

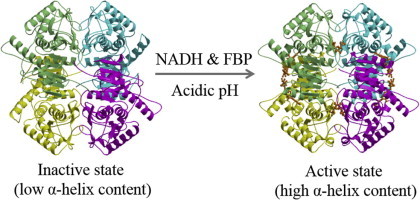
Graphical abstract
Abbreviations: B., Bacillus; Bf., Bifidobacterium; CD, circular dichroism; FBP, fluctose-1,6-bisphosphate; Ec., Enterococcus; E., Escherichia; LDH, lactate dehydrogenase; LAB, lactic acid bacteria; Lb., Lactobacillus; Lc., Lactococcus; rms, root mean square; VUV, vacuum-ultraviolet
Keywords: Allosteric regulation, Crystal structure, Circular dichroism, l-Lactate dehydrogenase, Lactic acid bacteria
Highlights
-
•
Enterococcus mundtii 15-1A harbors two kinds of l-lactate dehydrogenase (l-LDH).
-
•
One of the l-LDHs, named LDH-2, displays a maximal catalytic activity at pH 5.5.
-
•
To activate LDH-2 at pH 7.5, a high concentration of FBP is necessary.
-
•
LDH-2 takes a compact inactive form distinctive from that of other bacterial l-LDHs.
-
•
Difficulty in forming an α-helix is related to the regulation of the LDH-2 activity.
Abstract
A plant-derived Enterococcus mundtii 15-1A, that has been previously isolated from Brassica rapa L. subsp. nipposinica (L.H. Bailey) Hanelt var. linearifolia by our group, possesses two kinds of l-lactate dehydrogenase (l-LDH): LDH-1 and LDH-2. LDH-1 was activated under low concentration of fluctose-1,6-bisphosphate (FBP) at both pH 5.5 and 7.5. Although LDH-2 was also activated under the low concentration of FBP at pH 5.5, a high concentration of FBP is necessary to activate it at pH 7.5. The present study shows the crystal structures of the acidophilic LDH-2 in a complex with and without FBP and NADH. Although the tertiary structure of the ligands-bound LDH-2 is similar to that of the active form of other bacterial l-LDHs, the structure without the ligands is different from that of any other previously determined l-LDHs. Major structural alterations between the two structures of LDH-2 were observed at two regions in one subunit. At the N-terminal parts of the two regions, the ligands-bound form takes an α-helical structure, while the form without ligands displays more disordered and extended structures. A vacuum-ultraviolet circular dichroism analysis showed that the α-helix content of LDH-2 in solution is approximately 30% at pH 7.5, which is close to that in the crystal structure of the form without ligands. A D241N mutant of LDH-2, which was created by us to easily form an α-helix at one of the two parts, exhibited catalytic activity even in the absence of FBP at both pH 5.5 and 7.5.
1. Introduction
Lactic acid bacteria (LAB) have their natural habitats in rather different environments and interact differently with human beings. In spite of having their different lifestyles, LAB predominantly obtain energy by the lactic acid fermentation. In addition to providing a characteristic flavor, lactic acid confers important preservative properties to fermented products [1]. Lactic acid produced by LAB is also known to be a material essential to generate polylactic acid. The free energy generated during homolactic acid fermentation is 2 mol of ATP per 1 mol of glucose. The crucial enzyme in this pathway is lactate dehydrogenase (LDH), which is responsible for catalyzing the reversible reduction of pyruvate to l- or d-lactate [2]. d- and l-LDHs stereospecifically convert pyruvate to d- and l-lactate, respectively. This reaction serves to balance the redox potential by oxidation of NADH to NAD.
l-LDH (EC 1.1.1.27), which is widely distributed in nature, is a tetrameric enzyme composed of four subunits. The arrangement of the subunits displays 222 symmetry where three 2-fold axes, which we defined as X-, Y-, and Z-axes, are present. Each monomer has one active site, and the tetramer has two allosteric sites, each of which is situated at the Y-axis interface between two monomers. Bacterial l-LDHs are homotetrameric and usually allosteric enzymes, which require fructose-1,6-bisphosphate (FBP), an intermediate in the glycolysis pathway, for catalytic activities. On the other hand, vertebrate cells contain non-allosteric l-LDH isozymes.
The mechanism for the allosteric regulation of the bacterial l-LDHs was first elucidated from the structural studies of the enzyme from Bacillus (B.) stearothermophilus [3–5]. In the FBP-bound tetrameric structure, each FBP molecule interacts with four positively charged residues (Arg173 and His188 residues in each of two juxtaposed subunits). The tetramer of the B. stearothermophilus l-LDH dissociates into two inactive X-axis-related dimers in the absence of FBP [6,7]. FBP may play a role in stabilizing and maintaining the tetrameric structure.
Another mechanism for allosteric regulation was proposed by the structural studies of the Bifidobacterium (Bf.) longum l-LDH [8,9]. In this case, the inactive tetrameric structures without and with FBP and NADH were determined [8,9], as well as the active tetrameric structure with FBP, NADH, and oxamate, a pyruvate analog [9]. The quaternary structure of the enzyme is significantly different between the inactive and active states, in which two Y-axis-related dimers in a tetramer take open and closed conformations, respectively. Furthermore, the structure of the inactive dimer of the B. stearothermophilus l-LDH [5] resembles the structure of the X-axis-related dimer in the inactive tetramer of the Bf. longum l-LDH in many features. Particularly, Arg171 residue, which is essential for the binding of pyruvate, does not point toward the pyruvate-binding site in both inactive structures. Therefore, it was concluded that FBP plays an important role in inducing the inter-subunit rearrangement of the l-LDH tetramer from an inactive to an active state, which is accompanied by local intra-subunit conformational changes. However, the fact that the crystal structure of the FBP-bound inactive state is present indicates that FBP merely increases the ratio of the active state to the inactive one. The binding of pyruvate to the active site may complete the structural change from the inactive to the active state.
However, it is unclear what key structural feature is responsible for the distinct behavior of the allosteric and non-allosteric l-LDHs, since bacterial l-LDHs are highly divergent and evolutionarily distant from the vertebrate non-allosteric enzymes. Bacterial l-LDHs exhibit wide variation in their regulation properties, as well as in their primary structures [2]. Some of them absolutely require FBP for their catalytic activities, whereas some enzymes can exhibit their activities even without FBP.
Among the allosteric bacterial l-LDHs, the enzyme from Lactobacillus (Lb.) casei shows unique properties, exhibiting a great pH-dependence in the enzyme activation [10–12]. The enzyme absolutely requires a high concentration of FBP (about 10−1 M) for its maximum catalytic activity under the neutral pH condition, where it exhibits low affinity for the substrate pyruvate. In contrast, under a weak acidic condition, the enzyme exhibits activity even in the absence of FBP, despite the low affinity for pyruvate, while the addition of FBP to the enzyme increases the affinity for pyruvate. Structural and mutational analyses of the Lb. casei l-LDH indicated that two inter-subunit salt bridges formed between His20 and Asp264 and between His205 and Glu211 are important to form the active state [14]. The salt bridges seem to be efficiently formed under the acidic condition, since the His residues are sufficiently protonated.
On the other hand, the l-LDH from Lb. pentosus is known to be an exceptionally non-allosteric enzyme [15], which constitutively exhibits high catalytic activity independently of FBP, similar to the vertebrate enzymes. The Lb. pentosus l-LDH constitutionally takes an active state independently of the binding of FBP. Structural analysis of the Lb. pentosus l-LDH revealed that the subunit interfaces of this enzyme are specifically stabilized to adopt the active state by increased numbers of inter-subunit salt bridges and hydrogen bonds and high geometrical complementarity [16].
Some strains of LAB produce antibacterial peptides, called bacteriocins, which are ribosomally synthesized and inhibitory to closely related Gram-positive bacteria [17,18]. Our group has recently isolated a bacteriocin-producing LAB from Brassica rapa L. subsp. nipposinica (L.H. Bailey) Hanelt var. linearifolia in Japan. The isolated strain, designated 15-1A, was identified as Enterococcus (Ec.) mundtii [19]. Whole genome sequence analysis of the 15-1A strain, now in progress in our laboratory, indicates that the strain contains at least two l-LDH-like proteins, designated LDH-1 and LDH-2. Another strain classified as Ec. mundtii was demonstrated to produce high yield of l-lactic acid with high optical purity from cellobiose or xylose [20,21]. Therefore, elucidation of the regulation mechanism of the Ec. mundtii l-LDHs at the molecular level is important to know why this bacterium can efficiently produce l-lactic acid.
In the present study, we performed an enzyme kinetic analysis of the recombinant l-LDHs from Ec. mundtii. As a result, it was found that the Ec. mundtii LDH-2 exhibits acidophilic kinetic properties. Understanding the allosteric regulation mechanism of the Ec. mundtii LDH-2 will be useful for creating an artificial enzyme with an acidophilic feature in future work. To clarify the allosteric regulation mechanism of the acidophilic LDH-2, we performed crystallographic, vacuum-ultraviolet circular dichroism (VUVCD) and site-directed mutation analyses.
