Figure 6A). Furthermore, stimulation of HSCs with exogenous PDGF-BB (50 nM) or TGF-β1 (5 nM), could not overcome the inhibitory effects of amitriptyline. An even greater effect of amitriptyline was observed on PDGF-BB-induced proliferation. Treatment of HSCs with PDGF-BB (50 nM) resulted in an 8-fold increase in proliferation (Figure 6B). However, treatment with amitriptyline completely abrogated PDGF-BB-induced proliferation (Figure 6B).
Figure 6.

Amitriptyline (TCA) inhibits collagen expression and proliferation of activated hepatic stellate cells in vitro. (A) Treatment with 25μM TCA caused a marked reduction in collagen and alpha-smooth muscle actin (α-SMA) expression by activated HSCs, which was maintained despite stimulation with 5nM of transforming growth factor beta type one (TGF-β1) or 50 nM platelet-derived growth factor-beta, beta (PDGF-BB). *p<0.05 compared to untreated, TGF-β1, PDGF-BB alone groups. (B) Treatment with 25μM TCA caused a marked reduction in proliferation by activated HSCs as measured by BrdU incorporation. Stimulation of HSCs with 50nM of PDGF-BB markedly increased proliferation. Concomitant treatment with TCA completely abrogated this effect. Data are expressed as mean and SEM. *p<0.05 compared to untreated PDGF-BB alone groups; **p<0.05 compared to all other groups.
Discussion
In this study, we examined the impact of the pharmacologic inhibition of ASM with amitriptyline in a murine model of CCl4 induced hepatic fibrosis. In this model, we found that treatment with amitriptyline decreases hepatic ASM activity and limits CCl4 induced hepatic fibrosis. When given in a therapeutic fashion, i.e., in the setting of established fibrosis and activated HSCs, amitriptyline impaired the progression of hepatic fibrosis and abrogated the development of portal hypertension. Additionally, we found in our in vitro experiments that inhibition of ASM in HSCs significantly reduces collagen production and abrogates cell proliferation. Our data provide new information regarding the therapeutic utility of targeting ASM in reducing existing liver fibrosis and also provides new mechanistic details regarding the role of ASM in HSC activation and proliferation.
ASM has previously been implicated in both liver fibrogenesis and HSC activation. One study, using heterozygous ASM mice, showed that reduced ASM activity was protective in both cholestatic and CCl4 models of hepatic fibrosis through the regulation of HSC activation and proliferation via cathepsin B and D processing.19 However, this work did not determine if targeting ASM after established fibrosis could be an effective therapeutic strategy. Furthermore, the study focused on the function of ASM in regulating the expression of cathepsin B and the role of this mediator in the activation of HSCs. In contrast, our study demonstrates, for the first time, that therapeutic inhibition of ASM can reverse established liver fibrosis and prevent portal hypertension, and that these effects are likely linked to disruption of signaling events in HSCs that are essential for pro-fibrotic and cell proliferative signaling. Our data provide strong evidence that inhibition of ASM in HSCs results in reduced TGF-β1- and PGDF-ββ-mediated effects.
Another study showed, in a rat model of Wilson’s disease, that inhibition of ASM with daily intraperitoneal injections of amitriptyline, limited the development of hepatocyte injury and liver fibrosis.21 However, this study did not investigate the mechanisms by which ASM inhibition reduced liver fibrosis. In this regard, the study was limited in scope and examined ASM inhibition only in a prophylactic setting.
PDGF-BB is the most potent mitogen of HSCs and PDGF receptor-beta subunit (PDGFR-β) is required for PDGF induced proliferation of HSCs.31, 32 In vivo genetic silencing of PDGFR-β with a HSC-specific promoter demonstrated reduced liver injury and hepatic fibrosis in both a bile duct ligation and CCl4 model of hepatic fibrosis, suggestive of HSC-specific RNA interference as a treatment of hepatic fibrogenesis.32 Here we propose that inhibition of the ASM/ceramide axis with amitriptyline may represent an alternative, and more plausible, treatment with a similar mechanism of action.
