

Abstract
Mutations in SCN1A and other ion channel genes can cause different epileptic phenotypes, but the precise mechanisms underlying the development of hyperexcitable networks are largely unknown. Here, we present a multisystem analysis of an SCN1A mouse model carrying the NaV1.1-R1648H mutation, which causes febrile seizures and epilepsy in humans. We found a ubiquitous hypoexcitability of interneurons in thalamus, cortex, and hippocampus, without detectable changes in excitatory neurons. Interestingly, somatic Na+ channels in interneurons and persistent Na+ currents were not significantly changed. Instead, the key mechanism of interneuron dysfunction was a deficit of action potential initiation at the axon initial segment that was identified by analyzing action potential firing. This deficit increased with the duration of firing periods, suggesting that increased slow inactivation, as recorded for recombinant mutated channels, could play an important role. The deficit in interneuron firing caused reduced action potential-driven inhibition of excitatory neurons as revealed by less frequent spontaneous but not miniature IPSCs. Multiple approaches indicated increased spontaneous thalamocortical and hippocampal network activity in mutant mice, as follows: (1) more synchronous and higher-frequency firing was recorded in primary neuronal cultures plated on multielectrode arrays; (2) thalamocortical slices examined by field potential recordings revealed spontaneous activities and pathological high-frequency oscillations; and (3) multineuron Ca2+ imaging in hippocampal slices showed increased spontaneous neuronal activity. Thus, an interneuron-specific generalized defect in action potential initiation causes multisystem disinhibition and network hyperexcitability, which can well explain the occurrence of seizures in the studied mouse model and in patients carrying this mutation.
Keywords: epilepsy, genetics, ion channel, mouse model, network activity
Introduction
Voltage-gated ion channels play a pivotal role in initiation and conduction of action potentials (APs). Four subunits constitute the major brain isoforms of voltage-gated Na+ channels (NaVs). Among these, the most relevant gene affected in neuropsychiatric disorders is SCN1A, encoding the NaV1.1 subunit. SCN1A mutations cause a variety of symptoms including febrile seizures and epilepsy, ataxia, mental decline, and headache (Marini and Mantegazza, 2010). Disorders caused by SCN1A mutations include generalized/genetic epilepsy with febrile seizures plus (GEFS+; Escayg et al., 2000), Dravet syndrome (DS; Claes et al., 2001), and familial hemiplegic migraine (Dichgans et al., 2005). DS is a severe epileptic encephalopathy with pharmacoresistant seizures and mental decline starting in the first or second year of life, whereas GEFS+ is characterized by milder febrile and afebrile seizures, which mostly can be controlled by antiepileptic drugs. For these two disorders, a genotype–phenotype relationship has been established: DS is mainly associated with deleterious de novo mutations that induce haploinsufficiency; whereas, GEFS+ is associated with missense mutations altering NaV1.1 protein function in different ways (Zuberi et al., 2011).
Accordingly, gene-targeted Scn1a mouse models expressing truncated NaV1.1 proteins have been used as models for DS. Indeed, many clinical features of DS are mimicked by those models, such as severe epilepsy, ataxia, and premature death. The first two studies of such mice (Yu et al., 2006; Ogiwara et al., 2007) revealed evidence for a reduced Na+ channel expression in interneurons as a possible epileptogenic mechanism.
Despite these results, the pathomechanisms underlying increased excitability in human epilepsies caused by point mutations in the SCN1A gene are not understood. It is still unknown (1) whether NaV1.1 missense mutations cause a gain-of-function or loss-of-function with respect to neuronal activity, and in which neurons this effect is most prevalent; (2) in which neuronal compartments mutant channels exert their main pathophysiological effects; and (3) what impact such mutations have on complex network dynamics, which has been addressed only in a single study in NaV1.1 knock-out mice (Liautard et al., 2013).
We set out to study the consequences of an epileptogenic SCN1A mutation on a cellular and network level, using acute brain slices of a knock-in mouse model. To identify the most important consequences on the neuronal dysfunction, we chose a mutation with subtle changes of channel function, p.R1648H (Alekov et al., 2000; Spampanato et al., 2001; Lossin et al., 2002). This mutation has been previously identified in a large family with GEFS+ with 13 affected members exhibiting either febrile or afebrile generalized tonic–clonic or absence seizures (Escayg et al., 2000). Whereas homozygous (Scn1aRH/RH) animals exhibit spontaneous generalized seizures and premature death from postnatal day 16 (P16) on, heterozygous (Scn1aRH/+) animals present less frequent seizures and no change in life span, comparable to that in patients (Martin et al., 2010).
Our results reveal a widespread disinhibition with reduced intrinsic excitability of GABAergic inhibitory neurons, which is presumably due to impaired AP initiation in axon initial segments (AISs). These defects lead to an increased spontaneous neuronal network activity and can explain the generation of epileptic seizures.
Materials and Methods
Experimental animals.
The mouse strain used in this study, carrying the R1648H mutation in NaV1.1 on a C57BL/6 background, has been described previously (Martin et al., 2010). Wild-type (WT) littermates were used as controls. To label inhibitory neurons, we crossed the R1648H mouse strain with glutamate decarboxylase (GAD67)-GFP knock-in animals (same C57BL/6 background). In GAD67-GFP knock-in animals, GABAergic inhibitory neurons positive for parvalbumin, calretinin, and somatostatin are colocalized, with GFP with parvalbumin-positive cells being the most abundant in the neocortex (Tamamaki et al., 2003). Experiments were approved by the local Animal Care and Use Committee (Regierungspraesidium Tuebingen, Tuebingen, Germany; and Direction Départementale de la Protection des Populations Cote d'Azur, Nice, France).
Preparation and maintenance of slices.
Thalamocortical slices from P14 to P20 mice were obtained with a Microm HM 650 V vibratome (Thermo Fisher Scientific) using procedures previously described (Agmon and Connors, 1991). Horizontal hippocampal slices of the same age were used for recordings in the CA1 region. Animals were anesthetized with Sevorane (active ingredient Sevofluran, Abbott Laboratories) or isoflurane (Axience) and decapitated. Brains were removed quickly and cut in ice-cold artificial CSF (ACSF) with the following composition (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 7 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 10 glucose, pH 7.4, equilibrated with 95% O2/5% CO2. Slices were stored to recover at 36°C for 1 h before recordings. For storage and recording, the following solution was used (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 10 glucose, pH 7.4, equilibrated with 95% O2/5% CO2 (standard ACSF).
Immunohistochemistry.
For morphological studies, patched cells were filled with 0.2% biocytin or Lucifer yellow. Slices were fixed for 1 h with 4% paraformaldehyde and incubated for 1 h in block medium (PBS with 6% normal goat serum and 0.6% Triton X-100) at room temperature. After washing steps performed with PBS, slices were incubated with a monoclonal antibody against GAD67 (Millipore Bioscience Research Reagents; 1:500 dilution) for 1 h at room temperature and at 4°C overnight. After washing steps performed with PBS, slices were incubated for 1 h at room temperature with secondary Alexa Fluor 488-conjugated goat anti-mouse antibodies (dilution 1:500; Invitrogen) and cy3-conjugated streptavidin (Jackson ImmunoResearch; dilution 1:1000) when neurons were filled with biocytin or with secondary Alexa Fluor 568-conjugated goat anti-mouse antibodies only (dilution 1:500; Invitrogen) when cells were filled with Lucifer yellow. Slices were stained with DAPI (Sigma-Aldrich; dilution 1:10,000) to identify the nuclei. After washing, slices were air dried and mounted with Mowiol (polyvinyl alcohol; Sigma-Aldrich) and visualized on an Axiophot 2 microscope (Zeiss).
Mutagenesis.
Site-directed mutagenesis was performed to engineer the mutation into the human NaV1.1 channel (GenBank sequence NM_006920.4 subcloned into the pCDM8 vector; Cestèle et al., 2008) using QuikChange (Agilent Technologies; primers are available upon request). The mutant cDNA was fully resequenced before use in experiments to confirm the introduced mutation and exclude any additional sequence alterations. The human Na+ channel subunits hβ1 and hβ2 in the pCLH vector were provided by GlaxoSmithKline. We exchanged the Hygromycin coding region in the vector with the sequence coding for either enhanced GFP (EGFP) or CD8 marker genes to obtain pCLH-hβ1-EGFP and pCLH-hβ2-CD8 (Liao et al., 2010).
Transfection and expression in tsA201 cells.
Human tsA201 cells were cultured at 37°C, with 5% CO2 humidified atmosphere, and were grown in DMEM (Invitrogen) plus 10% (v/v) fetal bovine serum. Transfections using Mirus TransIT-LT1 Transfection Reagent were performed for transient expression of wild-type or mutant Na+ channel α-subunits together with β1- and β2-subunits in tsA201 cells. For the coexpression, 2.4 μg of total DNA was transfected in a molar ratio of 1:1:1 as described previously (Liao et al., 2010). Cells positive for CD8 antigen and EGFP fluorescence were used for electrophysiological recordings.
Primary cell culture.
Hippocampal and cortical neurons were isolated for multielectrode arrays (MEAs) or voltage-clamp recordings from embryonic day 17 (E17) R1648H or R1648H × GAD67 mouse brains, respectively. Pregnant females were killed using CO2, and embryos were quickly taken out and decapitated. Using microsurgical dissection methods, the hippocampus or the cerebral cortex was isolated. Tissues were washed three times with 4°C magnesium- and calcium-free HBSS (PAA Laboratories GmbH) before treatment for 15 min with 2.5% trypsin. Subsequently, tissues were rinsed in DMEM with fetal bovine serum (Biochrom AG), l-glutamine (Invitrogen) and penicillin/streptomycin (Invitrogen) to block the trypsin reaction. Single neurons were obtained by mechanical dissociation using a pipette and a cell strainer (Becton Dickinson). For MEA recordings, 150,000 cells in 110 μl of solution were plated on the arrays. MEAs were coated with poly-d-lysine solution (5 mg poly-d-lysine in 100 ml of HBSS, filtered with a 0.45 μm filter) and 500 μl of DMEM with fetal bovine serum (Biochrom AG), l-glutamine (Invitrogen) and penicillin/streptomycin (Invitrogen). One hundred ten microliters of the cells in medium were plated on MEAs and allowed to settle down for 4 h. After that time, MEAs were flooded with Neurobasal culture medium (Invitrogen) supplemented with B27 (Invitrogen), glutamine, and penicillin/streptomycin.
For nucleated patch-clamp experiments, the dissociated neurons were plated on 13 mm coverslips in 24-well cell culture plates (Greiner Bio-One). The coverslips were coated with a poly-d-lysine solution (5 mg of poly-d-lysine in 100 ml of HBSS, filtered by using a 0.45 μm filter) and 500 μl of DMEM with fetal bovine serum (Biochrom AG), l-glutamine (Invitrogen), and penicillin/streptomycin (Invitrogen). After allowing the neurons to settle down for 5 h in a 37°C incubator with 5% CO2 supply, the solution was replaced with Neurobasal culture medium (Invitrogen) supplemented with B27 (Invitrogen), glutamine, and penicillin/streptomycin.
Since homozygous animals with EGFP-labeled interneurons were not viable, we could use only Scn1aRH/+ animals with EGFP-labeled interneurons for our Na+ current analysis.
Electrophysiological recordings.
Whole-cell patch-clamp recordings of inhibitory and/or excitatory neurons within thalamus, cortex, or hippocampus were performed at 34°C using Axopatch 200B, Multiclamp 700B (Molecular Devices), or BVC 700A (Dagan) amplifiers; a DigiData 1420 digitizer (Molecular Devices); and pClamp 10.3 software (Molecular Devices). Slices were positioned in a submerged-type recording chamber (Luigs & Neumann or Warner Instruments), continuously superfused with ACSF, and visualized with an Axioskop 2FS (Zeiss) or an Eclipse FN1 (Nikon) microscope. Pipettes were pulled from borosilicate glass (Science Products GmbH) using a Sutter P97 Puller (Sutter Instruments), with resistances of 3–5 MΩ. For current-clamp experiments, cells were held at −70 mV, and the patch pipette solution contained the following (in mm): 5 KCl, 4 ATP-Mg, 10 phosphocreatine, 0.3 GTP-Na, 10 HEPES, 125 K-gluconate, 2 MgCl2, 10 EGTA, and 0.2% biocytin or Lucifer yellow with a final pH of 7.2 and an osmolarity of 290 mOsm/kg. To record spontaneous and miniature IPSCs or tonic GABAA current, the AMPA and kainate receptor antagonist DNQX (10 μm; Sigma-Aldrich) and the NMDA receptor antagonist APV (30 μm) were added to the standard ACSF; patch pipettes contained the following (in mm): 105 CsCl, 35 CsOH, 10 HEPES, 10 EGTA, 10 phosphocreatine, 4 ATP-Mg, 0.3 GTP-Na, 14 d-mannitol, 0.2% biocytin, or Lucifer yellow. Membrane voltage was clamped to −70 mV, and IPSCs were recorded over 5 min epochs. For current and voltage-clamp experiments, the sampling rate was 100 kHz and the data were low-pass filtered at 30 kHz. Series resistance (<20 MΩ) was monitored during the experiment. Cells showing unstable series resistance or resting membrane potential were discarded.