2. Results
2.1. Amino acid sequence of LDH-1 and LDH-2 from Ec. mundtii 15-1A
Ec. mundtii 15-1A has two genes encoding l-LDH (LDH-1 and LDH-2). Redundancy of the gene encoding l-LDH was also found in many LAB including Lactococcus (Lc.) lactis [22], Lb. plantarum [23], and Ec. faecalis [24]. There are two reports about the kinetic characterizations of two l-LDHs contained in one bacterium.
Lc. lactis FI9078, which is a derivative of the MG1363 strain carrying a disruption of the ldh gene, can convert glucose to l-lactate, since the strain overexpresses the ldhB gene [25]. The Kact value of LDHB, which is defined as the concentration of FBP necessary for activation at 50% of the maximum, was found to be strongly dependent on pH (700-fold increase in the pH range from 6.0 to 7.0), in contrast to LDH. That is, the Lc. lactis LDHB is active under the neutral pH condition only when the FBP concentration is high enough, as with the Lb. casei l-LDH. However, the Lc. lactis LDHB is different from the Lb. casei l-LDH, since the affinity of the former enzyme for NADH, but not for pyruvate, is lowered with the increase of pH even in the presence of a high concentration of FBP.
Ec. faecalis V583 also contains two l-LDH-encoding genes (ldh-1 and ldh-2). The roles of the two genes were studied using knockout mutants [26]. Deletion of ldh-1 caused a metabolic shift from homolactic fermentation to ethanol, formate, and acetoin production, with a high level of formate production even under aerobic conditions. On the other hand, deletion of ldh-2 kept the homolactic fermentation, suggesting that LDH-2 plays only a minor role in the lactate production. Kinetic study showed that the Km values of LDH-2 for both pyruvate and NADH are larger than those of LDH-1, indicating the minor role in lactate production [27].
The sequence identity between the Ec. mundtii LDH-1 and LDH-2 is 44.9% (Fig. 1). However, the Ec. mundtii LDH-1 shows a high sequence identity with the Ec. faecalis LDH-1 (86.2%) and the Lc. lactis LDH (68.7%), whereas the Ec. mundtii LDH-2 shows a high sequence identity with the Ec. faecalis LDH-2 (65.0%) and the Lc. lactis LDHB (71.0%). These results indicate that LDH-1 mainly plays a role in l-lactate production in Ec. mundtii, while LDH-2 plays an additional role.
Fig. 1.

Sequence alignment of the Ec. mundtii LDH-2 with other l-LDHs from various LAB species. Sequence alignment was done using the ClustalX program [44]. Em-LDH-2, Ec. mundtii LDH-2; Ef-LDH-2, Ec. faecalis LDH-2; Ll-LDHB, Lc. lactis LDHB; Lp-LDH, Lb. pentosusl-LDH; Bl-LDH, Bf. longuml-LDH; Lc-LDH, Lb. caseil-LDH; Em-LDH-1, Ec. mundtii LDH-1; Ef-LDH-1, Ec. faecalis LDH-1; Ll-LDH, Lc. lactis LDH. The fully conserved residues in all sequences are marked with black shading. The residues conserved among alternative l-LDHs (Em-LDH-2, Ef-LDH-2, and Ll-LDHB) are marked with gray shading. In addition, the residues conserved among general allosteric l-LDHs (Bl-LDH, Lc-LDH, Em-LDH-1, Ef-LDH-1, and Ll-LDH) are also marked with gray shading. The residues of l-LDHs are numbered according to the N-system for vertebrate l-LDHs proposed by Eventoff et al. [45].
2.2. Kinetic properties of LDH-1 and LDH-2 from Ec. mundtii 15-1A
To characterize the Ec. mundtii LDH-1 and LDH-2, we performed enzyme kinetic analysis using each recombinant enzyme, which was overexpressed in Escherichia (E.) coli and purified almost to homogeneity. At first, we investigated the effect of pH on the catalytic activities of LDH-1 and LDH-2. The pH profiles of the activities of LDH-1 and LDH-2 in the presence of 20 mM pyruvate, 1.5 mM NADH, and 0.1 mM FBP are depicted in Fig. 2. LDH-1 maintains a high level of activity between pH 4.0 and pH 7.5. On the other hand, the activity of LDH-2 was maximal at pH 5.5 and then gradually decreased with the increase of pH.
Fig. 2.

The effect of pH on LDH activity. The activities of the wild-type LDH-1 and LDH-2 and the E60Q and D241N mutants derived from LDH-2 were measured under the given pH conditions in the presence of 20 mM pyruvate, 1.5 mM NADH, and 0.1 mM FBP. Diamonds, circles, triangles, and squares indicate the results of LDH-1, LDH-2, and E60Q and D241N mutants of LDH-2, respectively.
The results of the detailed kinetic analysis, which was done at pH 5.5 and pH 7.5, are summarized in Table 1. In the presence of 3 mM FBP, both LDH-1 and LDH-2 showed hyperbolic kinetic responses to increasing concentrations of pyruvate or NADH independently of pH. The Km of LDH-1 for pyruvate increased substantially (4-fold) with the increase of pH from 5.5 to 7.5, while the Km for NADH decreased slightly along with pH. On the other hand, the Km of LDH-2 for pyruvate was scarcely altered with pH, while the Km of LDH-2 for NADH increased approximately 2-fold with the increase of pH.
Table 1.
Kinetic parameters of Ec. mundtii LDH-1, LDH-2, and D241N mutant of LDH-2.
| LDH-1 |
LDH-2 |
LDH-2 D241N |
||||
|---|---|---|---|---|---|---|
| pH 5.5 | pH 7.5 | pH 5.5 | pH 7.5 | pH 5.5 | pH 7.5 | |
| Kact for FBP (μM) | 3.9 ± 1.0 | 7.0 ± 2.0 | 2.4 ± 0.3 | 1300 ± 200 | 0.076 ± 0.045 | 1.9 ± 0.3 |
| With 3 mM FBP | ||||||
| kcat (s−1) | 1600 ± 100 | 1500 ± 100 | 1800 ± 100 | 1300 ± 100 | 1800 ± 100 | 1800 ± 1 00 |
| Km for NADH (mM) | 0.18 ± 0.03 | 0.11 ± 0.02 | 0.41 ± 0.10 | 0.93 ± 0.28 | 0.20 ± 0.04 | 0.26 ± 0.03 |
| kcat/Km for NADH (mM−1 s−1) | 9000 ± 1600 | 13,000 ± 3000 | 4400 ± 1100 | 1400 ± 400 | 9000 ± 1800 | 6800 ± 700 |
| Km for pyruvate (mM) | 0.93 ± 0.11 | 3.8 ± 0.6 | 2.2 ± 0.5 | 2.7 ± 0.5 | 2.3 ± 0.4 | 3.0 ± 0.3 |
| kcat/Km for pyruvate (M−1 s−1) | 1700 ± 200 | 390 ± 60 | 810 ± 180 | 480 ± 100 | 790 ± 160 | 610 ± 60 |
| Without FBP | ||||||
| Relative activity (%) | ND | ND | ND | ND | 36 ± 12 | 30 ± 5 |
| Km for NADH (mM) | ND | ND | ND | ND | 0.9 ± 0.2 | ND |
| kcat/Km for NADH (mM−1 s−1) | ND | ND | ND | ND | 1200 ± 300 | 200 ± 10 |
| Km for pyruvate (mM) | ND | ND | ND | ND | 15 ± 3 | ND |
| kcat/Km for pyruvate (M−1 s−1) | ND | ND | ND | ND | 59 ± 12 | 15 ± 1 |
ND means “not determined”.
In the presence of 1.5 mM NADH and 20 mM pyruvate, LDH-1 and LDH-2 showed hyperbolic kinetic responses to an increasing concentration of FBP at both pH 5.5 and 7.5. Both enzymes were found to be allosteric ones that require FBP for their catalytic activities. The Kact value of LDH-1 was not significantly altered with pH. In contrast, the Kact value of LDH-2 was increased 500-fold when pH was shifted from 5.5 to 7.5. Like most bacterial l-LDHs, the Ec. mundtii LDH-1 was activated by the low concentration of FBP (in a range of 10−7–10−5 M) at pH 5.5 and pH 7.5. On the other hand, although LDH-2 was activated under a low FBP concentration condition at pH 5.5, a high concentration condition is required for activation at pH 7.5. These results suggest that although the Ec. mundtii LDH-1 is classified into the general bacterial l-LDH, the LDH-2 from Ec. mundtii is an acidophilic enzyme, as are the Lc. lactis LDHB [25] and the Lb. casei l-LDH [10–12].
Since the Ec. mundtii LDH-2 has a higher sequence similarity to the Lc. lactis LDHB (71%) than to the Lb. casei l-LDH (46%), the regulation mechanism of the Ec. mundtii LDH-2 may be common with that of the Lc. lactis LDHB but different from that of the Lb. casei l-LDH. In fact, in the presence of a high concentration of FBP, the Ec. mundtii LDH-2 and the Lc. lactis LDHB commonly exhibit lowered affinities for NADH, but not for pyruvate, with the increase of pH. Furthermore, considering the amino acid sequence similarity, the Ec. faecalis LDH-2 is likely to exhibit kinetic properties similar to those of the Ec. mundtii LDH-2 and the Lc. lactis LDHB. However, the kinetic parameters of the Ec. faecalis LDH-2 [27] are obviously different from those of the Ec. mundtii LDH-2 and the Lc. lactis LDHB [25].