It is plausible that inhibition of ASM by amitriptyline alters PDGFR-β availability. Conversion of sphingomyelin to ceramide by ASM occurs within discrete plasma membrane microdomains enrichined in sphingolipids and cholesterol termed caveolae.33, 34 These localized high levels of ceramide help to form a more stabilized lipid matrix, regrouping receptors and signaling molecules, providing ordered support for receptor-mediated signaling events.9, 35 PDGFR-β has been localized within caveolae and PDGFR-β cofractionates with ASM from these ceramide enriched microdomains.35, 36 More importantly, these microdomains have been shown to be the major site of PDGF stimulated signal transduction.36 Our in vitro studies demonstrate that the inhibition of ASM with amitriptyline limits activated HSC proliferation, even in the presence of PDGF-BB stimulation. Inhibition of ASM may result in decreased stability of plasma membrane microdomains through the reduction in ceramide levels with ensuing loss of support for caveolae based receptor mediated signaling, namely through the PDGFR-β.
Likewise, a similar argument could be made for TGF-β1 receptor, which also has been found to colocalize and cofractionate with caveolae.37 We note that exogenously added TGF-β1 did not stimulate collagen synthesis over the basal value in activated HSCs indicating that the system is already fully activated. Indeed, constitutively increased nuclear Smad3/Smad4, which is coupled to TGF-β1-induced collagen production by activated HSCs 38, was observed in a cell line developed from HSCs from the cirrhotic liver, and exogenous treatment with TGF-β1 did not affect the increased collagen expression in them.39 Thus, a strong amilriptyline-induced inhibition of collagen synthesis by HSCs both in the absence and presence of exogenous TGF-β1 suggests that the inhibitor may interfere with constitutively activated intracellular signaling responsible for collagen synthesis.
In summary, the current study demonstrates that inhibition of ASM can limit the development of liver fibrosis and more importantly can reverse established liver fibrosis. These beneficial effects are mediated by inhibition of PDGF-BB and TGF-β1 signaling, possibly via decreased ceramide in caveolin-enriched domains in HSCs resulting in dissociation of PDGF-β receptors from these micrordomains or by interfering with the signaling coupled to profibrogenic activity. These findings suggest that the inhibition of ASM may be an effective therapeutic modality to improve liver fibrosis.
Acknowledgments
This work was supported in part by National Institutes Health grants DK56029 and AG025881 (A.B.L.) and AA020846 (C.R.G.) and VA Merit Review grant BX001174 (C.R.G.).
References
- 1.Bataller R, Brenner DA. Liver fibrosis. The Journal of clinical investigation. 2005 Feb;115:209–18. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merion RM, Schaubel DE, Dykstra DM, Freeman RB, Port FK, Wolfe RA. The survival benefit of liver transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2005 Feb;5:307–13. doi: 10.1111/j.1600-6143.2004.00703.x. [DOI] [PubMed] [Google Scholar]
- 3.Thuluvath PJ, Guidinger MK, Fung JJ, Johnson LB, Rayhill SC, Pelletier SJ. Liver transplantation in the United States, 1999–2008. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2010 Apr;10:1003–19. doi: 10.1111/j.1600-6143.2010.03037.x. [DOI] [PubMed] [Google Scholar]
- 4.Kim WR, Stock PG, Smith JM, et al. OPTN/SRTR 2011 Annual Data Report: liver. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2013 Jan;13( Suppl 1):73–102. doi: 10.1111/ajt.12021. [DOI] [PubMed] [Google Scholar]