Nucleated patch recordings were performed using an Axopatch 200B amplifier (in primary cortical cultures) and a multiclamp 700B amplifier (in brain slices), a DigiData 1420 digitizer, and pClamp 10 data acquisition software (Molecular Devices), as has been described previously (Liao et al., 2010; Lauxmann et al., 2013). For whole-cell patch-clamp recordings of tsA201 cells, an Axopatch 200B amplifier, a DigiData 1320A digitizer, and pCLAMP 8 data acquisition software were used (Molecular Devices), and cells were visualized with an inverted microscope (Axio Vert.A1, Zeiss). Leakage and capacitive currents were automatically subtracted using a prepulse protocol (−P4). Currents were filtered at 5 kHz and digitized at 20 kHz. All recordings were performed at room temperature of 21–23°C. Borosilicate glass pipettes were fire polished with a final tip resistance of 1–2 MΩ when filled with internal recording solution.
The extracellular solution used for nucleated patches of primary cortical cultures contained the following (in mm): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 20 tetraethylammonium (TEA)-Cl, 5 CsCl, 0.1 CdCl2, and 10 HEPES; pH was adjusted to 7.3 with CsOH. Pipettes were filled with an intracellular solution containing the following (in mm): 140 CsF, 10 NaCl, 1 EGTA, and 10 HEPES, with pH was adjusted to 7.3 with CsOH. The ACSF for recording Na+ currents in nucleated patches in acute slices contained the following (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 25 glucose, bubbled with 95% O2 and 5% CO2. TEA (20 mm) was added to block voltage-gated K+ channels. Pipettes were filled with a Cs+-rich internal solution, containing the following (in mm): 140 CsCl, 10 EGTA, 2 MgCl2, 2 ATP-Na2, and 10 HEPES, with pH adjusted to 7.3 with CsOH.
For recording the persistent sodium current (INaP) in hippocampal and nucleus reticularis of the thalamus (nRt) inhibitory neurons in acute slices, the ACSF contained the following (in mm): 50 NaCl, 90 TEA-Cl, 10 HEPES-free acid, 2 CaCl2, 2 MgCl2, 3.5 KCl, 3 CsCl, 0.2 CdCl2, 4 4-AP, and 25 glucose. pH was adjusted to 7.4 using NaOH. The intracellular solution contained the following (in mm): 110 CsF, 10 HEPES-Na, 11 EGTA, 2 MgCl2, 0.5 GTP-Na, and 2 ATP-Na2. pH was adjusted to 7.3 with CsOH. For eliciting persistent sodium currents, voltage ramps from −80 to 20 mV with a velocity of 25 mV/s were used. In each case, currents were recorded in ACSF and in the presence of TTX to subtract remaining potassium or calcium currents.
Na+ currents of 1–12 nA were recorded from transfected tsA201 cells at least 10 min after establishing the whole-cell configuration. The pipette solution contained the following (in mm): 105 CsF, 35 NaCl, 10 EGTA, and 10 HEPES, pH 7.4. The bath solution contained the following (in mm): 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.4.
Local extracellular field potentials were recorded using glass electrodes in slices continuously perfused with ACSF bubbled with 95% O2/5% CO2 and maintained in an interface chamber at 34°C (Campden Instruments) with Multiclamp 700B or DAGAN EX4–400 amplifiers. Electrical stimulations were applied with bipolar electrodes (FHC Inc.), a SIU91A isolator (Cygnus Technologies), pCLAMP software, and a DigiData 1420 digitizer, as in Liautard et al. (2013). Sampling rate was 10 kHz and data were low-pass filtered at 4 kHz. Recording electrodes were positioned in the S1 cortex [layer IV (LIV) and layer V (LV)] and in the thalamus [nRt and ventrobasal part of the thalamus (VB)]. Paired-pulse stimulations of the VB, at the intensity eliciting the maximal LIV response, consisted of the application of two pulse stimuli with increasing interpulse interval (IPI); analysis was performed on the monosynaptic response (Agmon and Connors, 1991). For coastline analysis, the coastline index (Korn et al., 1987) was the length of the line describing the monosynaptic responses, which was calculated by applying Pythagoras' theorem to each pair of data points, computed with Origin version 8.5 software (OriginLab) as follows:
with xi and yi being the time (in milliseconds) and amplitude (in millivolts), respectively, of each data point (i).
The paired-pulse ratio was calculated by dividing the amplitude, or the coastline index, of the second monosynaptic response in the pair by the amplitude, or the coastline index, of the first one. For displaying high-frequency oscillation (HFO) traces, data were bandpass filtered off-line at 40–200 and 200–600 Hz with pClamp Bessel eight-pole filters. For analysis of HFOs, we also subtracted the contribution of the noise to the filtered traces: for both 40–200 and 200–600 Hz filters, we computed the absolute area of the oscillations with Origin version 8.5 software and subtracted the absolute area of an eventless period of the same duration immediately preceding the activity under analysis. To take into account the possible bias in HFO areas induced by the amplitude of the spontaneous activity (HFOs may appear larger in slices of Scn1aRH/+ animals just because amplitudes of spontaneous activities are larger), we also compared normalized noise-subtracted HFO absolute areas, dividing the absolute area of the HFO by the absolute area of the corresponding spontaneous activity (0.1 Hz–1 kHz filtered). For some recordings, results were confirmed obtaining HFOs by filtering traces (40–200 and 200–600 Hz) with a fast Fourier transform bandpass filter (Origin version 8.5 software).
Voltage-clamp protocols and data analysis for transient Na+ currents recorded from neurons and tsA201 cells.
For the voltage-clamp protocols used, recorded currents from nucleated patches were averaged up to 10 times to obtain homogeneous results.
The activation curve (conductance–voltage relationship) was derived from the current–voltage relationship curves obtained by plotting the peak current over various step depolarizations [7.5 mV steps from the holding potential of −90/−140 mV (neurons/tsA-cells)] according to
 |
with g being the conductance, I the recorded peak current at test potential V, and Vrev the apparent observed Na+ reversal potential.
The voltage dependence of activation was fit with the following Boltzmann function:
 |
with gmax being the maximal conductance, V1/2 the voltage of half-maximal activation, and kV a slope factor. Steady-state inactivation was determined using 300 ms conditioning pulses to various potentials followed by the test pulse to −20 mV at which the peak current reflected the percentage of non-inactivated channels. A standard Boltzmann function was fit to the inactivation curves:
 |
with Imax being the maximal current amplitude.
Recovery from fast inactivation was recorded from cells depolarized to −20 mV for 100 ms to inactivate all Na+ channels and then repolarized to −90/−140 mV (neurons/tsA-cells) recovery potential for increasing duration. A second-order exponential function with an initial delay was best fit to the time course of recovery from inactivation. The faster time constant with the much larger relative amplitude, τrec, is shown for data evaluation in Figure 2G.
Figure 2.

Transient Na+ currents of hippocampal inhibitory neurons recorded in acute brain slices and of transfected tsA201 cells. A, Original recordings of Na+ currents from nucleated patches derived from inhibitory neurons in CA1 stratum oriens of WT (left), heterozygous (middle), and homozygous (right) animals. B, Gating parameters of Na+ currents as recorded in A. Left, Current density; middle, steady-state activation and fast inactivation curves; right, recovery from fast inactivation. Voltage-clamp protocols and data evaluation procedures are given in the Materials and Methods section. Data are shown as means ± SEM. There were no significant differences among the three genotypes. C, Representative whole-cell patch-clamp recordings of tsA cells transfected with SCN1A-WT DNA (top) and SCN1A-R1648H DNA (bottom), respectively. D, Current density–voltage plots for hNaV1.1-WT and R1648H. See also Table 2. E, Mean voltage dependence of steady-state Na+ channel activation and fast inactivation with lines revealing Boltzmann functions. F, Voltage dependence of the persistent currents showing a largely increased persistent current for the R1648H mutation. Current amplitudes were recorded at the end of a 70 ms depolarization to 0 mV and are normalized to the peak amplitude (steady-state current/initial peak current). G, Time course of recovery from fast inactivation determined at 100 mV revealing acceleration for R1648H mutant channels. Lines represent fits of exponential functions. H, Mean voltage dependence of steady-state slow inactivation fitted to a standard Boltzman function (lines): V1/2= −71.8 ± 0.7 mV for WT (n = 7) vs V1/2= −85.8 ± 0.9 mV for R1648H (n = 5); p < 0.05, Student's t test.
Entry into and steady-state slow inactivation for tSA cells were characterized using cumulative protocols (Alekov et al., 2000). For steady-state slow inactivation, 30 s conditioning pulses starting at a holding potential of −140 mV in 10 mV steps up to 10 mV were used. Each of these steps was followed by a 20 ms hyperpolarization to −140 mV to let channels recover from fast inactivation and a test pulse to −20 mV. Curves were fitted to a standard Boltzmann function as for fast inactivation. To measure the entry into slow inactivation, cells were held at −140 mV and depolarized to 0 mV for increasing durations, repolarized for 100 ms to −100 mV to let the channels recover from fast inactivation, and then depolarized again to −10 mV to determine the fraction of slow inactivated channels. The time course of slow inactivation was best fitted by a first-order exponential function.
MEAs.
MEAs were used to record activity from primary hippocampal neurons prepared from E17 mice after 3 weeks in culture. MEAs had a square grid of 60 planar Ti/TiN electrodes of 30 μm diameter and 200 μm spacing. Electrodes had an input impedance of 30–50 kΩ according to the specifications of the manufacturer (Multi Channel Systems). Signals from all 60 electrodes were simultaneously sampled at 25 kHz, visualized, and stored using the standard software MC_Rack provided by Multi Channel Systems. Spike detection was performed off-line by the SPANNER software suite (RESULT Medical; see also Illes et al., 2009). Synchronous network activity was analyzed by population burst (PB) detection using custom-built Matlab software.
Spikes from all electrodes were aggregated in nonoverlapping 5 ms bins and smoothed by a Gaussian kernel with a 100 ms SD to obtain the population firing rate (PFR; Schock et al., 2012). PB detection was performed in a three-step procedure. First, PB candidate intervals were identified whenever the PFR exceeded the slowly varying 1 s firing rate average. In a second step, the actual PBs from these candidate intervals were selected, if (1) their peak firing rate exceeded 3 SDs of the recording PFR, (2) their peak firing rate exceeded 10% of the average of the top five peaks, and (3) at least three electrodes contributed. Finally, neighboring PBs were merged if they were <200 ms apart. PB onset and termination were defined by the actual spike time stamps of the first and last spike in the resulting time interval. In this way, synchronous network activity on time scales of tens to several hundreds of milliseconds could reliably be captured. The peak firing rate (i.e., the maximum PFR within a PB interval, reported in spikes per second) and the number of PBs per minute were analyzed. For the quantification of firing synchrony across pairs of electrodes, spikes were collected in 10-ms-wide bins and subsequently dichotomized to either zero spikes or at least one spike. Consequently, both electrodes were active during a time bin (“coincident bin”), only one electrode was active, or both were silent. Cohen's κ statistic then captures the proportion of observed coincident bins exceeding the chance expected proportion of coincidences (Woolson and Clarke, 2002). Similar to Pearson's correlation coefficient, κ values lie in the range −1 to +1. The average κ value of all electrode pairs with a firing rate of at least 30 spikes/min was calculated as a measure of the overall synchrony of a recording.
Ca2+ dye loading and imaging.
An initial 5 mm OGB-1-AM (Oregon-Green 488 BAPTA −1, cell permeant; Invitrogen) stock solution dissolved in 20% Pluronic acid, and DMSO was mixed properly and diluted with HEPES to obtain the final concentration of 8 mm. The final solution was filtered and backloaded into a pipette for pressure injection. The pipette was placed in stratum oriens of the hippocampal CA1 region to load the slice with the Ca2+ dye. To image Ca2+ transients, slices were incubated in ACSF at 36°C for 30 min to ensure dye uptake. A custom-made selective planar illumination microscope was used for Ca2+ imaging (modified after Holekamp et al., 2008).