2.3. Overall structures of ligands-unbound and -bound LDH-2
We crystallographically analyzed the acidophilic LDH-2 from Ec. mundtii. Although we tried to crystallize the LDH-2 in various conditions, we could obtain the measurable diffraction data from only two crystals. One crystal was grown in the absence of any ligands, while the other was grown in the presence of FBP and NADH (Table 2). In both cases, an asymmetric unit in the crystal contains one tetramer (Fig. 3a and b). The atomic coordinates and structure factors of the ligands-unbound and -bound LDH-2 have been deposited in the Protein Data Bank with accession codes 3WSV and 3WSW, respectively. A secondary structure assignment based on the crystal structures is shown in Fig. 4. In the crystal structure of the ligands-bound LDH-2, two FBP and four NADH molecules were modeled (Fig. 3a). The electron densities for the FBP molecules are strong, while the densities for the NADH molecules are relatively weak. Specifically, the densities for the nicotinamide moiety of NADH were poorly defined, probably due to the high mobility.
Table 2.
Data collection and refinement statistics.
| Data set | Ligands-unbound | Ligands-bound |
|---|---|---|
| Data collectiona | ||
| Space group | P212121 | P21 |
| Cell dimensions | ||
| a (Å) | 104.43 | 67.77 |
| b (Å) | 127.82 | 123.54 |
| c (Å) | 133.80 | 85.79 |
| β (°) | – | 107.75 |
| Wavelength (Å) | 1.0000 | 1.0000 |
| Resolution (Å) | 100–2.38 | 100–2.30 |
| Unique reflection | 71,995 | 57,963 |
| Redundancy | 6.9 (4.8) | 2.9 (2.8) |
| Completeness (%) | 99.5 (96.3) | 97.3 (97.4) |
| Rmerge (%) | 8.3 (40.9) | 10.7 (41.3) |
| I/σ | 27.2 (2.5) | 8.2 (2.4) |
| Refinement | ||
| Resolution (Å) | 30–2.38 | 30–2.30 |
| Used reflections | 69,778 | 55,635 |
| No. of atoms | ||
| Protein | 9682 | 9655 |
| Ligand | 48 | 234 |
| Water | 408 | 507 |
| R (%) | 23.2 | 19.5 |
| Rfree (%) | 27.1 | 24.6 |
| Rms deviationsb | ||
| Bond length (Å) | 0.006 | 0.006 |
| Bond angle (°) | 1.1 | 1.2 |
| Mean B-factor (Å2) | ||
| Protein | 57.3 | 32.4 |
| Ligand | 81.6 | 65.8 |
| Solvent | 59.2 | 35.4 |
| Ramachandran plot (%) | ||
| Favored | 88.6 | 91.3 |
| Allowed | 11.1 | 8.4 |
| Disallowed | 0.4 | 0.3 |
| PDB code | 3WSV | 3WSW |
Values in parentheses are for the highest resolution bin.
Rms deviations are calculated by CNS.
Fig. 3.
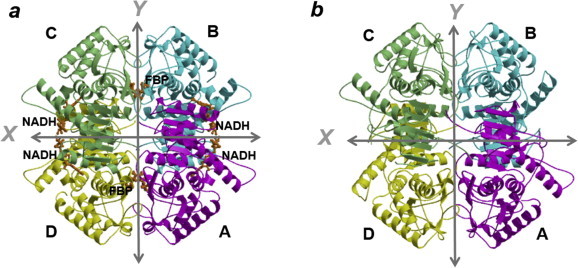
Ribbon diagrams of the tetrameric structures of the Ec. mundtii LDH-2. Structures of the Ec. mundtii LDH-2 with and without ligands are shown in a and b, respectively. The four subunits are colored cyan, magenta, lightgreen, and yellow. In a, four NADH and two FBP molecules bound to the enzyme are shown in the stick model colored orange. The X- and Y-axes are indicated by arrows. The structures are viewed along the Z-axis. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.
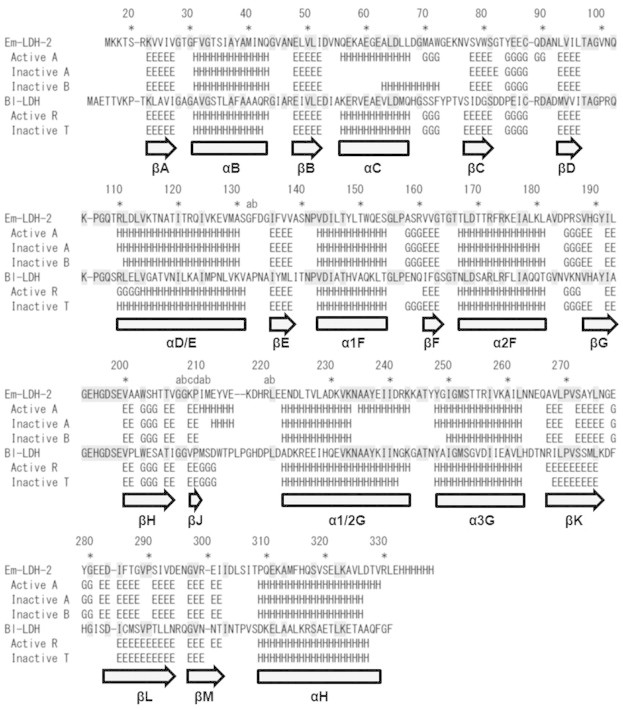
Structure-based sequence alignment between the Ec. mundtii LDH-2 (Em-LDH-2) and the Bf. longuml-LDH (Bl-LDH). Subunits in the active and inactive states of Bl-LDH were named R and T, respectively [9]. H, E, and G mean the residues assigned as α-helix, β-strand, and 310-helix, respectively. The secondary structure elements are systematically named according to the accepted nomenclature [46]. The residues conserved between Em-LDH-2 and Bl-LDH are marked with gray shading.
Four subunits in the ligands-bound tetramer of the Ec. mundtii LDH-2 are similar to one another. They can be superimposed with root mean square (rms) deviations in a range between 0.20 and 0.48 Å for the 312 Cα atoms. When compared with the active state of the Bf. longum l-LDH, the residues Val100–Leu112 and Val213–Arg220a of the Ec. mundtii LDH-2 are structurally different (Fig. 5a). The residues Val100–Leu112 protrude away from the main body of the subunit, as found in the inactive state of the Bf. longum l-LDH. Since this region is one side of the pyruvate-binding pocket, the binding of pyruvate to the active site may induce the structural change of this region. On the other hand, the residues Val213–Arg220a in the Ec. mundtii LDH-2 are structurally different from the corresponding residues in the Bf. longum l-LDH because former enzyme lacks two residues on the amino acid sequence (Figs. 1 and 4). However, without these residues, each subunit of the ligands-bound Ec. mundtii LDH-2 is structurally similar to that of the Bf. longum l-LDH in the active state with rms deviations in a range between 1.1 and 1.2 Å. It should be noted that an α-helix (α1/2G in Fig. 4) consisting of the residues Glu222–Lys243 is bent around Asn234, as found in the active state of the Bf. longum l-LDH.
Fig. 5.

Superposition of subunit structures of the Ec. mundtii LDH-2 and the Bf. longuml-LDH. Subunit A in the active state and subunits A and B in the inactive state of the Ec. mundtii LDH-2 are colored red, yellow, and darkgreen, respectively. Subunits of the Bf. longuml-LDH in the active and inactive states [9] are colored blue and cyan, respectively. In a, subunit A in the active state of the Ec. mundtii LDH-2 are compared with the subunits of the Bf. longuml-LDH in the active and inactive states. In b, subunit A in the active state and subunits A and B in the inactive state of the Ec. mundtii LDH-2 are superimposed. Superpositions were calculated using all Cα atoms (a) or Cα atoms in the catalytic domain (b). NADH molecule bound to subunit A in the active state of the Ec. mundtii LDH-2 is shown in the stick model colored orange. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
It has been suggested that the side-chain orientation of the Arg171 residue is important for discriminating between inactive and active states. The side chain of the Arg171 residue of the Ec. mundtii LDH-2, which seems to be important for the binding of pyruvate, points toward the pyruvate-binding site (Fig. 6a), as seen in the active state of other bacterial l-LDHs. Furthermore, the quaternary structure of the ligands-bound LDH-2 is similar to that of the other bacterial l-LDHs in the active state, in which a compact tetramer is formed by many inter-subunit interactions (Fig. 7a and c). Therefore, we defined the quaternary structure of the ligands-bound LDH-2 as an active state.
Fig. 6.

Structural differences of the Val54–Lys75 and Lys233–Gly249 residues in the Ec. mundtii LDH-2. Structure of subunit A in the active state and structures of subunits A and B in the inactive state, which are shown in the stereo view, are shown in a, b, and c, respectively. Glu57, Glu60, Arg171, Arg173, Glu175, His188, Glu238, Asp241, and Tyr248 residues are shown in the stick model. Hydrogen bonds are shown by broken lines.
Fig. 7.