- 5.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annual review of pathology. 2011;6:425–56. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 6.Ghiassi-Nejad Z, Friedman SL. Advances in antifibrotic therapy. Expert review of gastroenterology & hepatology. 2008 Dec;2:803–16. doi: 10.1586/17474124.2.6.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X, Xu J, Brenner DA, Kisseleva T. Reversibility of Liver Fibrosis and Inactivation of Fibrogenic Myofibroblasts. Current pathobiology reports. 2013 Sep;1:209–14. doi: 10.1007/s40139-013-0018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kornhuber J, Tripal P, Reichel M, et al. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell Physiol Biochem. 2010;26:9–20. doi: 10.1159/000315101. [DOI] [PubMed] [Google Scholar]
- 9.Bollinger CR, Teichgraber V, Gulbins E. Ceramide-enriched membrane domains. Biochim Biophys Acta. 2005 Dec 30;1746:284–94. doi: 10.1016/j.bbamcr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Shea BS, Tager AM. Sphingolipid regulation of tissue fibrosis. The open rheumatology journal. 2012;6:123–9. doi: 10.2174/1874312901206010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008 Feb;9:139–50. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 12.Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annual review of biochemistry. 2004;73:321–54. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 13.Rivera R, Chun J. Biological effects of lysophospholipids. Reviews of physiology, biochemistry and pharmacology. 2008;160:25–46. doi: 10.1007/112_0507. [DOI] [PubMed] [Google Scholar]
- 14.Pagadala M, Kasumov T, McCullough AJ, Zein NN, Kirwan JP. Role of ceramides in nonalcoholic fatty liver disease. Trends in endocrinology and metabolism: TEM. 2012 Aug;23:365–71. doi: 10.1016/j.tem.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith EL, Schuchman EH. The unexpected role of acid sphingomyelinase in cell death and the pathophysiology of common diseases. FASEB J. 2008 Oct;22:3419–31. doi: 10.1096/fj.08-108043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henry B, Moller C, Dimanche-Boitrel MT, Gulbins E, Becker KA. Targeting the ceramide system in cancer. Cancer Lett. 2013 May 28;332:286–94. doi: 10.1016/j.canlet.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 17.Kornhuber J, Tripal P, Gulbins E, Muehlbacher M. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs) Handbook of experimental pharmacology. 2013;215:169–86. doi: 10.1007/978-3-7091-1368-4_9. [DOI] [PubMed] [Google Scholar]
- 18.Becker KA, Tummler B, Gulbins E, Grassme H. Accumulation of ceramide in the trachea and intestine of cystic fibrosis mice causes inflammation and cell death. Biochem Biophys Res Commun. 2010 Dec 17;403:368–74. doi: 10.1016/j.bbrc.2010.11.038. [DOI] [PubMed] [Google Scholar]
- 19.Moles A, Tarrats N, Morales A, et al. Acidic sphingomyelinase controls hepatic stellate cell activation and in vivo liver fibrogenesis. Am J Pathol. 2010 Sep;177:1214–24. doi: 10.2353/ajpath.2010.091257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nahrlich L, Mainz JG, Adams C, et al. Therapy of CF-Patients with Amitriptyline and Placebo - a Randomised, Double-Blind, Placebo-Controlled Phase IIb Multicenter, Cohort-Study. Cell Physiol Biochem. 2013;31:505–12. doi: 10.1159/000350071. [DOI] [PubMed] [Google Scholar]
- 21.Lang PA, Schenck M, Nicolay JP, et al. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nature medicine. 2007 Feb;13:164–70. doi: 10.1038/nm1539. [DOI] [PubMed] [Google Scholar]
- 22.Domenicali M, Caraceni P, Giannone F, et al. A novel model of CCl4-induced cirrhosis with ascites in the mouse. J Hepatol. 2009 Dec;51:991–9. doi: 10.1016/j.jhep.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Teichgraber V, Ulrich M, Endlich N, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med. 2008 Apr;14:382–91. doi: 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- 24.Brand V, Koka S, Lang C, et al. Influence of amitriptyline on eryptosis, parasitemia and survival of Plasmodium berghei-infected mice. Cell Physiol Biochem. 2008;22:405–12. doi: 10.1159/000185482. [DOI] [PubMed] [Google Scholar]