As an excitation light source, a 488 nm optically pumped semiconductor laser (Sapphire 488 LP, Coherent) with a maximal output power of 20 mW was used. The laser intensity in the light sheet focus was ∼3% of the intensity emitted by the laser. To image the illuminated tissue a 16× water-immersion objective (CFI75 LWD, Nikon) was used mounted to a piezoelectrical objective positioning system (Mipos 500, Piezo Sytems). To filter the wavelength emitted by the calcium dye, a 536/40 emission filter (536/40, BrightLine) was used, and the filtered picture was focused with a 0.5× tube lens (InfiniTube FM-100, Infinity Photo-Optical Company). Images were recorded using a 1 megapixel electron multiplying charge-coupled device camera (iXon EM+, Andor).
ROIs were detected using a Gauss-filtering- and contour-filtering-based cell detection algorithm written in MatLab (Allène et al., 2008). Ca2+ traces were recorded as changes in mean pixel intensity in each ROI over 20 min. Ca2+ events were detected using a custom-made detection algorithm written in MatLab. Background was subtracted and photo bleaching was removed by dividing a double exponential function. Traces were high-pass filtered by subtracting the low-pass-filtered signal using a Butterworth low-pass filter. The signal over threshold was depicted as a possible event and confirmed if the unfiltered event showed an exponential decay with a time constant 500 ± 200 ms (Grewe et al., 2010). The maximum of the second derivative of the Ca2+ event was chosen as the time point for the underlying cell activity (Henze et al., 2000). Readout data were processed using Origin (version 6.1, OriginLab).
Data and statistical analysis.
Traces were displayed off-line with the Clampfit software pClamp version 10.0 (Molecular Devices). Graphics were generated using Excel (Microsoft) and Origin (version 6.1 or 8.5), statistics were performed using SigmaStat 3.1 (STATCON). All data were tested for normal distribution. For statistical evaluation, the following tests were used as indicated in the respective figure legends or in text: (1) Mann–Whitney rank-sum test for unpaired datasets of WT and Scn1aRH/+ animals; (2) Wilcoxon signed-rank nonparametric test for paired datasets (e.g., before and after the application of drugs); (3) ANOVA on ranks (Kruskal–Wallis test) with Dunn's post hoc test for comparing more than two groups; or (4) t test or one-way ANOVA when two datasets of unpaired groups were normally distributed. All data are shown as the means ± SEM. Box-and-whisker plots show medians (lines), means (square), lower and upper quartiles, minimums, and maximums. Data are shown as the mean ± SEM; n gives the number of cells or activities. For all statistical tests, significance with respect to the control is indicated on the figures using the following symbols: *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Epileptic phenotypes in patients carrying SCN1A mutations including p.R1648H are variable, including febrile, generalized tonic–clonic and absence seizures. This indicates that the human mutation functionally impairs different neuronal systems, potentially including neocortical, allocortical, and thalamic structures. Therefore, we performed a comprehensive electrophysiological analysis examining the intrinsic firing properties of inhibitory and excitatory neurons, as well as synaptic and network activity in thalamus, cortex, and hippocampus. We used acute brain slices of WT and mainly heterozygous mice (Scn1aRH/+), since they should best mimic the situation in patients. To better understand Na+ channel dysfunction, we also used homozygous Scn1aRH/RH mice in some conditions. Since seizures in GEFS+ patients with SCN1A mutations start in early childhood (Weber and Lerche, 2008), we studied the effects of the mutation in slices of animals between P14 and P20.
Intrinsic firing properties of inhibitory and excitatory thalamic neurons
In the thalamocortical network (Fig. 1A), which is involved in the generation of generalized seizures (Steriade, 2005; Beenhakker and Huguenard, 2009), we studied the neurons that were mainly involved: cortical LIV and LV inhibitory and excitatory neurons, inhibitory neurons within the nRt, as well as thalamocortical relay neurons in the VB of P14–P20 mice.
Figure 1.

Firing behavior of neurons within thalamocortical and hippocampal network. A, Schematic of thalamocortical circuit with reciprocal interconnections between thalamus and cortex. Thalamocortical relay neurons receive excitatory input and subsequently activate cortical pyramidal neurons and adjacent inhibitory GABAergic neurons. Cortical pyramidal neurons are reciprocally connected to thalamic relay neurons and excite GABAergic neurons in the nRt, which in turn inhibit each other as well as relay neurons. Right, Pictures of recorded cells in the different areas filled with Lucifer yellow or biocytin (red). Scale bars, 100 μm. B, Top, Representative whole-cell current-clamp recordings of AP series in GABAergic nRt neurons of WT (+/+, left) and heterozygous (RH, right) animals. Original recordings, top, Voltage traces upon injection of −0.05, −0.025, 0, and 0.1 nA; middle: 0.2 nA, bottom: 0.3 nA (current-clamp protocol shown below). Dashed lines show holding potential of −70 mV. All current steps are given in plots below showing the number of APs plotted vs current injection. Bottom, Number of APs is plotted vs current injection. WT: n = 23 cells from 14 animals; Scn1aRH/+: n = 37 cells from 22 animals. C, Top, Whole-cell current-clamp recordings of excitatory relay neurons of WT (left) and Scn1aRH/+ (right) animals. Original recordings are selected as in A. Bottom, Number of APs is plotted vs current injection. WT mice: n = 12 cells from six animals; Scn1aRH/+ mice: n = 12 cells from seven animals. D, Top, Whole-cell current-clamp recordings of the postinhibitory rebound in nRt neurons of WT (left) and heterozygous (right) animals during and after −400 pA current injection. Insets show amplifications highlighted with dashed boxes. Number of APs during the postinhibitory rebound (PIR) plotted vs different preceding hyperpolarizing current injections. E, Number of APs recorded in cortical layer IV (top) and hippocampal (CA1 region, stratum oriens; bottom) FS inhibitory neurons plotted vs current injections. Cortex: WT mice, n = 7 cells from five animals; Scn1aRH/+ mice, n = 9 cells from six animals. Hippocampus: WT mice, n = 7 cells from three animals; Scn1aRH/+ mice, n = 11 cells from six animals; Scn1aRH/RH mice, n = 6 cells from three animals. F, Number of APs recorded in cortical layer V (top) and hippocampal (CA1 region, stratum pyramidale; bottom) pyramidal cells plotted vs current injections. Cortex: WT mice, n = 17 cells from 11 animals; Scn1aRH/+ mice, n = 26 cells from 10 animals. Hippocampus: WT mice, n = 15 cells from six animals; Scn1aRH/+ mice, n = 14 cells from seven animals; Scn1aRH/RH mice, n = 8 cells from three animals. *p < 0.05; **p < 0.01; ***p < 0.001, Mann–Whitney test and ANOVA on ranks (hippocampus). Data are shown as means ± SEM.
In the nRt, we found two types of inhibitory neurons. Most of them displayed an initial burst discharge of APs as a response to current injections (Fig. 1B). Others, predominantly in the dorsal part of the nRt, showed only tonic activity (Contreras et al., 1992; Lee et al., 2007). Firing was recorded upon different current injections. Both bursting and tonically active nRt neurons of heterozygous animals showed significantly fewer APs in response to the same current injections compared with WT littermates (number of spikes during 250 pA current injection: bursting neurons: WT mice, 47 ± 5, n = 23; Scn1aRH/+ mice, 32 ± 3, n = 37; p < 0.001, Mann–Whitney test; Fig. 1B; tonically active neurons: WT mice, 87 ± 13, n = 7; Scn1aRH/+ mice, 53 ± 5, n = 7; p < 0.01, Mann–Whitney test).
A characteristic feature of inhibitory nRt cells is a postinhibitory rebound after a hyperpolarization characterized by a low-threshold calcium spike and a burst of APs (Fig. 1D, top; Contreras et al., 1992; Llinás and Steriade, 2006). Neurons from heterozygous animals showed a significantly reduced number of APs within the rebound burst confirming their reduced excitability, when compared with WT mice (Fig. 1D).
In contrast, the firing rates of excitatory thalamocortical relay neurons within the thalamic VB were not significantly different between heterozygous and WT animals (number of spikes during 250 pA current injection: WT mice, 20.0 ± 3.1, n = 11; Scn1aRH/+ mice, 21.5 ± 5.1, n = 13); p = NS, Mann–Whitney test; Fig. 1C). These findings indicate that the mutation selectively influences firing of thalamic inhibitory, but not of excitatory neurons.
Intrinsic firing properties of cortical and hippocampal inhibitory and excitatory neurons
We next recorded from cortical neurons in the S1 region. GABAergic neurons were labeled by crossing R1648H knock-in mice with a line expressing EGFP in interneurons (Tamamaki et al., 2003). Similar to the thalamus, LIV fast-spiking (FS) inhibitory neurons, but not LV pyramidal cells, fired fewer APs in response to current injections in Scn1aRH/+ mice compared with WT mice, as shown in Figure 1, E and F, top (number of spikes during 250 pA current injection: interneurons: WT mice, 93 ± 16, n = 7; Scn1aRH/+ mice, 59 ± 11, n = 9; p < 0.05; pyramidal cells: WT mice, 26 ± 5, n = 17; Scn1aRH/+ mice, 22 ± 3, n = 26; p = NS, Mann–Whitney test).
For recordings in the hippocampal CA1 region, we used WT, Scn1aRH/+, and also homozygous Scn1aRH/RH mice, since we later also used hippocampal neurons for Na+ current recordings for which homozygous animals were important (see below). Interneurons and pyramidal neurons were identified by morphology and localization, and were confirmed by biocytin or Lucifer yellow labeling. For analysis, we used only FS neurons with homogenous electrophysiological characteristics (AP width, <0.6 ms; APs are followed by a large afterhyperpolarization) as well as morphological properties (located in the stratum oriens, close to the stratum pyramidale with main projections in the pyramidal cell layer), which were presumed to represent basket cells. We did not include interneurons showing regular or stuttering spiking. As GABAergic neurons in thalamus and cortex, FS interneurons in stratum oriens of the hippocampal CA1 region showed reduced firing for mutant mice (number of spikes during 250 pA current injection: WT mice, 96 ± 10, n = 7; Scn1aRH/+ mice, 48 ± 7, n = 11; p < 0.001), which was more pronounced for homozygous animals (Scn1aRH/RH mice, 20 ± 10, n = 6; p < 0.001, ANOVA on ranks; Fig. 1E, bottom). Excitatory pyramidal cells in CA1 stratum pyramidale did not show significant differences in firing properties (WT mice, 40 ± 5, n = 15; Scn1aRH/+ mice, 43 ± 8, n = 14; Scn1aRH/RH mice, 35 ± 5, n = 8; p = NS, ANOVA on ranks; Fig. 1F, bottom). We also examined the passive membrane properties in the neuron types discussed above showing no significant differences between WT and mutant animals in any type of neuron (Table 1).
Table 1.