Molecular surfaces of the Ec. mundtii LDH-2 and the Bf. longuml-LDH. The surfaces of the Ec. mundtii LDH-2 in the active and inactive states are shown in a and b, respectively. The surfaces of the Bf. longuml-LDH in the active and inactive states [9] are shown in c and d, respectively. Each subunit in the tetramer is colored the same as in Fig. 3. In a, c and d, four NADH and two FBP molecules bound to the enzyme are shown in the stick model colored orange. In c, four oxamate (a pyruvate analog) molecules bound to the active-state Bf. longuml-LDH are shown in the stick model colored blue. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
For the general allosteric l-LDHs, FBP is speculated to shift the equilibrium toward an active state [9], although the sensitivities to FBP are different among the enzymes. In the inactive state (Fig. 7d), the Y-axis-related dimer takes an open conformation, resulting in the expanded shape of the tetramer. In the presence of FBP, the ratio of active state tetramer, in which the Y-axis-related dimer is stabilized to take a closed conformation in accordance with the rearrangement of the α-helices in each subunit (Fig. 7c), is increased. The conformational change from an inactive to an active state of l-LDH is not significantly associated with the changes of the secondary structure in each subunit, as shown in Table 3 and Fig. 4.
Table 3.
Contents of α-helix and β-strand calculated from crystal structure and VUVCD analysis.
| α-Helix (%) | β-Strand (%) | |
|---|---|---|
| From crystal structure | ||
| Ligands-bound Ec. mundtii LDH-2 | ||
| Subunit A | 42.2 | 18.0 |
| Subunit B | 42.5 | 20.2 |
| Subunit C | 41.6 | 20.2 |
| Subunit D | 42.2 | 18.0 |
| Average | 42.1 | 19.1 |
| Ligands-unbound Ec. mundtii LDH-2 | ||
| Subunit A | 33.9 | 18.3 |
| Subunit B | 35.1 | 18.0 |
| Subunit C | 32.9 | 17.7 |
| Subunit D | 35.7 | 18.0 |
| Average | 34.4 | 18.0 |
| Bf. longuml-LDH in the active state [9] | 39.4 | 18.1 |
| Bf. longuml-LDH in the inactive state [9] | 39.7 | 17.8 |
| From VUVCD analysis | ||
| Wild type | 28.5 ± 1.3 | 16.3 ± 2.3 |
| Wild type + 20 mM FBP | 29.6 ± 1.5 | 15.8 ± 2.2 |
| D241N mutant | 30.1 ± 1.4 | 16.3 ± 1.8 |
| D241N mutant + 20 mM FBP | 32.6 ± 1.8 | 14.3 ± 2.8 |
The molecular shape of the LDH-2 tetramer without the binding of ligands (Fig. 7b), determined in the present study, is more compact than that of the ligands-bound tetramer (Fig. 7a) or that of the other bacterial l-LDHs in the active state (Fig. 7c). For examples, the distances between Cα atoms of Arg171 in subunits A and B and between the atoms in subunits A and D in the ligands-unbound tetramer are shorter by 1.8 and 1.2 Å than those in the ligands-bound tetramer, respectively. In the four subunits of the ligands-unbound LDH-2, subunits A and C, shown in Fig. 3b, can be superimposed well with an rms deviation of 0.34 Å for the 309 Cα atoms; subunits B and D can be also be superimposed well with an rms deviation of 0.15 Å for the 312 Cα atoms. On the other hand, rms deviations between the other pairs of subunits were calculated to be large values in a range between 2.3 and 2.4 Å.
When the structure of subunit A (or C) is compared with that of subunit B (or D) in the ligands-unbound form of the Ec. mundtii LDH-2, three regions including the residues Val54–Lys75, Val100–Leu112, and Lys233–Gly249 are different (Fig. 5b). Atoms in these regions have high B-factors commonly. Specifically, the residues Glu62–Leu66 in subunit A (or C) in the ligands-unbound form are invisible in the electron density map. The structures of the residues Val100–Leu112 in subunits A (or C) and B (or D) are similar to those of the corresponding residues of the Bf. longum l-LDH in the inactive and active states, respectively (Fig. 5). The Val100–Leu112 residues may be flexible due to the absence of pyruvate.
In addition, both subunits A (or C) and B (or D) in the ligands-unbound form are structurally different from the subunits in the ligands-bound form due to the differences in some regions in the subunit (Fig. 5b). In detail, rms deviations between subunit A (or C) in the ligands-unbound form and subunits in the ligands-bound form were calculated to be large values in a range between 2.2 and 2.4 Å, while rms deviations between subunit B (or D) in the ligands-unbound form and subunits in the ligands-bound form ranged from 2.9 to 3.2 Å. However, when calculated without the residues Val54–Lys75, Val100–Leu112, and Lys233–Gly249, both subunits A (or C) and B (or D) in the ligands-unbound form can be superimposed on the subunits in the ligands-bound form with rms deviations in a range between 0.74 and 0.82 Å. It should be noted that the regions Val54–Lys75 and Lys233–Gly249 in the ligands-unbound form also take different structures from those in the ligands-bound form (Fig. 5b). That is, these two regions in the LDH-2 can take three different structures.
Subunit structure of l-LDH can be divided into N-terminal NADH-binding domain and C-terminal catalytic domain. When the superposition was calculated using the Cα atoms in the catalytic domain (The165–Arg330) of the Ec. mundtii LDH-2 without the residues Lys233–Gly249, rms deviations were ranged between 0.66 and 0.75 Å, which are slightly smaller than a global superposition without the residues Val54–Lys75, Val100–Leu112, and Lys233–Gly249. The superposition shows an apparent rocking motion of the NADH-binding domain with respect to the catalytic domain (Fig. 5b). However, although the arrangement of subdomains may contribute to the binding of NADH in a small extent, conformational change of the residues Val54–Lys75 is primarily required for the binding.
Based on the orientation of the side chain of Arg171, subunits A and C are assumed to be in an active state, in which Arg171 points toward the pyruvate-binding site without intra-subunit interactions (Fig. 6b). On the other hand, subunits B and D are assumed to be in an inactive state, in which Arg171 does not point toward the pyruvate-binding site due to the intra-subunit interactions with Glu175 and Tyr248 (Fig. 6c). Especially, torsion angles between Cα and Cβ atoms of Arg171 in subunits A and C are different by about 90°, when compared with those in subunits B and D. However, the structure of each subunit in the ligands-unbound LDH-2 is quite different from that of the subunit in the other bacterial l-LDHs in either the inactive or active state (Fig. 5). In fact, even when calculated without the residues Val100–Leu112 and Val213–Arg220a of the Ec. mundtii LDH-2, the subunits in the ligands-unbound form are structurally different from the subunit of the Bf. longum l-LDH in the active state with rms deviations in a range between 2.4 and 2.7 Å, and from the subunit of the Bf. longum l-LDH in the inactive state with rms deviations in a range between 2.4 and 2.8 Å. Importantly, secondary structure analysis using the DSSP program indicated that α-helix content in the ligands-unbound LDH-2 is about 34% on average (Table 3), which is smaller than that in the ligands-bound form or those in the inactive and active states of the other l-LDHs. The structure of ligands-unbound LDH-2 seems to be an inactive form, since the NADH- and pyruvate-binding pockets are different from those of the other bacterial l-LDHs in the active state, as described below. Therefore, we defined the compact quaternary structure of the ligands-unbound LDH-2 as another inactive state.
2.4. Substrate-binding sites
In the Val54–Lys75 region of the active state LDH-2, Gln56–Trp72 contains helical structures including an α-helix (αC) and following 310-helix (Figs. 4 and 6a). On the other hand, the corresponding region of subunit B in the inactive state has a shorter α-helix consisting of Ala63–Trp72, and the helix is differently oriented from that in the active state (Fig. 6c). The corresponding region of subunit A in the inactive state is more extended (Fig. 6b), and some of the residues are invisible in the electron density map. Significantly, the N-terminal parts of this region in both subunits A and B commonly take the disordered and extended structures in the inactive state and occupy the binding site for the adenosine moiety of NADH (Fig. 5b). Therefore, the current inactive state of LDH-2 likely is unable to accommodate NADH, which may be related to the kinetic result that LDH-2 exhibits low affinity for NADH even under the high concentration of FBP (Table 1).
The Lys233–Gly249 region is positioned at one side of the pyruvate-binding pocket. In the active state, the N-terminal part of this region (Ala235–Lys243) is a C-terminal part of the bent α-helix (α1/2G), and the following part takes a rigid loop structure (Figs. 4 and 6a). On the other hand, in the inactive state, the corresponding regions of subunits A and B have no helical structures (Fig. 6b and c), suggesting that the structures of the pyruvate-binding pockets of subunits A and B in the inactive state are clearly different from those in the active state. Therefore, this compact inactive state likely is unable to bind to pyruvate, as with NADH.
In summary, the N-terminal parts of the two regions (Val54–Lys75 and Lys233–Gly249) form the α-helices in the active state LDH-2, while the parts take more disordered and extended structures in the compact inactive state LDH-2. It should be noted that the residues Lys233–Gly249 of one subunit are positioned near the residues Val54–Lys75 of the X-axis-related subunit (Fig. 8a and b), indicating that the binding of NADH to one subunit facilitates the formation of the pyruvate-binding pocket in the X-axis-related subunit.
Fig. 8.