- 25.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005 Jun;41:1313–21. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 26.Sumpter TL, Dangi A, Matta BM, et al. Hepatic stellate cells undermine the allostimulatory function of liver myeloid dendritic cells via STAT3-dependent induction of IDO. J Immunol. 2012 Oct 15;189:3848–58. doi: 10.4049/jimmunol.1200819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dangi A, Sumpter TL, Kimura S, et al. Selective expansion of allogeneic regulatory T cells by hepatic stellate cells: role of endotoxin and implications for allograft tolerance. J Immunol. 2012 Apr 15;188:3667–77. doi: 10.4049/jimmunol.1102460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ziobro R, Henry B, Edwards MJ, Lentsch AB, Gulbins E. Ceramide mediates lung fibrosis in cystic fibrosis. Biochem Biophys Res Commun. 2013 Mar 21; doi: 10.1016/j.bbrc.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Meijer VE, Sverdlov DY, Popov Y, et al. Broad-spectrum matrix metalloproteinase inhibition curbs inflammation and liver injury but aggravates experimental liver fibrosis in mice. PLoS One. 2010;5:e11256. doi: 10.1371/journal.pone.0011256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dumont AE, Amorosi E, Stahl WM. Significance of splenomegaly in patients with hepatic cirrhosis and bleeding esophageal varices. Ann Surg. 1970 Apr;171:522–6. doi: 10.1097/00000658-197004000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kinnman N, Goria O, Wendum D, et al. Hepatic stellate cell proliferation is an early platelet-derived growth factor-mediated cellular event in rat cholestatic liver injury. Laboratory investigation; a journal of technical methods and pathology. 2001 Dec;81:1709–16. doi: 10.1038/labinvest.3780384. [DOI] [PubMed] [Google Scholar]
- 32.Chen SW, Chen YX, Zhang XR, Qian H, Chen WZ, Xie WF. Targeted inhibition of platelet-derived growth factor receptor-beta subunit in hepatic stellate cells ameliorates hepatic fibrosis in rats. Gene therapy. 2008 Nov;15:1424–35. doi: 10.1038/gt.2008.93. [DOI] [PubMed] [Google Scholar]
- 33.Liu P, Anderson RG. Compartmentalized production of ceramide at the cell surface. The Journal of biological chemistry. 1995 Nov 10;270:27179–85. doi: 10.1074/jbc.270.45.27179. [DOI] [PubMed] [Google Scholar]
- 34.Shaul PW, Anderson RG. Role of plasmalemmal caveolae in signal transduction. Am J Physiol. 1998 Nov;275:L843–51. doi: 10.1152/ajplung.1998.275.5.L843. [DOI] [PubMed] [Google Scholar]
- 35.Zundel W, Swiersz LM, Giaccia A. Caveolin 1-mediated regulation of receptor tyrosine kinase-associated phosphatidylinositol 3-kinase activity by ceramide. Mol Cell Biol. 2000 Mar;20:1507–14. doi: 10.1128/mcb.20.5.1507-1514.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu P, Ying Y, Ko YG, Anderson RG. Localization of platelet-derived growth factor-stimulated phosphorylation cascade to caveolae. The Journal of biological chemistry. 1996 Apr 26;271:10299–303. doi: 10.1074/jbc.271.17.10299. [DOI] [PubMed] [Google Scholar]
- 37.Razani B, Zhang XL, Bitzer M, von Gersdorff G, Bottinger EP, Lisanti MP. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. The Journal of biological chemistry. 2001 Mar 2;276:6727–38. doi: 10.1074/jbc.M008340200. [DOI] [PubMed] [Google Scholar]
- 38.Furukawa F, Matsuzaki K, Mori S, et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003 Oct;38:879–89. doi: 10.1053/jhep.2003.50384. [DOI] [PubMed] [Google Scholar]
- 39.Inagaki Y, Mamura M, Kanamaru Y, et al. Constitutive phosphorylation and nuclear localization of Smad3 are correlated with increased collagen gene transcription in activated hepatic stellate cells. J Cell Physiol. 2001 Apr;187:117–23. doi: 10.1002/1097-4652(2001)9999:9999<00::AID-JCP1059>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]