Membrane properties of different neurons recorded in brain slices of WT and mutant animals
| Animals | Vm (mV) | Rin (MΩ) | AP threshold for eliciting 1 AP (mV) | 1 AP width (ms) | Rheobase (pA)* | Cells (n) |
|---|---|---|---|---|---|---|
| Nucleus reticularis, burster | ||||||
| WT | −68.6 ± 3.5 | 181.4 ± 14.9 | −50.1 ± 4.1 | 0.65 ± 0.03 | 47.6 ± 4.5 | 23 |
| Scn1aRH/+ | −69.6 ± 3.6 | 156.8 ± 13.0 | −49.1 ± 2.5 | 0.61 ± 0.02 | 54.1 ± 4.3 | 37 |
| Scn1aRH/RH | −69.9 ± 0.8 | 225.5 ± 17.9 | −52.0 ± 0.8 | 0.64 ± 0.02 | 57.5 ± 5.8 | 10 |
| Nucleus reticularis, non-burster | ||||||
| WT | −72.3 ± 1.7 | 303.0 ± 47.9 | −53.1 ± 0.9 | 1.04 ± 0.07 | 59.3 ± 7.4 | 7 |
| Scn1aRH/+ | −72.3 ± 1.5 | 306.2 ± 69.5 | −55.0 ± 5.3 | 1.02 ± 0.08 | 79.3 ± 10.9 | 7 |
| VB, relay neuron | ||||||
| WT | −72.1 ± 1.0 | 104.5 ± 4.1 | −57.3 ± 1.8 | 1.03 ± 0.02 | 97.7 ± 8.6 | 11 |
| Scn1aRH/+ | −70.9 ± 1.8 | 134.75 ± 16.2 | −55.3 ± 1.3 | 1.01 ± 0.03 | 87.5 ± 11.7 | 13 |
| Scn1aRH/RH | −69.3 ± 1.4 | 153.82 ± 17.48 | −52.6 ± 1.0 | 1.02 ± 0.04 | 87.5 ± 12.5 | 6 |
| Cortex, inhibitory neurons | ||||||
| WT | −70.1 ± 1.4 | 200.1 ± 16.8 | −54.1 ± 1.8 | 0.44 ± 0.04 | 92.9 ± 13.0 | 7 |
| Scn1aRH/+ | −69.9 ± 1.5 | 196.7 ± 14.6 | −52.4 ± 1.3 | 0.54 ± 0.03 | 105.0 ± 16.2 | 9 |
| Cortex, pyramidal cells | ||||||
| WT | −72.9 ± 1.1 | 140.7 ± 11.2 | −54.7 ± 1.8 | 1.02 ± 0.05 | 107.8 ± 9.9 | 17 |
| Scn1aRH/+ | −74.1 ± 1.0 | 152.0 ± 8.9 | −57.2 ± 1.5 | 1.03 ± 0.04 | 101.0 ± 7.8 | 26 |
| Hippocampus, Stratum oriens | ||||||
| WT | −74.5 ± 5.1 | 174.9 ± 11.4 | −54.4 ± 1.0 | 0.57 ± 0.07 | 50.0 ± 9.5 | 7 |
| Scn1aRH/+ | −73.4 ± 4.8 | 163.7 ± 16.9 | −53.1 ± 1.9 | 0.57 ± 0.05 | 79.6 ± 16.8 | 11 |
| Scn1aRH/RH | −69.3 ± 1.1 | 206.4 ± 14.2 | −52.8 ± 1.1 | 0.58 ± 0.03 | 64.2 ± 7.7 | 6 |
| Hippocampus, Stratum pyramidale | ||||||
| WT | −74.8 ± 1.2 | 226.2 ± 14.0 | −53.4 ± 1.2 | 1.04 ± 0.02 | 50.0 ± 5.9 | 15 |
| Scn1aRH/+ | −71.0 ± 1.2 | 190.8 ± 19.1 | −55.8 ± 1.6 | 1.06 ± 0.04 | 55.6 ± 12.3 | 14 |
| Scn1aRH/RH | −70.7 ± 2.1 | 183.4 ± 16.9 | −54.0 ± 1.7 | 1.08 ± 0.02 | 43.8 ± 6.3 | 8 |
Data are presented as means ± SEM; Mann–Whitney rank-sum test and ANOVA on ranks with Dunn's post hoc test.
*Current injection of 800 ms. Vm, resting membrane potential; Rin, input resistance.
In summary, these results demonstrate a widespread dysfunction of inhibitory, but not of excitatory neurons with respect to AP firing. The reduced firing indicated a loss-of-function of mutant Na+ channels expressed in interneurons.
Properties of somatic fast Na+ currents in inhibitory neurons
Na+ currents from native neurons are ideally recorded from nucleated patches, which permit recording from unequivocally identified neurons, and allow adequate voltage control (Martina and Jonas, 1997). Nucleated patches were obtained from inhibitory neurons of stratum oriens and primary cortical cultures. Recorded Na+ currents had amplitudes of 100–400 pA. There were no significant differences between heterozygous and WT animals, both in interneurons in stratum oriens (Fig. 2A,B; Table 2) and primary cortical cultures (Table 2), although slight alterations in a direction similar to that recorded using recombinant human channels could be observed (see below; reduced current density and decreased slope of the steady-state fast inactivation curve; Fig. 2).
Table 2.
Activation and inactivation of Na+ currents in nucleated patches of neurons and whole cell recordings of tsA cells
| Current–density (pA/pF) | Steady-state activation |
Steady-state inactivation |
Trec at −100 mV | |||||
|---|---|---|---|---|---|---|---|---|
| V1/2 (mV) | kV | n | V1/2 (mV) | kV | n | |||
| Nucleated patches of inhibitory neurons in primary cortical neurons | ||||||||
| WT | −684.6 ± 93.2 | −29.6 ± 0.5 | −6.7 ± 0.1 | 22 | −63.1 ± 0.8 | 5.2 ± 0.2 | 22 | 4.1 ± 0.4 |
| Scn1aRH/+ | −757.4 ± 142.1 | −29.4 ± 0.8 | −6.5 ± 0.2 | 20 | −63.6 ± 1.1 | 5.6 ± 0.4 | 20 | 4.4 ± 0.5 |
| Nucleated patches of inhibitory CA1 neurons in acute slices | ||||||||
| WT | −143.3 ± 29.5 | −33.0 ± 1.4 | −6.8 ± 0.2 | 10 | −68 ± 1.1 | 9.1 ± 0.4 | 9 | 3.9 ± 1.0 |
| Scn1aRH/+ | −116.9 ± 12.0 | −33.3 ± 1.1 | −6.4 ± 0.4 | 15 | −66.6 ± 1.4 | 9.6 ± 0.4 | 15 | 5.1 ± 1.3 |
| Scn1aRH/RH | −106.7 ± 17.5 | −34.4 ± 1.1 | −6.6 ± 0.4 | 9 | −71.0 ± 1.1 | 11.8 ± 1.5 | 8 | 6.6 ± 1.6 |
| Whole cell patch-clamp recordings of transfected tsA cells | ||||||||
| WT | −421.8 ± 54.6 | −35.1 ± 2.2 | 6.2 ± 0.4 | 9 | −73.0 ± 1.3 | 4.6 ± 0.1 | 8 | 5.3 ± 0.6 |
| R1648H | −251.2 ± 53.5* | −34.4 ± 1.7 | 7.1 ± 0.3 | 11 | −73.6 ± 0.8 | 7.1 ± 0.5* | 11 | 1.8 ± 0.2* |
Data are presented as means ± SEM. n, Number of recorded neurons. TRec, time constant of recovery from fast inactivation.
*p < 0.001, WT vs R1648H. Mann–Whitney rank-sum test for cases with two groups compared (WT vs Scn1aRH/+ or R1648H mutation) and ANOVA on ranks with Dunn's post hoc test for comparing WT, Scn1aRH/+, and Scn1aRH/RH mice.
Thus, we re-examined the properties of the R1648H mutation in a heterologous expression system. In tsA201 cells, the R1648H mutation in hNaV1.1 showed results similar to those reported previously (Fig. 2C–H; Table 2; Alekov et al., 2000; Spampanato et al., 2001; Lossin et al., 2002). Compared with the WT mice, mutant channels showed defects in fast inactivation with decreased slope and slight shift to more hyperpolarized potentials of the fast steady-state inactivation curve, accelerated recovery from fast inactivation (Table 2), and increased persistent current (Fig. 2F; see below). Additional findings were a reduced current density and a large hyperpolarizing shift in steady-state slow inactivation (Fig. 2H; Table 2).
Properties of persistent Na+ currents in interneurons
An important functional consequence of the R1648H mutation in the human NaV1.1 channel described in tsA201 cells (Lossin et al., 2002) was an increased persistent Na+ current, which we could confirm using a different clone of the human NaV1.1 channel (Fig. 2F; Rusconi et al., 2007), but which was not detected in studies using the human skeletal muscle channel hNaV1.4 in tsA201 cells (Alekov et al., 2000), the rat NaV1.1 channel in Xenopus oocytes (Spampanato et al., 2001), or hippocampal neurons dissociated from transgenic mice expressing R1648H in mouse NaV1.1 (Tang et al., 2009). To assess the persistent current in native neurons in brain slices, we used voltage ramps in whole-cell recordings of nRt and hippocampal GABAergic neurons. However, we were not able to identify significant differences in persistent currents among the three genotypes in hippocampal (Fig. 3A,B) and thalamic nRt neurons (Fig. 3B, right). The peak amplitudes as well as the voltage dependence of the persistent sodium currents were similar in WT and mutant animals (Fig. 3B; hippocampus: WT mice, V1/2 = −39.3 ± 2.7 mV, n = 11; Scn1aRH/+ mice, V1/2 = −38.7 ± 1.6 mV, n = 9); Scn1aRH/RH mice, V1/2 = −38.9 ± 2.9 mV, n = 12; p = NS; nRt: WT mice, V1/2 = 41.6 ± 6.3 mV, n = 15; Scn1aRH/+ mice, V1/2 = −40.6 ± 2.8 mV, n = 24; Scn1aRH/RH mice, V1/2 = −43.8 ± 3.1 mV, n = 11; one-way ANOVA).
Figure 3.

Persistent Na+ currents recorded in inhibitory nRt and hippocampal neurons. A, Representative whole-cell patch-clamp recordings of persistent currents of hippocampal FS neurons elicited by voltage ramps from −80 to 0 mV. B, Box plots of peak persistent currents recorded in inhibitory hippocampal (left) and nRt neurons (right). The peak INaP does not significantly differ between WT and mutant animals. ANOVA on ranks with Dunn's post hoc test.
Analysis of spike initiation
The results so far clearly demonstrate a deficit in interneuron firing, but did not reveal any detectable changes in fast or persistent Na+ currents. Nucleated patch recordings report on the properties of somatic Na+ channels, but much higher densities of Na+ channels are observed at the AIS, the place of spike initiation (Palmer and Stuart, 2006; Meeks and Mennerick, 2007). Changes at the AIS can clearly change spike initiation and input–output properties of excitatory neurons (Royeck et al., 2008; Wimmer et al., 2010). We hypothesized that spike initiation is altered in interneurons, due to changes in AIS Na+ channels. During AP trains, we calculated the second derivation of the voltage trace (d2V/dt2; Fig. 4A, first and last APs in a train during 200 pA current injections for all three genotypes). In these traces, two components could be identified, representing a first phase for the axonal spike component, and a second phase for the somatic spike, with a delay indicating the time from the initiation at the AIS and the arrival at the soma (Royeck et al., 2008; Wimmer et al., 2010).
Figure 4.

Analysis of AP initiation. A, Representative traces of first (top block) and last (bottom block) spikes (top traces) and their corresponding first (middle trace) and second (bottom trace) derivations of hippocampal FS inhibitory neurons of WT, Scn1aRH/+, and Scn1aRH/RH mice (CA1 stratum oriens). Black dotted lines illustrate the delay between the first (axonal) and the second (somatic) peaks; gray dotted lines illustrate the increased delay between the two peaks for the last spike recorded from mutated animals. B, Representative phase plots of dV/dt vs V for first (top) and last (bottom) spikes generated from recordings, as in A. Asterisks mark the beginning of the two phases of action potential initiation in the axon initial segment (green asterisk) and in the soma (black asterisk). C, Left, Amplitudes of the first axonal peaks in the second derivation of the first and last occurring spikes within AP trains, *p < 0.05. Right, Amplitudes of the second somatic peaks in the second derivation of first and last occurring spikes within AP trains, *p < 0.05. D, Delay between the first (axonal) and second (somatic) peaks of the first and last APs. One-way ANOVA with multiple comparisons, with the WT group serving as control (Dunnett's method) and t test for analysis within the same genotype. WT mice, white, n = 15; Scn1aRH/+ mice, gray, n = 14; Scn1aRH/RH mice, red, n = 8.