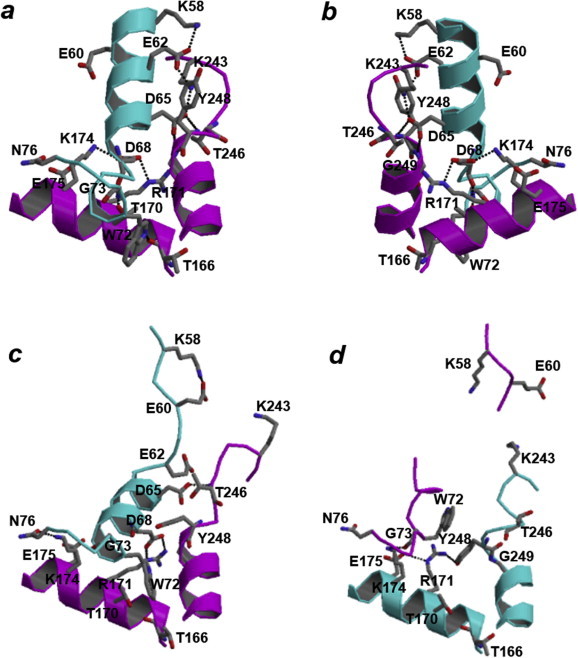
Inter-subunit interactions around the Arg171 residue found in the Ec. mundtii LDH-2. The structures around the Arg171 residue of subunit A in the active state and of subunits A and B in the inactive state are shown in a, c, and d, respectively. Ribbon models of subunits A and B are shown in magenta and cyan, respectively. The residues Lys58, Glu60, Glu62, Asp65, Asp68, Trp72, Gly73, and Asn76 in one subunit and Thr166, Thr170, Arg171, Lys174, Glu175, Lys243, Thr246, Tyr248, and Gly249 in the other subunit are shown in the stick model. Hydrogen bonds are shown by broken lines. View directions are opposite in a and b.
Interestingly, these two parts in the Ec. mundtii LDH-2 contain two acidic residues separated by two residues in common (Glu57 and Glu60 in the former part and Glu238 and Asp241 in the latter, Figs. 4 and 6). These two parts seem to have difficulty forming α-helices under neutral to alkaline pH conditions because the positions of the negatively charged residues are three-dimensionally close after the formation of α-helices. The difficulty in forming the α-helices at the two parts may be related to the pH dependence of the enzyme activation.
2.5. Site-directed mutation analysis
To elucidate the effects of acidic residues on catalytic activity, site-directed mutational analysis was performed. Residues selected to generate the mutation were Glu60 and Asp241. The former and latter residues were replaced by Gln and Asn, respectively. The site-directed mutants were named E60Q and D241N, respectively. The pH profiles of the activities of E60Q and D241N in the presence of 20 mM pyruvate, 1.5 mM NADH, and 0.1 mM FBP are depicted in Fig. 2. The activity of the mutant E60Q was maximal at pH 5.5 and then gradually decreased in accordance with the increase of pH, similar to the wild-type enzyme. On the other hand, the activity of D241N was consistently high between pH 4.5 and 7.5, similar to the Ec. mundtii LDH-1 and other bacterial l-LDHs. These results indicate that electrostatic repulsion between Glu238 and Asp241 of the Ec. mundtii LDH-2 is an important factor for the regulation of catalytic activity, whereas the repulsion between Glu57 and Glu60 is not.
Detailed kinetic analysis of D241N, which was done at pH 5.5 and pH 7.5, is summarized in Table 1. The Km values for NADH and pyruvate and the kcat value of the D241N mutant were determined in the presence of 3 mM FBP. The Km values for NADH were substantially lower than those of the parent enzyme (2-fold at pH 5.5 and 4-fold at pH 7.5). Other kinetic parameters of D241N were not significantly different from those of the parent enzyme. Next, we investigated the effect of FBP on the catalytic activity of the D241N mutant in the presence of 1.5 mM NADH and 20 mM pyruvate. Although the wild-type LDH-2 absolutely requires FBP for catalytic activity, its D241N mutant displays enzyme activity even in the absence of FBP, which is approximately 30% of the maximum at either pH 5.5 or pH 7.5. Although the Kact value was increased approximately 25-fold by the change of pH from 5.5 to 7.5, the mutant was activated by a rather low concentration of FBP under the neutral pH condition when compared with the wild-type enzyme. These results indicate that, if the region including Glu238 and Asp241 can form the α-helix easily, the enzyme has a stronger affinity for NADH and is active in the absence or in the presence of a low concentration of FBP. We also determined the kinetic parameters of the D241N mutant in the absence of FBP (Table 1). At pH 5.5, the kcat/Km values for NADH and pyruvate were 7.5- and 13-fold lower than those obtained in the presence of FBP, respectively. The kcat/Km values were further decreased at pH 7.5 (for NADH and pyruvate, 34- and 41-fold lower than those obtained in the presence of FBP, respectively). It is considered that the allosteric regulation of the D241N mutant is still dependent on the pH conditions, although the sensitivity to FBP was greatly increased by the mutation.
2.6. VUVCD spectroscopic analysis
The secondary structure contents of wild-type LDH-2 and its D241N mutant at pH 7.5 were estimated by VUVCD analysis, and the results are listed in Table 3. As shown in this table, the α-helix content of wild-type LDH-2 was calculated to be 28.5%, which is close to that in the crystal structure of the ligands-unbound form (the average α-helical content for four subunits is 34.4%). The D241N mutation was found to slightly increase the α-helix content (30.1%). If the residues Ala235–Lys243 form an α-helix, the helix content is expected to increase by 2.8%. Formation of an α-helix at this region seems to be not completely induced by the mutation. Since the wild-type LDH-2 and its D241N mutant with a concentration above 0.1 mg mL−1 had the aggregations in the buffer at a pH lower than 6, VUVCD analysis could not be performed under the weak acidic pH conditions.
In the CD measurements, absorbance of the solution should be kept as low as possible. The effect of NADH could not be investigated due to the high absorption of the compound. Therefore, we only examined the effect of FBP on the secondary structure contents of the wild-type LDH-2 and its D241N mutant (Table 3). As a result, it was found that the addition of FBP to the D241N mutant slightly increased the α-helix content (30.1% and 32.6% in the absence and presence of 20 mM FBP, respectively, Table 3). The effect of the addition of FBP to wild type (1.1% increase in the α-helix content) is smaller as compared with the case of the wild-type enzyme (2.5%). These observations strongly indicate that the sensitivity to FBP was increased by the mutation. However, the α-helix content of the D241N mutant in the presence of 20 mM FBP, which is the maximum value obtained in the present study, is about 10% lower than that estimated from the crystal structure of the ligands-bound form. Structural change from the inactive to the active state seems to be not completely induced by the addition of FBP.
3. Discussion
More than two l-LDH-encoding genes exist in the genome of LAB: one exhibits a major role, whereas the others have minor roles [25–27]. In the present study, we also found that the Ec. mundtii LDH-1 is a general l-LDH. On the other hand, Ec. mundtii LDH-2 is an “alternative l-LDH”, like the Lc. lactis LDHB [25] and the Ec. faecalis LDH-2 [26,27]. At this point, it is still unclear why Ec. mundtii can efficiently produce l-lactic acid. Alternative l-LDHs seem to have in common a low affinity for NADH [25,27]. In addition, the Ec. mundtii LDH-2 and the Lc. lactis LDHB [25] have an acidophilic feature.
The quaternary structure of the ligands-bound form of LDH-2 is similar to that of the active state of the other bacterial l-LDHs [4,9,14,16]. On the other hand, the crystal structure of the ligands-unbound LDH-2 may be an inactive form, although its quaternary structure is very compact compared to that of other l-LDHs in the inactive state [5,9,14]. In addition, we found that electrostatic repulsion between Glu238 and Asp241 of the Ec. mundtii LDH-2 is an important factor for the regulation of catalytic activity. A similar mechanism may be present in the Lc. lactis LDHB and the Ec. faecalis LDH-2, since the Glu238 and Asp241 residues are conserved in these two alternative l-LDHs (Fig. 1). In the general or non-allosteric bacterial l-LDHs, at least one of two residues that correspond to Glu238 and Asp241 in the Ec. mundtii LDH-2 is not acidic.
Considering that the D241N mutant partially exhibits catalytic activity in the absence of FBP, a small part of the Ec. mundtii LDH-2 seems to adopt an active state without the help of FBP in the solution state. In fact, the active state of the Ec. mundtii LDH-2 is stabilized by an abundance of inter-subunit hydrophilic interactions (120 interactions within a tetramer), although the inactive state has only 48 interactions. The number of inter-subunit interactions is larger than that in the other allosteric bacterial l-LDHs (64 interactions in the case of active state of Bf. longum l-LDH) but smaller than that in the non-allosteric Lb. pentosus l-LDH (128 interactions).
It is important to note that most of the inter-subunit interactions in the active state of the Ec. mundtii LDH-2 are formed by the residues in the regions Val54–Lys75 and Lys233–Gly249 (Fig. 8a and b), which have different structures between inactive and active states. In the inter-subunit interactions, although some of them are common with the other bacterial l-LDHs, some are considered to be characteristic to alternative l-LDHs. Specifically, in the active state of the Ec. mundtii LDH-2, the side chain of Asp68 in one subunit interacts with those of Arg171 and Lys174 in the X-axis-related subunit (Fig. 8a and b), indicating that the Asp68 and Lys174 residues are important in making Arg171 protrude toward the pyruvate-binding pocket. Furthermore, the side chains of Trp72 and Thr170 in one subunit interact with the main-chain carbonyls of Thr166 and Asp68 in the X-axis-related subunit, respectively (Fig. 8a and b). These inter-subunit interactions are not formed in the inactive state except for the hydrogen bonds between Arg171 in subunit A and Asp68 in subunit B (Fig. 8c and d). Furthermore, it is important to note that the Asp68, Trp72, Thr170, and Lys174 residues are conserved in the alternative l-LDHs but not in the other bacterial allosteric l-LDHs (Fig. 1).
In the inactive tetramer of LDH-2, the Z-axis-related subunits superimpose better than the X- or Y-axis-related subunits. In addition, the Val54–Lys75 and Lys233–Gly249 regions, which take different structures between inactive and active states, are positioned at the X-axis interface. These observations indicate that the Val54–Lys75 and Lys233–Gly249 regions in each subunit can deform the structure to increase the geometric complementarity at the X-axis interface. As far as we know, this is the first repot for the asymmetric tetrameric structure of l-LDH. Probably, in the solution state, an inactive tetramer where two AB dimers are related by Z-axis is coexist with another tetramer where two AB dimers are related by Y-axis. Current structure, which corresponds to the former tetramer, is likely derived from selective crystallization.
The mechanism of the allosteric regulation of the Ec. mundtii LDH-2 can be summarized as follows: under the weak acidic condition, since the electrostatic repulsion between Glu238 and Asp241 is lowered, the Ala235–Lys243 residues can form an α-helix easily. The formation of the α-helix at the Ala235–Lys243 residues may induce the structural change to generate a long α-helix at residues Gln56–Leu67 in the X-axis-related subunit, which are three-dimensionally close to the Ala235–Lys243 residues, leading to the formation of a pocket for the accommodation of NADH. This proposition is in agreement with the experimental results that the Km value of the Ec. mundtii LDH-2 for NADH was lowered with the decrease of pH (Table 1) and that the D241N mutation further decreased the Km values. The formation of α-helices in the two regions may induce the change from the compact inactive state to the expanded inactive or the compact active state, although the expanded inactive state of the Ec. mundtii LDH-2 is structurally undetermined.
The Ec. mundtii LDH-2 may dominantly take a compact inactive state under the neutral pH condition, whereas the ratio of the expanded inactive or compact active state would be increased with the decrease of pH. Among the three states, only the expanded inactive state may allow the invasion of FBP, whereas the compact inactive or active state might not be bound to FBP due to the molecular shape (Fig. 7). Therefore, FBP may act only on the expanded inactive tetramer and induce the change of the quaternary structure to the active state. This proposition explains the result that a higher concentration of FBP is required for catalytic activity in accordance with the increase of pH. However, the CD analysis indicated that induction of the structural change by the addition of only FBP is difficult. Perhaps, in addition to FBP, high concentration of NADH may be needed for the structural change. A long α-helix at the Gln56–Leu67 residues would be completely generated after the binding of NADH, although the formation of an α-helix at the Ala235–Lys243 residues in the X-axis-related subunit may induce the helix formation at the Gln56–Leu67 residues.
The D241N mutant loses the electrostatic repulsion between Glu238 and Asp241, thus the Ala235–Lys243 residues can much more easily form the α-helix. As a result, the D241N mutant shows catalytic activity even without FBP or by the addition of a low concentration of FBP. However, the CD analysis also indicated that the ratio of the active state was only slightly increased, even with the addition of a high concentration of FBP. This result again suggests that the high concentration of NADH is needed for the complete structural change.
Like the Ec. mundtii LDH-2, the Lb. casei l-LDH is also known to be an acidophilic enzyme showing a low sensitivity to FBP under a neutral pH condition [12,13]. Another research group has suggested that the acidophilicity of the Lb. casei l-LDH is caused by the tendency to adopt an expanded inactive state [14]. They insisted that the inter-subunit interactions formed between His20 and Asp264 and between His205 and Glu211, which seem necessary in order to form a compact active state, are not generated in the enzyme under a neutral pH condition. The His205 and Glu211 residues are also present in the Ec. mundtii LDH-2 (Fig. 1), which may be a reason for the acidophilicity found in the D241N mutant in the absence of FBP. However, the His20, His205, Glu211, and Asp264 residues are completely conserved in Ec. mundtii LDH-1 (Fig. 1), which was classified as a general l-LDH in this study. Probably, the extraordinary low sensitivity of Lb. casei l-LDH to FBP has another cause that is currently undetermined.
The exact role of the acidophilic Ec. mundtii LDH-2 is currently unknown. It may act only under a low pH condition or in the presence of a high concentration of FBP and NADH. However, in the present study, we could clarify the allosteric regulation mechanism of the acidophilic LDH-2. Another research group has recently suggested that the computational studies are useful to predict the catalytic properties of l-LDHs [27]. However, the study seems to be meaningless, since their conclusions are based on the homology models built using the active state structures as a reference. To accurately predict the catalytic properties of l-LDHs, consideration of the compact inactive state, shown in Fig. 3b, is necessary. Furthermore, the knowledge obtained from the present study may be applied to protein engineering if one wishes to add the acidophilic feature to a specific protein.
4. Materials and methods
4.1. Gene cloning, expression, and purification of LDH-1 and LDH-2
The ldh-1 gene was amplified by PCR from Ec. mundtii 15-1A genomic DNA with the forward primer, 5′-GGAATTCCATATGACTGCAAACGCAGAAAAAAAAG-3′ (the underline indicates an NdeI site), and the reverse primer, 5′-TCCGCTCGAGTTCAGCATCCAATTTAGC-3′ (the underline indicates an XhoI site). The ldh-2 gene was amplified by PCR with the forward primer, 5′-GGAATTCCATATGAAAAAAACAAGTCG-3′ (the underline indicates an NdeI site), and the reverse primer, 5′-TCCGCTCGAGGCGTACAGTATCAAGTAC-3′ (the underline indicates an XhoI site). The amplified DNA fragment including ldh-1 or ldh-2 was digested with NdeI and XhoI and inserted into the pET-21a(+) vector (Novagen) to generate an expression plasmid for LDH-1 or LDH-2, respectively. Each expression vector was introduced into the E. coli BL21(DE3) pLysS strain. To overproduce the enzymes with His6-tag at the C-terminus, the E. coli cells were grown at 28 °C in the LB medium, and the production was induced by 1 mM isopropyl-β-d-thiogalactopyranoside. The cells were harvested by centrifugation and frozen until being lysed by sonication. Recombinant LDH-1 and LDH-2 were purified using a Ni(II)-chelated His-Bind Resin (Novagen) according to the standard protocol.
4.2. Mutation
A QuikChange Site-Directed Mutagenesis Kit (Stratagene) was used. To create the E60Q mutant of LDH-2, an LDH-2-expression vector was amplified by PCR with the mutagenesis primer, 5′-CGATGTCAACCAAGAAAAAGCACAAGGAGAAGCTTTAGAC-3′ (the underline indicates the mutation site), and its complement chain. To create the D241N mutant of LDH-2, the vector was amplified with the mutagenesis primer, 5′-GCAGCTTATGAAATCATTAACCGTAAAAAAGC-3′ (the underline indicates the mutation site), and its complement chain. The confirmation of the mutation was done by DNA-sequencing analysis. Each of the mutated vectors was introduced into the E. coli BL21(DE3) pLysS strain. Overproduction and purification of the mutated product were done as described above.
4.3. X-ray crystallography
Prior to crystallization, the LDH-2 protein was concentrated to 12 mg mL−1 using Amicon Ultra (Millipore). Crystals of a ligands-unbound form of LDH-2 were obtained using the sitting-drop vapor diffusion method, with a 1:1 (v/v) ratio of protein solution to precipitant solution. Crystals were successfully formed at 25 °C when the 0.1 M MES–NaOH buffer (pH 6.2) containing 7% (w/v) PEG 20,000 was used as a precipitant solution. Crystals of the ligands-bound form were grown in the presence of 1.5 mM NADH and 1 mM FBP. Initial crystals were obtained by using the 0.1 M sodium citrate buffer (pH 5.3) containing 6% (w/v) PEG 4000 and 0.2 M ammonium acetate as a precipitant solution. The quality of the crystals was improved by repeating the micro-seeding method.
The diffraction intensities of the crystals were obtained using synchrotron radiation from the BL26B2 station at SPring-8, Japan. Each crystal was captured in a loop and then frozen in liquid nitrogen. After the delivery of the crystals to the facility, they were placed on goniometer head in a stream of nitrogen gas cooled to 100 K automatically. We obtained the diffraction intensity data of ligands-unbound and -bound crystals up to 2.38 and 2.30 Å resolutions, respectively. The diffraction intensities of ligands-unbound crystal were integrated and scaled by HKL2000 [28], while those of ligands-bound crystal were processed by the combination of Mosflm and Scala in the CCP4 program suite [29]. The three-dimensional structure of the ligands-unbound LDH-2 was solved by the molecular replacement method using the program Molrep in the CCP4 program suite [29]. The starting model used in this study was one subunit in the active state of the B. stearothermophilus l-LDH (PDB code 1LDN) [4]. The model was further refined with simulated annealing and conventional restrained refinement methods using the CNS program [30]. A subset of 5% of the reflections was used to monitor the free R factor (Rfree) [31]. Each refinement cycle includes the refinement of the positional parameters, individual isotropic B-factors, and revision of the model visualized by the program Xfit in the XtalView software package [32]. The ligands-bound crystal structure was also determined by molecular replacement using Molrep and refined by CNS. The details of the data collection and refinement statistics are shown in Table 2.