These two phases were also identified in phase plots in which the first derivation, dV/dt, is plotted versus voltage (Fig. 4B, asterisks). Phase plots of the last APs showed a strong alteration in the portion of the phase plot with a less abrupt initiation phase (green asterisk) in heterozygous and homozygous mice compared with phase plots of the first spike (Fig. 4B). This initiation phase in the AIS can also be quantified as a maximum of the d2V/dt2. Indeed, when the magnitude of the first peak in the d2V/dt2 was measured for the first and last AP in a train of APs, there was a significant decrease in membrane potential acceleration for both heterozygous and homozygous mice (WT mice: first, 1203 ± 201; last, 1136 ± 299; n = 15; Scn1aRH/+ mice: first, 1058 ± 215; last, 487 ± 80; n = 14; Scn1aRH/RH mice: first, 628 ± 63; last, 375 ± 82; n = 8). One-way ANOVA with multiple comparisons with the WT group serving as a control (Dunnett's method and t test for analysis within the same genotype; Fig. 4C, left axis). Also, the second peak was smaller in mutant animals, but there was no significant change from the first to the last AP in a train (Fig. 4C, right axis). Additionally, the latency was prolonged between the first spike (AIS initiation) and the second peak (somatic invasion) for both heterozygous and homozygous animals, which could be due to both a slower propagation or a shift in the spike initiation site (WT mice: first, 0.10 ± 0.02 ms; last, 0.09 ± 0.01 ms; n = 15; Scn1aRH/+ mice: first, 0.14 ± 0.04 ms; last, 0.26 ± 0.07 ms; n = 14; Scn1aRH/RH mice: first, 0.18 ± 0.03 ms; last, 0.26 ± 0.04 ms; n = 8. *WT versus Scn1aRH/+/Scn1aRH/RH mice, p < 0.05; one-way ANOVA with multiple comparisons with the WT group serving as control (Dunnett's method and t test for analysis within the same genotype; Fig. 4D). None of these changes were observed between genotypes when hippocampal pyramidal neurons were studied with identical techniques (data not shown; for similar analysis of hippocampal pyramidal neurons, see also Royeck et al., 2008). In GABAergic nRt neurons, we discovered a similar effect: the latency between AIS and somatic AP peaks was prolonged in heterozygous mice (WT mice: first, 0.17 ± 0.02 ms; last, 0.20 ± 0.02 ms; n = 23; Scn1aRH/+ mice: first, 0.21 ± 0.03 ms; last, 0.3 ± 0.03 ms; n = 37; p < 0.05). Together with the lack of significant changes in somatic fast Na+ currents, these data suggest that the R1648H mutation specifically causes a dysfunction of the AIS and spike initiation in GABAergic interneurons.
Recordings of spontaneous synaptic activity in thalamic and cortical neurons
We next examined the postsynaptic effects of the reduced activity of inhibitory neurons. We recorded spontaneous IPSCs (sIPSCs) in different neurons of the thalamocortical circuit receiving inhibitory input, namely, in cortical LV pyramidal cells, in GABAergic nRt neurons inhibiting themselves reciprocally, and in excitatory thalamocortical relay neurons (Fig. 1A). For all three subtypes, we found significantly reduced frequencies of sIPSCs in heterozygous compared with WT animals (cortical pyramidal neurons: WT mice, 9.7 ± 0.3 Hz, n = 5; Scn1aRH/+ mice, 6.7 ± 0.5 Hz, n = 10; p < 0.05; nRt neurons: WT mice, 2.7 ± 0.5 Hz, n = 10; Scn1aRH/+ mice, 1.6 ± 0.3 Hz, n = 10; p < 0.05; relay neurons: WT mice, 10.5 ± 2.3 Hz, n = 12; Scn1aRH/+ mice, 5.1 ± 1.1 Hz, n = 12; p < 0.05, Mann–Whitney test; Fig. 5A–F), whereas the amplitudes were similar (cortical pyramidal neurons: WT mice, −64.7 ± 7.5 pA, n = 5; Scn1aRH/+ mice, −58.5 ± 2.0 pA, n = 10; nRt neurons: WT mice, −56.9 ± 3.6 pA, n = 10; Scn1aRH/+ mice, −53.0 ± 3.6 pA, n = 10; relay neurons: WT mice, 38.8 ± 5.6 pA, n = 12; Scn1aRH/+ mice, 32.6 ± 3.6 pA, n = 12; Mann–Whitney test). When APs were blocked by 1 μm tetrodotoxin (TTX) application to record miniature IPSCs (mIPSCs), there was no longer a difference between nRt neurons of WT and Scn1aRH/+ mice (nRt neurons: WT mice: frequency, 2.5 ± 0.4 Hz; amplitude, −42.8 ± 5.2 pA, n = 8; Scn1aRH/+ mice: frequency, 2.6 ± 0.5 Hz; amplitude, −47.7 ± 2.2 pA; n = 9; VB neurons: WT mice: frequency, 3.44 ± 0.67 Hz; amplitude, −24.7 ± 4.1 pA; n = 10; Scn1aRH/+ mice: frequency, 2.7 ± 0.5 Hz; amplitude, −17.4 ± 0.9 pA; n = 9; Mann–Whitney rank-sum test). This clearly indicates a normal number of functional GABAergic synapses and an intact release machinery, and points toward a firing deficit of inhibitory neurons with a failure of orthodromic action potential propagation along the axons as the major source of reduced GABAergic activity.
Figure 5.
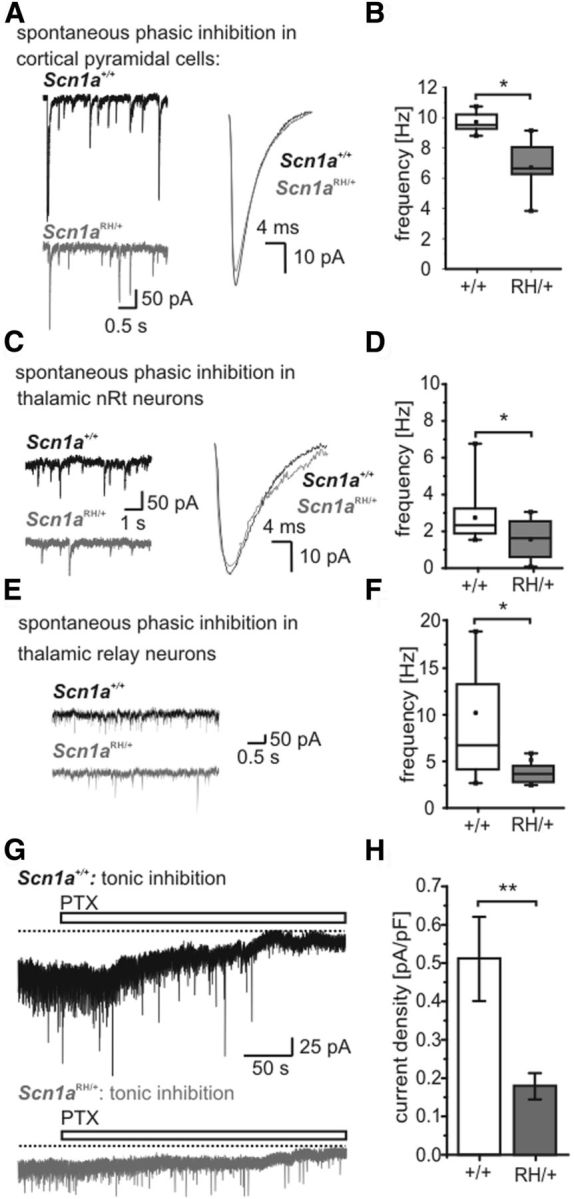
Phasic and tonic inhibition. A, C, E, Left, Representative whole-cell current traces recorded from cortical pyramidal neurons (layer V; A), nRt neurons (C), and thalamic relay neurons (E) in thalamocortical slices from WT (top traces) and Scn1aRH/+ (bottom traces) animals. Membrane voltage clamped to −70 mV. Right, Superimposed average sIPSCs from WT (cortex, n = 821 sIPSCS; nRt, n = 219 sIPSCs) and Scn1aRH/+ animals (cortex, n = 519 sIPSCs; nRt, n = 173 sIPSCs). B, D, F, The sIPSC frequency as recorded in A, C, and E was reduced for Scn1aRH/+ animals in all three neuronal subtypes. Cortical pyramidal neurons (B): WT mice, n = 5; Scn1aRH/+ mice, n = 10; nRt neurons (D): WT mice, n = 10; Scn1aRH/+ mice, n = 10; relay neurons (F): WT mice, n = 12; Scn1aRH/+ mice, n = 12; *p < 0.05; Mann–Whitney test. G, Whole-cell patch-clamp recordings of tonic GABA-sensitive current in thalamic relay neurons from a WT (top) and a heterozygous animal (bottom). Tonic inhibition could be blocked by application of the GABA receptor blocker picrotoxin (PTX). H, Current density (pA/pF) was significantly decreased in neurons of Scn1aRH/+ animals: WT mice, n = 9; Scn1aRH/+ mice, n = 7, **p < 0.01, Student's t test.
Synaptic GABA release activates synaptic as well as extrasynaptic GABA receptors, which gives rise to a tonic GABA-mediated current. This current is prominent in thalamocortical VB neurons (Jia et al., 2005) and is enhanced in several animal models of absence epilepsy (Cope et al., 2009). Both WT and heterozygous mice showed typical tonic inhibitory currents, which could be blocked by picrotoxin (Fig. 5G), but the magnitude of the tonic GABA current was significantly reduced in heterozygous mice compared with control mice (WT mice: 0.51 ± 0.11 pA/pF, n = 9 neurons in 9 slices; Scn1aRH/+ mice: 0.18 ± 0.03 pA/pF, n = 7 neurons in 7 slices, p < 0.01; Fig. 5H).
Assessment of spontaneous network activity
To determine how mutant NaV1.1 channels affect neuronal network activity, we applied three different techniques. As a first screening experiment, we used primary hippocampal cultures from WT and heterozygous animals, and plated them on 60-channel MEAs. Neuronal activities were illustrated using spike raster plots, in which spikes recorded on each electrode are plotted over time (Fig. 6A). In contrast to cultures derived from WT mice, cultures from heterozygous animals showed intermittent periods of low activity, interspersed with long periods of highly synchronous activity (Fig. 6A, blue boxes indicating population bursts). This could be quantified as an increased intraburst spike frequency (Fig. 6B, PFR of population bursts), a reduced number of bursts (Fig. 6C) with a longer duration (Fig. 6D), and longer interburst intervals (Fig. 6E). However, the relative number of spikes occurring within or outside a population burst was similar between cultures from WT and heterozygous animals (WT mice: 96.3 ± 0.8%; n = 12; Scn1aRH/+ mice: 97.2 ± 1.3%; n = 9; p = NS), due to the many short population bursts in cultures from WT animals. These results thus show an altered organization of discharges in neurons from mutant animals, with increased occurrence of high-frequency, long-lasting synchronized activity.
Figure 6.

Network activities of primary neuronal cultures recorded on multielectrode arrays. A, Spike raster plots of all 60 recorded electrodes with spikes represented as “+” over time of primary cultures of WT (top) and heterozygous animals (bottom), each shown without (left) and after application of bicuculline (Bic.; 25 μm, right). The y-axis represents the electrode number within an MEA configuration of 8 × 8 electrodes. Area within red dashed box in spike raster plot of primary culture of a WT animal (top left) and a heterozygous animal (bottom left) is shown enlarged to the right. Blue boxes highlight examples of population bursts within the different spike raster plots. Note that the spike raster plot of the culture from a heterozygous animal shows a high variability in the duration of the population bursts. B, Box plots of PFR of population bursts before and after Bic. application for WT (white) and heterozygous animals (gray). WT mice, n = 12; Scn1aRH/+ mice, n = 9; *p < 0.05, ANOVA on ranks. C, Box plots of number of population bursts for cultures from heterozygous (gray) and WT (white) animals. WT mice, n = 12; Scn1aRH/+ mice, n = 9; *p < 0.05, **p < 0.01, ANOVA on ranks. D, Box plots of burst duration for cultures from heterozygous (gray) and WT (white) animals. WT mice, n = 12; Scn1aRH/+ mice, n = 9; *p < 0.05, Mann–Whitney test. E, Box plots of interburst interval for cultures from heterozygous (gray) and WT (white) animals. WT mice, n = 12; Scn1aRH/+ mice, n = 9; *p < 0.05, ANOVA on ranks. F, Box plots of κ ratio 25 μm Bic/baseline for recordings from cultures of heterozygous (gray) and WT (white) animals. WT mice, n = 12; Scn1aRH/+ mice, n = 9; *p < 0.05, Mann–Whitney test.
If these differences between WT and heterozygous animals are due to a deficit in GABAergic synaptic transmission in the latter group, then pharmacological inhibition of GABA receptors should exert larger effects in cultures from WT animals. We therefore applied the GABAA receptor antagonist bicuculline on MEAs plated with neurons from WT and heterozygous animals. Bicuculline application on cultures from WT mice led to significant changes in bursting behavior, causing a reorganization of discharge behavior in the form of higher frequency, fewer burst periods with increased interburst intervals, but not significantly longer bursts (Fig. 6A–E). In addition, the percentage of spikes occurring within population bursts increased significantly with bicuculline in cultures from WT animals (from 96.3 ± 0.8% before to 99.7 ± 0.1% after bicuculline application; n = 12; p < 0.001). In stark contrast, changes in population bursts from cultures of Scn1aRH/+ animals were much smaller, and not statistically significant in any of these parameters (Fig. 6B–E; 97.2 ± 1.3% of spikes within bursts before bicuculline application; 99.5 ± 0.5% spikes within bursts after bicuculline application; n = 9).
To quantify the firing synchrony of the plated neuronal networks, we evaluated Cohen's κ statistic across all pairs of active electrodes. The network activity recorded from cultures of WT animals showed a 1.8-fold increase of κ values upon application of bicuculline, indicating a higher synchrony (from 0.38 ± 0.05 at baseline to 0.56 ± 0.03 in bicuculline; p < 0.01; n = 12). In cultures of Scn1aRH/+ animals, the κ statistic was already 0.51 ± 0.04 at baseline without a significant increase in bicuculline (0.53 ± 0.04; n = 9). Using the ratio of κ values after and before bicuculline application, we found that the increase in synchrony was significantly larger in cultures from WT animals compared with ones from heterozygous animals (Fig. 6F). Altogether, our findings indicate that cultures from heterozygous animals show hyperexcitable networks with increased synchrony compared with the WT. Since blocking GABAA receptors has significant effects only in neurons from WT mice, but not in those from heterozygous mice, these results are consistent with the idea that increased synchrony in Scn1aRH/+ networks is due to a deficit in GABAergic synaptic transmission.
Second, we tested the impact of the R1648H mutation on network activity in acute thalamocortical slices. We recorded extracellular field potentials in LIV and LV of the neocortical S1 region, in the thalamic VB and nRt. The presence of functional thalamocortical connections was tested by stimulating the VB and recording the monosynaptic response in LIV (Agmon and Connors, 1991), showing no significant differences between heterozygous and WT mice (Fig. 7A). Paired-pulse stimulations did not disclose statistically significant differences either (Fig. 7A). However, we found spontaneous discharges in 16 of 17 slices obtained from heterozygous mice (Fig. 7B), which were observed in only 5 of 12 slices from WT littermates (Fig. 7C; p = 0.003, Fisher's exact test). Multielectrode recordings in cortex and thalamus showed simultaneous spontaneous activity spreading in different areas of the thalamocortical loop only in slices from heterozygous mice (9 of the 16 slices showed spontaneous activity at all four electrodes; Fig. 7B,E; Table 3). Furthermore, spontaneous activities were more frequent in slices from heterozygous mice compared with WT mice (Fig. 7; Table 3). The area in which discharges were first generated during simultaneous activity was variable (Fig. 7B,E; Table 3). In slices from heterozygous mice, simultaneous activities were larger than nonsimultaneous ones, with the latter having amplitudes similar to those observed in slices from WT mice. Few activities in slices from WT mice had amplitudes comparable to those of simultaneous heterozygous ones, but they did not spread to other areas (Fig. 7C; Table 3). Analysis of spontaneous cortical sharp waves showed HFOs in slices of Scn1aRH/+ mice in both the physiological (40–200 Hz) and the pathological (200–600 Hz) range (Fig. 7B,D; Table 3). In contrast, HFOs were hardly seen in slices from WT mice, including activities with amplitudes comparable to those seen in slices of mutant mice (Fig. 7C,D; Table 3).
Figure 7.
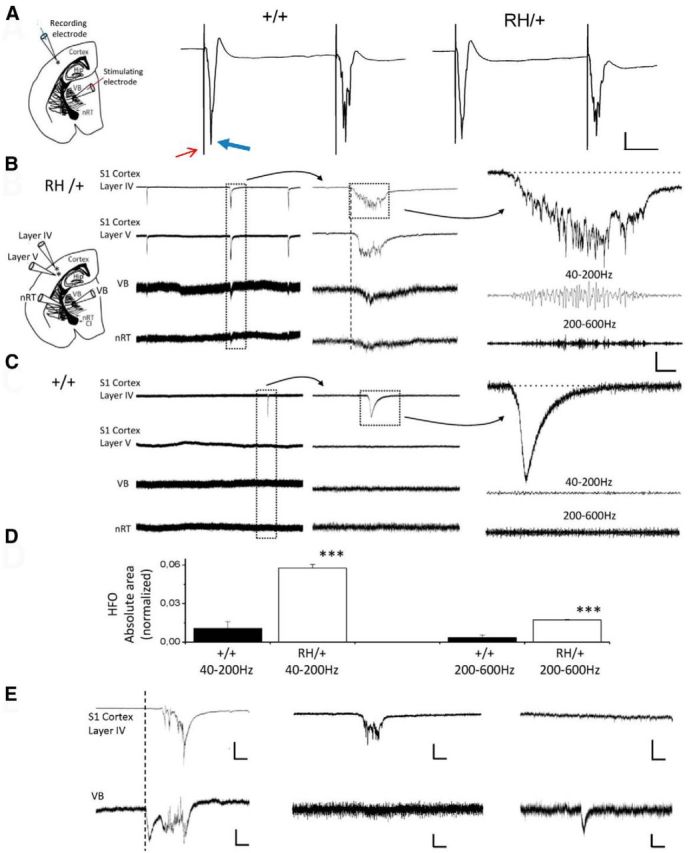
Field potential recordings of network activity in thalamocortical slices. A, Representative voltage traces of cortical layer IV responses to paired-pulse stimulations of thalamus (VB) with IPI of 100 ms in slices of Scn1a+/+ (left) and Scn1aRH/+ (right) animals; schematic shows electrode position. The thin red arrow highlights the stimulus artifact; the thick blue arrow highlights the monosynaptic response. Maximum amplitude: 2.7 ± 0.6 mV (n = 12) for Scn1a+/+ mice; 3.4 ± 0.7 mV (n = 17) for Scn1aRH/+ mice. Half-width: 3.1 ± 0.5 ms (n = 12) for WT mice; 3.8 ± 0.7 ms (n = 17) for Scn1aRH/+ mice. Coastline index: 13.68 ± 0.52 (n = 12) for Scn1a+/+ mice; 14.55 ± 0.94 (n = 17) for Scn1aRH/+ mice. Paired-pulse ratio computed using maximum amplitudes: 50 ms IPI: 0.80 ± 0.12 (n = 5) for Scn1a+/+ mice; 0.91 ± 0.09 (n = 5) for Scn1aRH/+ mice; 100 ms IPI: 0.85 ± 0.19 (n = 5) for Scn1a+/+ mice; 0.91 ± 0.09 (n = 5) for Scn1aRH/+ mice; 150 ms IPI: 0.74 ± 0.03 (n = 5) for Scn1a+/+ mice; 0.77 ± 0.16 (n = 5) for Scn1aRH/+ mice; 200 ms IPI: 0.71 ± 0.05 (n = 5) for Scn1a+/+ mice; 0.78 ± 0.15 (n = 5) Scn1aRH/+ mice. Paired-pulse ratio computed using coastline index: 50 ms IPI: 1.18 ± 0.07 (n = 5) for Scn1a+/+ mice; 1.05 ± 0.14 (n = 5) for Scn1aRH/+ mice; 100 ms IPI: 1.48 ± 0.13 (n = 5) for Scn1a+/+ mice; 1.49 ± 0.18 (n = 5) for Scn1aRH/+ mice; 150 ms IPI: 1.19 ± 0.12 (n = 5) for Scn1a+/+ mice; 1.16 ± 0.20 (n = 5) for Scn1aRH/+ mice; 200 ms IPI: 1.62 ± 0.18 (n = 5) for Scn1a+/+ mice; 1.41 ± 0.31 (n = 5) for Scn1aRH/+ mice. Calibration: 1 mV, 25 ms. B, C, Representative voltage traces of spontaneous extracellular activity in slices of Scn1aRH/+ (B) and Scn1a+/+ (C) animals recorded in cortical layers IV and V, thalamic VB, and nRt. Placement of the recording electrodes is shown in the schematic on the left (CI, capsula interna). Middle traces, Time expansions of the trace segments outlined by the dotted box in the left traces, showing that in this slice (B, Scn1aRH/+) simultaneous activities began in cortical layers (vertical dashed line; see E for activities of thalamic origin). Right traces, Cortical sharp waves outlined by the dotted box in the middle traces and the corresponding 40–200 and 200–600 Hz HFOs, which were hardly observed in WT mice. Calibration: left traces, 900 μV, 1.1 s; middle traces, 900 μV, 500 ms; right traces, 200 μV, 100 ms. D, Absolute areas of HFOs in the 40–200 and 200–600 Hz ranges. HFOs were noise subtracted and normalized to the area of the corresponding sharp wave (see Materials and Methods). E, Simultaneous recordings in the cortical layer IV (top) and the VB (bottom) of slices from Scn1aRH/+ animals displaying spontaneous activities. Left traces, Synchronous activities with thalamic origin; middle traces, activity generated in cortex without spread to thalamus; right traces, activity generated in thalamus without spread to cortex. Calibration: top left and middle traces, 500 μV, 250 ms; top right trace, 100 μV, 250 ms; bottom left trace, 150 μV, 250 ms; bottom middle and right traces, 50 μV, 250 ms.
Table 3.
Properties of spontaneous extracellular field potential activities in thalamocortical slices
| WT mice | n | Scn1a RH/+ mice | n | |
|---|---|---|---|---|
| Spontaneous total activity | 12 slices | 17 slices | ||
| Frequency, cortex LIV | 0.16 ± 0.08 min−1 | 21 | 0.52 ± 0.08 min−1* | 139 |
| Frequency, thalamus VB | 0.14 ± 0.11 min−1 | 11 | 0.65 ± 0.13 min−1* | 137 |
| Nonsimultaneous activities (mV) | ||||
| Amplitude, cortex LIV | 0.26 ± 0.03 | 0.28 ± 0.04 | ||
| Amplitude, thalamus VB | 0.11 ± 0.01 | 0.09 ± 0.01 | ||
| Simultaneous corticothalamic activities | 86 | |||
| Cortical generation (%) | 29.1% | |||
| Thalamic generation (%) | 32.6% | |||
| Unclear (%) | 39.5% | |||
| Amplitude, cortex LIV (mV) | 1.24 ± 0.06**,a | |||
| Amplitude, thalamus VB (mV) | 0.21 ± 0.05**,b | |||
| Cortical HFOs 40–200 Hz | 21 | 100 | ||
| Absolute area (mV/ms)c | 1.2 ± 0.4 | 30.6 ± 1.8** | ||
| Normalized absolute aread | 0.011 ± 0.005 | 0.058 ± 0.003** | ||
| Cortical HFOs 200–600 Hz | 21 | 100 | ||
| Absolute area (mV/ms)c | 0.51 ± 0.17 | 7.25 ± 0.47** | ||
| Normalized absolute aread | 0.003 ± 0.002 | 0.017 ± 0.001** |
Data are presented as means ± SEM. n, Number of analyzed activities.
aIn comparison to layer IV non-simultaneous activities (Wilcoxon signed rank test).
bIn comparison to thalamic VB non-simultaneous activities (Wilcoxon signed rank test).
cNoise in absolute area subtracted from HFOs absolute area (see Materials and Methods).
dHFO absolute area noise subtracted normalized to the absolute area of the spontaneous activity (0.1 Hz–1 kHz filtered; see Materials and Methods).
*p < 0.05 and
**p < 0.001, Mann–Whitney rank-sum test or, when noted, Wilcoxon signed rank test.
Third, we studied the network activity of neuronal populations in the hippocampal CA1 region using multineuron Ca2+ imaging in hippocampal slices of WT, heterozygous, and homozygous animals. Following a bulk-loading technique with the Ca2+ indicator OGB-1AM (Fig. 8A), we captured spontaneous network activity in simultaneous recordings of hundreds of neurons (159 ± 94 cells per slice; Fig. 8A,B, examples). Ca2+ traces were converted to raster plots to determine the dynamics of neuronal spiking activity. Figure 8, C and D, shows that the average spontaneous discharge rate of neurons in the population was increased in heterozygous and homozygous animals compared with WT animals (WT mice: 0.0020 ± 0.0002 Hz; n = 294 cells; Scn1aRH/+ mice: 0.0117 ± 0.0007 Hz; n = 1502 cells; Scn1aRH/RH mice: 0.0139 ± 0.0008 Hz; n = 640 cells; p < 0.05, one-way ANOVA with multiple comparisons, with the WT group serving as the control (Dunnett's method; Fig. 8D). Evaluation of firing frequencies summarized from all recorded cells and plotted in a double logarithmic histogram revealed frequencies >0.01 Hz in heterozygous and homozygous mice (Fig. 8C, gray and red lines), which was not observed in WT mice (Fig. 8C, black line). Altogether, these findings demonstrate that neuronal networks in different brain areas are spontaneously more active in mutant mice compared with WT mice.