4.4. Activity assay of LDH
The LDH activity was determined by measuring the rate of NADH oxidation at 340 nm (ε = 6220 M−1 cm−1) in a quartz cell with a path-length of 0.1 or 0.2 mm. All measurements were based on at least duplicate determinations of the reaction rates. To measure the pH profiles of activity, reactions were carried out in a broad-range buffer (50 mM 3,3-dimethylglutaric acid, 50 mM Tris, and 50 mM 2-amino-2-methyl-1,3-propandiol), adjusted at a given pH, containing 5 mM magnesium chloride, 20 mM pyruvate, 1.5 mM NADH, and 0.1 mM FBP at 37 °C. Detailed kinetic analysis was performed using 100 mM MES–NaOH (pH 5.5) or HEPES–NaOH (pH 7.5) buffer. To determine the Km value for NADH, reactions were carried out in the buffer containing 20 mM pyruvate, 3 mM FBP, and a given concentration of NADH (0–1.5 mM). To determine the Km value for pyruvate, reactions were carried out in the buffer containing 1.5 mM NADH, 3 mM FBP, and a given concentration of pyruvate (0–20 mM). To determine the Kact value, reactions were carried out in the buffer containing 1.5 mM NADH, 20 mM pyruvate, and a given concentration of FBP (0–3 mM).
In any case, the enzymes exhibited a hyperbolic kinetic response to increasing concentrations of pyruvate, NADH, or FBP. Therefore, kinetic data were fitted to Eq. (1):
| (1) |
In the equation, v is the initial velocity, kcat is the catalytic constant, A and B are constant parameters, Et is the concentration of the enzyme, and X is the concentration of pyruvate, NADH, or FBP. In general, B was fixed to zero, and kcat and A, which means Km (for pyruvate or NADH) or Kact (for FBP), were determined by the non-linear least square method. Exceptionally, since the mutant enzyme D241N exhibited catalytic activity in the absence of FBP, B was also determined in addition to kcat and A. In this case, the ratio of activity in the absence of FBP to the maximum activity is correspondent with B/A, and Kact is expressed as A − 2B.
In the case of the D241N mutant, we also determined the kcat and Km values in the absence of FBP. At pH 7.5, the kcat and Km values for NADH and pyruvate could not be determined due to the fact that the enzyme activities did not reach the plateau in the concentration range of substrates. Since the enzymatic activity of the mutant increased linearly related to the increasing concentration of substrates, data for the steady-state kinetics were fitted to Eq. (2):
| (2) |
4.5. VUVCD measurements
CD spectroscopy is obviously useful for the estimation of the secondary-structure content of proteins, and the accuracy is improved by the extension of CD measurements to the VUV region below 190 nm [33–39]. The VUVCD spectra of the Ec. mundtii LDH-2 in a 20 mM sodium phosphate buffer (pH 7.5) were measured from 260 to 175 nm using the synchrotron-radiation VUVCD spectrophotometer constructed at Hiroshima Synchrotron Radiation Center and an assembled-type optical cell [40,41] at 25 °C. The path length of the cell was adjusted with a Teflon spacer to 10 μm. The details of the optical devices of the spectrophotometer and the optical cell are available elsewhere [41]. All of the VUVCD spectra were recorded with a 0.25-mm slit, a 4-s time constant, a 20-nm min−1 scan speed, and 4 accumulations. The ellipticity was reproducible within an error margin of 5%, which was mainly attributable to noise and to inaccuracy in the optical path length. The secondary structures of LDH-2 were analyzed using the SELCON3 program [42], which was improved by using the VUVCD spectra down to 160 nm of the 31 reference proteins with known X-ray structures [36,37]. The secondary structures in the crystal form were assigned using the DSSP program [43] based on the numbers and positions of hydrogen bonds between peptide groups. In this analysis, the 310-helix was classified as an unordered structure.
Author contribution statement
Y.M. planned experiments, performed experiments, analyzed data, and wrote the paper; M.M. performed experiments; K.M. performed experiments, analyzed data, and wrote the paper; K.O., M.N., F.H. and T.K. analyzed data; M.S. planned experiments, analyzed data, and wrote the paper.
Acknowledgements
We are grateful to Professor S. Kuramitsu, Osaka University, and his colleagues for data collection at the BL26B2 beamline in SPring-8 with the mail-in data collection system. We wish to thank the Analysis Center of Life Science, Hiroshima University for the use of their facilities. This study has been funded by the ANDERSEN Institute of Bread & Life (Y.M.). The VUVCD measurements were done at the Hiroshima Synchrotron Radiation Center with the approval of the institution (No. 13-B-40).
Appendix A. Supplementary data
This document contains PDB file.
This document contains PDB file.
References
- 1.Lindgren S.E., Dobrogosz W.J. Antagonistic activities of lactic acid bacteria in food and feed fermentations. FEMS Microbiol. Rev. 1990;7:149–163. doi: 10.1111/j.1574-6968.1990.tb04885.x. [DOI] [PubMed] [Google Scholar]
- 2.Garvie E.I. Bacterial lactate dehydrogenases. Microbiol. Rev. 1980;44:106–139. doi: 10.1128/mr.44.1.106-139.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piontek K., Chakrabarti P., Schär H.P., Rossmann M.G., Zuber H. Structure determination and refinement of Bacillus stearothermophilus lactate dehydrogenase. Proteins. 1990;7:74–92. doi: 10.1002/prot.340070108. [DOI] [PubMed] [Google Scholar]
- 4.Wigley D.B., Gamblin S.J., Turkenburg J.P., Dodson E.J., Piontek K., Muirhead H., Holbrook J.J. Structure of a ternary complex of an allosteric lactate dehydrogenase from Bacillus stearothermophilus at 2.5 Å resolution. J. Mol. Biol. 1992;223:317–335. doi: 10.1016/0022-2836(92)90733-z. [DOI] [PubMed] [Google Scholar]
- 5.Cameron A.D., Roper D.I., Moreton K.M., Mirhead H., Holbrook J.J., Wigley D.B. Allosteric activation in Bacillus stearothermophilus lactate dehydrogenase investigated by an X-ray crystallographic analysis of a mutant designed to prevent tetramerization of the enzyme. J. Mol. Biol. 1994;238:615–625. doi: 10.1006/jmbi.1994.1318. [DOI] [PubMed] [Google Scholar]
- 6.Clarke A.R., Waldman A.D.B., Munro I., Holbrook J.J. Changes in the state of subunit association of lactate dehydrogenase from Bacillus stearothermophilus. Biochim. Biophys. Acta. 1985;828:375–379. doi: 10.1016/0167-4838(85)90319-x. [DOI] [PubMed] [Google Scholar]
- 7.Clarke A.R., Atkinson T., Campbell J.W., Holbrook J.J. The assembly mechanism of the lactate dehydrogenase tetramer from Bacillus stearothermophilus; the equilibrium relationships between quaternary structure and the binding of fructose 1,6-bisphosphate, NADH and oxamate. Biochim. Biophys. Acta. 1985;829:387–396. [Google Scholar]
- 8.Iwata S., Ohta T. Molecular basis of allosteric activation of bacterial l-lactate dehydrogenase. J. Mol. Biol. 1993;230:21–27. doi: 10.1006/jmbi.1993.1122. [DOI] [PubMed] [Google Scholar]
- 9.Iwata S., Kamata K., Minowa T., Ohta T. T and R states in the crystals of bacterial l-lactate dehydrogenase reveal the mechanism for allosteric control. Nat. Struct. Biol. 1994;1:176–185. doi: 10.1038/nsb0394-176. [DOI] [PubMed] [Google Scholar]
- 10.Holland R., Pritchard G.G. Regulation of the l-lactate dehydrogenase from Lactobacillus casei by fructose-1,6-diphosphate and metal ions. J. Bacteriol. 1975;121:777–784. doi: 10.1128/jb.121.3.777-784.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon G.L., Doelle H.W. Purification, properties and immunological relationship of l(+)-lactate dehydrogenase from Lactobacillus casei. Eur. J. Biochem. 1976;67:543–555. doi: 10.1111/j.1432-1033.1976.tb10720.x. [DOI] [PubMed] [Google Scholar]
- 12.Hensel R., Mayr U., Stetter K.O., Kandler O. Comparative studies of lactic acid dehydrogenases in lactic acid bacteria. I. Purification and kinetics of the allosteric l-lactic acid dehydrogenase from Lactobacillus casei ssp. casei and Lactobacillus curvatus. Arch. Microbiol. 1977;112:81–93. doi: 10.1007/BF00446658. [DOI] [PubMed] [Google Scholar]
- 13.Hensel R., Mayr U., Woenckhause C. Affinity labeling of the allosteric site of the l-lactate dehydrogenase of Lactobacillus casei. Eur. J. Biochem. 1983;135:359–365. doi: 10.1111/j.1432-1033.1983.tb07662.x. [DOI] [PubMed] [Google Scholar]