Figure 8.

Multineuron Ca2+ imaging in the hippocampal CA1 region. A, Samples of slices loaded with the Ca2+ dye OGB-1-AM of WT (top) and Scn1aRH/+ (bottom) animals. Active cells filled with OGB-1-AM are marked in green, inactive cells filled with the dye are marked in white. B, Ca2+ transients of 20 representative spontaneously active cells recorded in the slices shown in A. C, Double logarithmic histogram showing the binned frequency of Ca2+ spikes normalized to the overall number of active cells (bin width at 0.001 Hz). Note that for slices from WT animals (black), spike frequencies of >0.01 Hz were absent, but that higher frequencies were frequently seen for mutant animals (gray and red). Dashed lines represent the maximum frequencies observed in slices of WT (black), Scn1aRH/+ (gray), and Scn1aRH/RH (red) animals. D, Logarithmic presentation of Ca2+ spike frequency as box plots plotted for all three genotypes (WT mice, 294 cells; Scn1aRH/+ mice, 1502 cells; Scn1aRH/RH mice, 640 cells). Box-and-whisker plots show medians (lines), means (square), lower and upper quartiles, 5% and 95%; percentiles represent the minimum and maximum. p < 0.05, ANOVA on ranks.
Discussion
We here performed a comprehensive neurophysiological analysis of an epilepsy-causing NaV1.1 mutation in different brain regions of a gene-targeted mouse model. This model recapitulates the human GEFS+ phenotype, showing spontaneous generalized seizures and a reduced threshold to thermally induced seizures (Martin et al., 2010). Recordings from mutant mice compared with WT mice at P14–P20 revealed that the mutation reduces firing in all studied inhibitory interneurons, but did not affect the examined excitatory neurons. These results show that at this age NaV1.1 is a major Na+ channel of interneurons but does not contribute to AP firing in excitatory neurons. This corresponds to previous immunohistochemical studies (Ogiwara et al., 2007), recordings from dissociated neurons (Yu et al., 2006; Mistry et al., 2014), and preferential inactivation of NaV1.1 in hippocampal and cortical parvalbumin-positive interneurons causing spontaneous seizures, in contrast to NaV1.1 knockout in pyramidal cells (Dutton et al., 2012). Even though recent studies suggest a modifying protective role of NaV1.1 in glutamatergic neurons (Ogiwara et al., 2013) and increased Na+ current density in dissociated pyramidal neurons at older ages (Mistry et al., 2014), our data suggest that interneurons in this model play a crucial role for epileptogenesis, even though there might be additional factors sustaining the epileptic state.
Relevant biophysical defect and involved neuronal compartments
Studies in heterologous expression systems have revealed both loss-of-function and gain-of-function mechanisms for this mutation (Alekov et al., 2000; Spampanato et al., 2001; Lossin et al., 2002). Our analysis of the human NaV1.1 mutation in tsA201 cells reveals the following three loss-of-function defects, which could explain the observed reduced firing: (1) a small but significant change in the slope of the fast steady-state inactivation curve; (2) probably most importantly, an enhanced slow inactivation; and (3) a reduced current density.
However, we did not find significant differences between WT and mutant animals in channel gating and in the persistent Na+ current in interneurons, in contrast with the differences observed in tsA201 cells. Although the biophysical properties of Na+ channels in neurons may differ from those in tsA201 cells and between species (mouse vs human), the observed changes in neuronal firing are difficult to explain on the basis of our neuronal Na+ channel recordings. Rather, they suggest that the somatic channel population in nucleated patches does not fully represent mutated loss-of-function channels and that other Na+ channels might carry a major part of the current. For example, NaV1.6 is expressed in hippocampal interneurons (Van Wart et al., 2007; Lorincz and Nusser, 2008; Mistry et al., 2014), and NaV1.3 may be compensatorily upregulated (Yu et al., 2006). Most of the functionally relevant NaV1.1 channels may in fact reside in the AIS and along whole axons where Na+ channels are concentrated (Rasband, 2010; Hu and Jonas, 2014). Accordingly, our data indicated a mainly axonal dysfunction increasing with prolonged AP series in GABAergic neurons. This could be explained by enhanced slow inactivation of mutated Na+ channels (as recorded in tsA201 cells), reducing the number of available channels with prolonged firing. The longer latencies between spike initiation and somatic invasion could be due to impaired propagation or a shift of the spike initiation site. Our results thus suggest that the inhibitory AIS (or whole axon according to Hu and Jonas, 2014) is the major site of NaV1.1 channel dysfunction in our model, and that enhanced slow inactivation represents the most important gating defect of R1648H mutant channels.
R1648H causes a reduction of inhibitory inputs
GABAergic inhibition controls neuronal excitability and a reduction in inhibition plays an important pathophysiological role in genetic and acquired epilepsies (Redecker et al., 2000; Reid et al., 2009; Ben-Ari and Dudek, 2010). Assessing the impact of reduced firing of inhibitory neurons on postsynaptic neurons and network dysfunction, our recordings of sIPSCs demonstrate that reduced AP firing of interneurons indeed translates into reduced GABAergic synaptic activity by failure of AP propagation from the AIS to the synaptic terminals in mutant animals compared with WT animals. The synaptic release machinery, however, was unaffected, as revealed by normal mIPSCs in the presence of TTX. Additionally, we found a decreased tonic inhibition mediated by activation of GABAA receptors located outside the synaptic cleft, which can be explained by a reduced spontaneous GABA spillover from presynaptic terminals. Interestingly, tonic inhibition is increased in several rodent models of absence epilepsy (Cope et al., 2009), which is consistent with a different mechanism of thalamocortical hyperexcitability in Scn1aRH/+ mice. Although resting membrane potentials in VB neurons were normal, this tonic disinhibition can destabilize the membrane potential, thereby contributing to the shift between different firing modes (tonic vs bursting). Our results thus suggest that a combination of reduced phasic and tonic inhibition contributes to a loss of inhibitory control of neuronal networks, triggering seizures in our mouse model.
Impaired network activity in mutant mice
Our simplified model using cultured neurons on MEAs revealed that the presynaptic SCN1A defect affecting fast-spiking interneurons alters the firing behavior of the network. Furthermore, the missing effect of blocking GABAA receptors in cultures from mutant mice is consistent with the disinhibition caused by the mutation. Other types of interneurons not studied here, like somatostatin-positive neurons regulating the gain of pyramidal cell input–output transformations (Lovett-Barron et al., 2012), could also be involved in causing this firing pattern. Further assessment of neuronal network activity in native brains revealed a widespread spontaneous hyperexcitability. Using multineuron Ca2+ imaging, we found an increased frequency of the spontaneous activity of single neurons in the CA1 pyramidal cell layer in mutant compared with WT mice. Multielectrode recordings revealed frequent spontaneous activity that spreads in the entire thalamocortical loop and was generated in variable areas, consistent with a generalized hyperexcitability within the loop. Moreover, cortical activity in heterozygous animals was characterized by the presence of HFOs in both the physiological (40–200 Hz) and the pathological (200–600 Hz) range not observed in WT mice. Human and rodent epileptic tissues exhibit pathological HFOs not found in nonepileptic controls, and HFOs have been proposed to be a biomarker of epileptic foci (Jefferys et al., 2012). Hence, the R1648H-induced disinhibition not only generates hyperexcitability, but also specific epileptic activity. Notably, partial block of GABAA receptors can also enhance neocortical and hippocampal pathological HFOs (Jones and Barth, 2002; Behrens et al., 2007) and increase the size of HFO-generating areas (Bragin et al., 2002).
Proper firing of inhibitory neurons is essential for correct network function. Cortical and hippocampal FS basket cells innervate the perisomatic regions of pyramidal cells, and can phase and synchronize the activity of large populations of pyramidal cells. These interneurons thus play an essential role in generating and maintaining theta- and gamma-frequency oscillations, for which a rapid system like phasic GABAA receptor activation is essential to synchronize at high frequencies (Buzsáki and Chrobak, 1995; Cobb et al., 1995). Basket cell maturation is involved in the development of synchronous oscillations, and an impairment of GABAergic interneurons during development can cause neurological disorders like autism, seizures, or schizophrenia (Le Magueresse and Monyer, 2013). The R1648H mutation may therefore also cause developmental alterations, which could contribute to seizure generation, although this effect should be limited to late developmental phases, since NaV1.1 expression begins at approximately P10 (Ogiwara et al., 2007). Additionally, it has been proposed that dendritic properties of different interneuron subtypes (low Na+ channel density in FS basket cells and high Na+ channel density in somatostatin-positive interneurons) could be involved in setting their differential activity in a neuronal network (Martina et al., 2000; Hu et al., 2010). Alterations in dendritic NaV1.1 channels in different interneuron types could additionally impact the activity and synchronization of neuronal networks.
In the thalamic nRt, a disturbance of the inhibitory neurons is involved in spike-and-wave discharges during generalized absence seizures (Danober et al., 1998). Likewise, a reduction of GABAA receptor-mediated inhibition in reticular neurons (by knockout of the GABAA receptor β3-subunit or modification of benzodiazepine binding to the α3-subunit) leads to pathological synchronization of thalamocortical oscillations, as seen in absence seizures (Huntsman et al., 1999; Christian et al., 2013). Our observed disinhibition can thus disturb the synchronization and function of whole networks and can explain the generation of epileptic seizures.