- 14.Arai K., Ishimitsu T., Fushinobu S., Uchikoba H., Matsuzawa H., Taguchi H. Active and inactive state structures of unliganded Lactobacillus casei allosteric l-lactate dehydrogenase. Proteins. 2010;78:681–694. doi: 10.1002/prot.22597. [DOI] [PubMed] [Google Scholar]
- 15.Taguchi H., Ohta T. Unusual amino acid substitution in the anion-binding site of Lactobacillus plantarum non-allosteric l-lactate dehydrogenase. Eur. J. Biochem. 1992;209:993–998. doi: 10.1111/j.1432-1033.1992.tb17373.x. [DOI] [PubMed] [Google Scholar]
- 16.Uchikoba H., Fushinobu S., Wakagi T., Konno M., Taguchi H., Matsuzawa H. Crystal structure of non-allosteric l-lactate dehydrogenase from Lactobacillus pentosus at 2.3 Å resolution: specific interactions at subunit interfaces. Proteins. 2002;46:206–214. doi: 10.1002/prot.1165. [DOI] [PubMed] [Google Scholar]
- 17.Klaenhammer T.R. Genetics of bacteriocins produced by lactic acid bacteria. FEMS Microbiol. Rev. 1993;12:39–85. doi: 10.1111/j.1574-6976.1993.tb00012.x. [DOI] [PubMed] [Google Scholar]
- 18.Jack R.W., Tagg J.R., Ray B. Bacteriocins of gram-positive bacteria. Microbiol. Rev. 1995;59:171–200. doi: 10.1128/mr.59.2.171-200.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeon H.J., Noda M., Matoba Y., Kumagai T., Sugiyama M. Crystal structure and mutagenic analysis of a bacteriocin immunity protein, Mun-im. Biochem. Biophys. Res. Commun. 2009;378:574–578. doi: 10.1016/j.bbrc.2008.11.093. [DOI] [PubMed] [Google Scholar]
- 20.Abdel-Rahman M.A., Tashiro Y., Zendo T., Shibata K., Sonomoto K. Isolation and characterisation of lactic acid bacterium for effective fermentation of cellobiose into optically pure homo l-(+)-lactic acid. Appl. Microbiol. Biotechnol. 2011;89:1039–1049. doi: 10.1007/s00253-010-2986-4. [DOI] [PubMed] [Google Scholar]
- 21.Abdel-Rahman M.A., Tashiro Y., Zendo T., Hanada K., Shibata K., Sonomoto K. Efficient homofermentative l-(+)-lactic acid production from xylose by a novel lactic acid bacterium, Enterococcus mundtii QU 25. Appl. Environ. Microbiol. 2011;77:1892–1895. doi: 10.1128/AEM.02076-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolotin A., Wincker P., Mauger S., Jaillon O., Malarme K., Weissenbach J., Ehrlich S.D., Sorokin A. The complete genome sequence of the lactic acid bacterium Lactococcus lactis ssp. lactis IL1403. Genome Res. 2001;11:731–753. doi: 10.1101/gr.169701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleerebezem M., Boekhorst J., van Kranenburg R., Molenaar D., Kuipers O.P., Leer R., Tarchini R., Peters S.A., Sandbrink H.M., Fiers M.W., Stiekema W., Lankhorst R.M., Bron P.A., Hoffer S.M., Groot M.N., Kerkhoven R., de Vries M., Ursing B., de Vos W.M., Siezen R.J. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl. Acad. Sci. U.S.A. 2003;100:1990–1995. doi: 10.1073/pnas.0337704100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourgogne A., Garsin D.A., Qin X., Singh K.V., Sillanpaa J., Yerrapragada S., Ding Y., Dugan-Rocha S., Buhay C., Shen H., Chen G., Williams G., Muzny D., Maadani A., Fox K.A., Gioia J., Chen L., Shang Y., Arias C.A., Nallapareddy S.R., Zhao M., Prakash V.P., Chowdhury S., Jiang H., Gibbs R.A., Murray B.E., Highlander S.K., Weinstock G.M. Large scale variation in Enterococcus faecalis illustrated by the genome analysis of strain OG1RF. Genome Biol. 2008;9:R110. doi: 10.1186/gb-2008-9-7-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaspar P., Neves A.R., Shearman C.A., Gasson M.J., Baptista A.M., Turner D.L., Soares C.M., Santos H. The lactate dehydrogenases encoded by the ldh and ldhB genes in Lactococcus lactis exhibit distinct regulation and catalytic properties – comparative modeling to probe the molecular basis. FEBS J. 2007;274:5924–5936. doi: 10.1111/j.1742-4658.2007.06115.x. [DOI] [PubMed] [Google Scholar]
- 26.Jönsson M., Saleihan Z., Nes I.F., Holo H. Construction and characterization of three lactate dehydrogenase-negative Enterococcus faecalis V583 mutants. Appl. Environ. Microbiol. 2009;75:4901–4903. doi: 10.1128/AEM.00344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feldman-Salit A., Hering S., Messiha H.L., Veith N., Cojocaru V., Sieg A., Westerhoff H.V., Kreikemeyer B., Wade R.C., Fiedler T. Regulation of the activity of lactate dehydrogenases from four lactic acid bacteria. J. Biol. Chem. 2013;288:21295–21306. doi: 10.1074/jbc.M113.458265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Collaborative Computational Project Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. Sect. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 30.Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., Read R.J., Rice L.M., Simonson T., Warren G.L. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 31.Brünger A.T. The free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 32.McRee D.E. A visual protein crystallographic software system for X11/XView. J. Mol. Graph. 1992;10:44–46. [Google Scholar]
- 33.Toumadje A., Alcorn S.W., Johnson W.C., Jr. Extending CD spectra of proteins to 168 nm improves the analysis for secondary structure. Anal. Biochem. 1992;200:321–331. doi: 10.1016/0003-2697(92)90473-k. [DOI] [PubMed] [Google Scholar]
- 34.Jones G.R., Clarke D.T. Applications of extended ultra-violet circular dichroism spectroscopy in biology and medicine. Faraday Discuss. 2004;126:223–236. doi: 10.1039/b305743g. [DOI] [PubMed] [Google Scholar]
- 35.Wallace B.A., Wien F., Miles A.J., Lees J.G., Hoffmann S.V., Evans P., Wistow G.J., Slingsby C. Biomedical applications of synchrotron radiation circular dichroism spectroscopy: identification of mutant proteins associated with disease and development of a reference database for fold motifs. Faraday Discuss. 2004;126:237–243. doi: 10.1039/b306055c. [DOI] [PubMed] [Google Scholar]
- 36.Matsuo K., Yonehara R., Gekko K. Secondary-structure analysis of proteins by vacuum-ultraviolet circular dichroism spectroscopy. J. Biochem. 2004;135:405–411. doi: 10.1093/jb/mvh048. [DOI] [PubMed] [Google Scholar]
- 37.Matsuo K., Yonehara R., Gekko K. Improved estimation of the secondary structures of proteins by vacuum-ultraviolet circular dichroism spectroscopy. J. Biochem. 2005;138:79–88. doi: 10.1093/jb/mvi101. [DOI] [PubMed] [Google Scholar]
- 38.Matsuo K., Watanabe H., Tate S., Tachibana H., Gekko K. Comprehensive secondary-structure analysis of disulfide variants of lysozyme by synchrotron-radiation vacuum-ultraviolet circular dichroism. Proteins. 2009;77:191–201. doi: 10.1002/prot.22430. [DOI] [PubMed] [Google Scholar]
- 39.Matsuo K., Namatame H., Taniguchi M., Gekko K. Membrane-induced conformational change of α1-acid glycoprotein characterized by vacuum-ultraviolet circular dichroism spectroscopy. Biochemistry. 2009;48:9103–9111. doi: 10.1021/bi901184r. [DOI] [PubMed] [Google Scholar]
- 40.Ojima N., Sakai K., Matsuo K., Matsui T., Fukazawa T., Namatame H., Taniguchi M., Gekko K. Vacuum-ultraviolet circular dichroism spectrophotometer using synchrotron radiation: optical system and on-line performance. Chem. Lett. 2001;30:522–523. [Google Scholar]
- 41.Matsuo K., Sakai K., Matsushima Y., Fukuyama T., Gekko K. Optical cell with a temperature-control unit for a vacuum-ultraviolet circular dichroism spectrophotometer. Anal. Sci. 2003;19:129–132. doi: 10.2116/analsci.19.129. [DOI] [PubMed] [Google Scholar]
- 42.Sreerama N., Woody R.W. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 43.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 44.Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eventoff W., Rossmann M.G., Taylor S.S., Torff H.J., Meyer H., Keil W., Kiltz H.H. Structural adaptations of lactate dehydrogenase isozymes. Proc. Natl. Acad. Sci. U.S.A. 1977;74:2677–2681. doi: 10.1073/pnas.74.7.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abad-Zapatero C., Griffith J.P., Sussman J.L., Rossmann M.G. Refined crystal structure of dogfish M4 apo-lactate dehydrogenase. J. Mol. Biol. 1987;198:445–467. doi: 10.1016/0022-2836(87)90293-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This document contains PDB file.
This document contains PDB file.