The variability in seizure types occurring with mutations in the same gene is particularly high in GEFS+ including febrile and afebrile generalized tonic–clonic seizures, absences, and even focal seizures originating from the temporal lobe (Scheffer and Berkovic, 1997; Weber and Lerche, 2008; Marini and Mantegazza, 2010). Specifically, individuals carrying the R1648H mutation exhibited febrile, afebrile generalized tonic–clonic and absence seizures (Baulac et al., 1999). Many circumstances may contribute to the phenotypic variability, including genetic modifiers and environmental factors (Glasscock et al., 2007; Martin et al., 2007). The widespread functional alterations we observed here may be a prerequisite for this phenomenon.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Grants Le1030/10-1/2 to H.L. and SFB 1089 to H.B.), the Bundesministerium für Bildung und Forschung (National Genome Research Network/Epilepsy and Migraine integrated network Grants 01GS08123 and 01GS08122 to H.L. and H.B.; EuroTransBio/ESSENCE Grant FKZ0315641A to M.D., S.T., and J.S.), the European Commission (Grant EPICURE, LSHM-CT-2006-037315 to H.L., M.M., and H.B.), Laboratory of Excellence Ion Channel Science and Therapeutics (to M.M.), and the Fondation Recherche Medicale (to M.M.). Research for this publication was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under Grant R01-NS072221 (to A.E.). We thank Drs. Olga Garaschuk, Henner Koch, Snezana Maljevic, and Thomas Wuttke for helpful discussions on the manuscript; and Snezana Maljevic for managing and coordinating the mouse transfer and preparation of neuronal cultures.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-J. [DOI] [PubMed] [Google Scholar]
- Alekov A, Rahman MM, Mitrovic N, Lehmann-Horn F, Lerche H. A sodium channel mutation causing epilepsy in man exhibits subtle defects in fast inactivation and activation in vitro. J Physiol. 2000;529:533–539. doi: 10.1111/j.1469-7793.2000.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allène C, Cattani A, Ackman JB, Bonifazi P, Aniksztejn L, Ben-Ari Y, Cossart R. Sequential generation of two distinct synapse-driven network patterns in developing neocortex. J Neurosci. 2008;28:12851–12863. doi: 10.1523/JNEUROSCI.3733-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac S, Gourfinkel-An I, Picard F, Rosenberg-Bourgin M, Prud'homme J-F, Baulac M, Brice A, LeGuern E. A second locus for familial generalized epilepsy with febrile seizures plus maps to chromosome 2q21–q33. J Hum Genet. 1999;65:1078–1085. doi: 10.1086/302593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron. 2009;62:612–632. doi: 10.1016/j.neuron.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens CJ, van den Boom LP, Heinemann U. Effects of the GABAA receptor antagonists bicuculline and gabazine on stimulus-induced sharp wave-ripple complexes in adult rat hippocampus in vitro. Eur J Neurosci. 2007;25:2170–2181. doi: 10.1111/j.1460-9568.2007.05462.x. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Dudek FE. Primary and secondary mechanisms of epileptogenesis in the temporal lobe: there is a before and an after. Epilepsy Curr. 2010;10:118–125. doi: 10.1111/j.1535-7511.2010.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragin A, Mody I, Wilson CL, Engel J., Jr Local generation of fast ripples in epileptic brain. J Neurosci. 2002;22:2012–2021. doi: 10.1523/JNEUROSCI.22-05-02012.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Chrobak JJ. Temporal structure in spatially organized neuronal ensembles: a role for interneuronal networks. Curr Opin Neurobiol. 1995;5:504–510. doi: 10.1016/0959-4388(95)80012-3. [DOI] [PubMed] [Google Scholar]
- Cestèle S, Scalmani P, Rusconi R, Terragni B, Franceschetti S, Mantegazza M. Self-limited hyperexcitability: functional effect of a familial hemiplegic migraine mutation of the NaV1.1 (SCN1A) Na+ channel. J Neurosci. 2008;28:7273–7283. doi: 10.1523/JNEUROSCI.4453-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian CA, Herbert AG, Holt RL, Peng K, Sherwood KD, Pangratz-Fuehrer S, Rudolph U, Huguenard JR. Endogenous positive allosteric modulation of GABA(A) receptors by diazepam binding inhibitor. Neuron. 2013;78:1063–1074. doi: 10.1016/j.neuron.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. J Hum Genet. 2001;68:1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- Contreras D, Curró Dossi R, Steriade M. Bursting and tonic discharges in two classes of reticular thalamic neurons. J Neurophysiol. 1992;68:973–977. doi: 10.1152/jn.1992.68.3.973. [DOI] [PubMed] [Google Scholar]
- Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lorincz ML, Gould TM, Carter DA, Crunelli V. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–1398. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Prog Neurobiol. 1998;55:27–57. doi: 10.1016/S0301-0082(97)00091-9. [DOI] [PubMed] [Google Scholar]
- Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari MD, Herzog J, van den Maagdenberg AM, Pusch M, Strom TM. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371–377. doi: 10.1016/S0140-6736(05)66786-4. [DOI] [PubMed] [Google Scholar]
- Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, Escayg A. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol Dis. 2012;49C:211–220. doi: 10.1016/j.nbd.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- Glasscock E, Qian J, Yoo JW, Noebels JL. Masking epilepsy by combining two epilepsy genes. Nat Neurosci. 2007;10:1554–1558. doi: 10.1038/nn1999. [DOI] [PubMed] [Google Scholar]
- Grewe BF, Langer D, Kasper H, Kampa BM, Helmchen F. High-speed in vivo calcium imaging reveals neuronal network activity with near-millisecond precision. Nat Methods. 2010;7:399–405. doi: 10.1038/nmeth.1453. [DOI] [PubMed] [Google Scholar]
- Henze DA, González-Burgos GR, Urban NN, Lewis DA, Barrionuevo G. Dopamine increases excitability of pyramidal neurons in primate prefrontal cortex. J Neurophysiol. 2000;84:2799–2809. doi: 10.1152/jn.2000.84.6.2799. [DOI] [PubMed] [Google Scholar]
- Holekamp TF, Turaga D, Holy TE. Fast three-dimensional fluorescence imaging of activity in neural populations by objective-coupled planar illumination microscopy. Neuron. 2008;57:661–672. doi: 10.1016/j.neuron.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Hu H, Jonas P. A supercritical density of Na+ channels ensures fast signaling in GABAergic interneuron axons. Nat Neurosci. 2014;17:686–693. doi: 10.1038/nn.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Martina M, Jonas P. Dendritic mechanisms underlying rapid synaptic activation of fast-spiking hippocampal interneurons. Science. 2010;327:52–58. doi: 10.1126/science.1177876. [DOI] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- Illes S, Theiss S, Hartung HP, Siebler M, Dihné M. Niche-dependent development of functional neuronal networks from embryonic stem cell-derived neural populations. BMC Neurosci. 2009;10:93. doi: 10.1186/1471-2202-10-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferys JG, Menendez de la Prida L, Wendling F, Bragin A, Avoli M, Timofeev I, Lopes da Silva FH. Mechanisms of physiological and epileptic HFO generation. Prog Neurobiol. 2012;98:250–264. doi: 10.1016/j.pneurobio.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia F, Pignataro L, Schofield CM, Yue M, Harrison NL, Goldstein PA. An extrasynaptic GABAA receptor mediates tonic inhibition in thalamic VB neurons. J Neurophysiol. 2005;94:4491–4501. doi: 10.1152/jn.00421.2005. [DOI] [PubMed] [Google Scholar]
- Jones MS, Barth DS. Effects of bicuculline methiodide on fast (>200 Hz) electrical oscillations in rat somatosensory cortex. J Neurophysiol. 2002;88:1016–1025. doi: 10.1152/jn.2002.88.2.1016. [DOI] [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R. Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA-mediated inhibition. J Neurophysiol. 1987;57:325–340. doi: 10.1152/jn.1987.57.1.325. [DOI] [PubMed] [Google Scholar]
- Lauxmann S, Boutry-Kryza N, Rivier C, Mueller S, Hedrich UB, Maljevic S, Szepetowski P, Lerche H, Lesca G. An SCN2A mutation in a family with infantile seizures from Madagascar reveals an increased subthreshold Na current. Epilepsia. 2013;54:e117–e121. doi: 10.1111/epi.12241. [DOI] [PubMed] [Google Scholar]
- Lee SH, Govindaiah G, Cox CL. Heterogeneity of firing properties among rat thalamic reticular nucleus neurons. J Physiol. 2007;582:195–208. doi: 10.1113/jphysiol.2007.134254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Magueresse C, Monyer H. GABAergic interneurons shape the functional maturation of the cortex. Neuron. 2013;77:388–405. doi: 10.1016/j.neuron.2013.01.011. [DOI] [PubMed] [Google Scholar]
- Liao Y, Anttonen AK, Liukkonen E, Gaily E, Maljevic S, Schubert S, Bellan-Koch A, Petrou S, Ahonen VE, Lerche H, Lehesjoki AE. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology. 2010;75:1454–1458. doi: 10.1212/WNL.0b013e3181f8812e. [DOI] [PubMed] [Google Scholar]
- Liautard C, Scalmani P, Carriero G, de Curtis M, Franceschetti S, Mantegazza M. Hippocampal hyperexcitability and specific epileptiform activity in a mouse model of Dravet syndrome. Epilepsia. 2013;54:1251–1261. doi: 10.1111/epi.12213. [DOI] [PubMed] [Google Scholar]
- Llinás RR, Steriade M. Bursting of thalamic neurons and states of vigilance. J Neurophysiol. 2006;95:3297–3308. doi: 10.1152/jn.00166.2006. [DOI] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008;28:14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL., Jr Molecular basis of an inherited epilepsy. Neuron. 2002;34:877–884. doi: 10.1016/S0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- Lovett-Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH, Nicoud JF, Zemelman BV, Sternson SM, Losonczy A. Regulation of neuronal input transformations by tunable dendritic inhibition. Nat Neurosci. 2012;15:423–430. S1–S3. doi: 10.1038/nn.3024. [DOI] [PubMed] [Google Scholar]
- Marini C, Mantegazza M. Na+ channelopathies and epilepsy: recent advances and new perspectives. Expert Rev Clin Pharmacol. 2010;3:371–384. doi: 10.1586/ecp.10.20. [DOI] [PubMed] [Google Scholar]
- Martina M, Jonas P. Functional differences in Na+ channel gating between fast-spiking interneurones and principal neurones of rat hippocampus. J Physiol. 1997;505:593–603. doi: 10.1111/j.1469-7793.1997.593ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina M, Vida I, Jonas P. Distal initiation and active propagation of action potentials in interneuron dendrites. Science. 2000;287:295–300. doi: 10.1126/science.287.5451.295. [DOI] [PubMed] [Google Scholar]
- Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. 2007;16:2892–2899. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- Martin MS, Dutt K, Papale LA, Dubé CM, Dutton SB, de Haan G, Shankar A, Tufik S, Meisler MH, Baram TZ, Goldin AL, Escayg A. Altered function of the SCN1A voltage-gated sodium channel leads to γ-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J Biol Chem. 2010;285:9823–9834. doi: 10.1074/jbc.M109.078568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Action potential initiation and propagation in CA3 pyramidal axons. J Neurophysiol. 2007;97:3460–3472. doi: 10.1152/jn.01288.2006. [DOI] [PubMed] [Google Scholar]
- Mistry AM, Thompson CH, Miller AR, Vanoye CG, George AL, Jr, Kearney JA. Strain-and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol Dis. 2014;65:1–11. doi: 10.1016/j.nbd.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, Yanagawa Y, Tamamaki N, Hensch TK, Itohara S, Yamakawa K. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet. 2013;22:4784–4804. doi: 10.1093/hmg/ddt331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Stuart GJ. Site of action potential initiation in layer 5 pyramidal neurons. J Neurosci. 2006;26:1854–1863. doi: 10.1523/JNEUROSCI.4812-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN. The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci. 2010;11:552–562. doi: 10.1038/nrn2852. [DOI] [PubMed] [Google Scholar]
- Redecker C, Luhmann HJ, Hagemann G, Fritschy JM, Witte OW. Differential downregulation of GABAA receptor subunits in widespread brain regions in the freeze-lesion model of focal cortical malformations. J Neurosci. 2000;20:5045–5053. doi: 10.1523/JNEUROSCI.20-13-05045.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009;87:41–57. doi: 10.1016/j.pneurobio.2008.09.016. [DOI] [PubMed] [Google Scholar]
- Royeck M, Horstmann MT, Remy S, Reitze M, Yaari Y, Beck H. Role of axonal NaV1.6 sodium channels in action potential initiation of CA1 pyramidal neurons. J Neurophysiol. 2008;100:2361–2380. doi: 10.1152/jn.90332.2008. [DOI] [PubMed] [Google Scholar]
- Rusconi R, Scalmani P, Cassulini RR, Giunti G, Gambardella A, Franceschetti S, Annesi G, Wanke E, Mantegazza M. Modulatory proteins can rescue a trafficking defective epileptogenic Nav1.1 Na+ channel mutant. J Neurosci. 2007;27:11037–11046. doi: 10.1523/JNEUROSCI.3515-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. 1997;120:479–490. doi: 10.1093/brain/120.3.479. [DOI] [PubMed] [Google Scholar]
- Schock SC, Jolin-Dahel KS, Schock PC, Theiss S, Arbuthnott GW, Garcia-Munoz M, Staines WA. Development of dissociated cryopreserved rat cortical neurons in vitro. J Neurosci Methods. 2012;205:324–333. doi: 10.1016/j.jneumeth.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Escayg A, Meisler MH, Goldin AL. Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. J Neurosci. 2001;21:7481–7490. doi: 10.1523/JNEUROSCI.21-19-07481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M. Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci. 2005;28:317–324. doi: 10.1016/j.tins.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Tang B, Dutt K, Papale L, Rusconi R, Shankar A, Hunter J, Tufik S, Yu FH, Catterall WA, Mantegazza M, Goldin AL, Escayg A. A BAC transgenic mouse model reveals neuron subtype-specific effects of a Generalized Epilepsy with Febrile Seizures Plus (GEFS+) mutation. Neurobiol Dis. 2009;35:91–102. doi: 10.1016/j.nbd.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wart A, Trimmer JS, Matthews G. Polarized distribution of ion channels within microdomains of the axon initial segment. J Comp Neurol. 2007;500:339–352. doi: 10.1002/cne.21173. [DOI] [PubMed] [Google Scholar]
- Weber YG, Lerche H. Genetic mechanisms in idiopathic epilepsies. Dev Med Child Neurol. 2008;50:648–654. doi: 10.1111/j.1469-8749.2008.03058.x. [DOI] [PubMed] [Google Scholar]
- Wimmer VC, Reid CA, Mitchell S, Richards KL, Scaf BB, Leaw BT, Hill EL, Royeck M, Horstmann MT, Cromer BA, Davies PJ, Xu R, Lerche H, Berkovic SF, Beck H, Petrou S. Axon initial segment dysfunction in a mouse model of genetic epilepsy with febrile seizures plus. J Clin Invest. 2010;120:2661–2671. doi: 10.1172/JCI42219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolson R, Clarke W. Statistical methods for the analysis of biomedical data. Ed 2. New York: Wiley-Interscience; 2002. [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Zuberi SM, Brunklaus A, Birch R, Reavey E, Duncan J, Forbes GH. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology. 2011;76:594–600. doi: 10.1212/WNL.0b013e31820c309b. [DOI] [PubMed] [Google Scholar]