

Abstract
Abnormal accumulation of β-amyloid (Aβ) is the major neuropathological hallmark of Alzheimer's disease (AD). However, the mechanisms underlying aberrant Aβ formation in AD remain unclear. We showed previously that inhibition of monoacylglycerol lipase (MAGL), the primary enzyme that metabolizes the endocannabinoid 2-arachidonoylglycerol (2-AG) in the brain, robustly reduces Aβ by inhibiting β-site amyloid precursor protein cleaving enzyme 1 (BACE1), a key enzyme responsible for Aβ formation. However, the molecular mechanisms responsible for suppression of BACE1 by inhibition of 2-AG metabolism are largely unknown. We demonstrate here that expression of the noncoding small RNA miR-188-3p that targets BACE1 was significantly downregulated both in the brains of AD humans and APP transgenic (TG) mice, a mouse model of AD. The downregulated miR-188-3p expression was restored by MAGL inhibition. Overexpression of miR-188-3p in the hippocampus reduced BACE1, Aβ, and neuroinflammation and prevented deteriorations in hippocampal basal synaptic transmission, long-term potentiation, spatial learning, and memory in TG mice. 2-AG-induced suppression of BACE1 was prevented by miR-188-3p loss of function. Moreover, miR-188-3p expression was upregulated by 2-AG or peroxisome proliferator-activated receptor-γ (PPARγ) agonists and suppressed by PPARγ antagonism or NF-κB activation. Reducing Aβ and neuroinflammation by MAGL inhibition was occluded by PPARγ antagonism. In addition, BACE1 suppression by 2-AG and PPARγ activation was eliminated by knockdown of NF-κB. Our study provides a novel molecular mechanism underlying improved synaptic and cognitive function in TG mice by 2-AG signaling, which upregulates miR-188-3p expression through PPARγ and NF-κB signaling pathway, resulting in suppressions of BACE1 expression and Aβ formation.
Keywords: endocannabinoids, miRNA sponge, monoacylglycerol lipase, NF-κB, noncoding small RNA, PPARγ
Introduction
2-Arachidonoyl glycerol (2-AG), the most abundant endogenous cannabinoid, is primarily hydrolyzed by monoacylglycerol lipase (MAGL) in the brain, resulting in the release of the free fatty acid arachidonic acid, a precursor of prostaglandins and leukotrienes (Sugiura, 2009; Blankman et al., 2007; Long et al., 2009a). 2-AG exhibits anti-inflammatory and neuroprotective properties (Panikashvili et al., 2001; Zhang and Chen, 2008; Bisogno and Di Marzo, 2010; Chen et al., 2011), whereas its metabolites prostaglandins and leukotrienes are proinflammatory and neurtoxic and involved in neurodegenerative diseases, such as Alzheimer's disease (AD) (Salmon and Higgs, 1987; Hein and O'Banion, 2009; Hensley, 2010). This information suggests that regulation of 2-AG signaling by MAGL plays an important role in neuroinflammatory and neurodegenerative processes (Mulvihill and Nomura, 2013; Xu and Chen, 2014). Recent studies demonstrate that pharmacological or genetic inhibition of MAGL reduces β-amyloid (Aβ) formation, neuroinflammation, and neurodegeneration and improves LTP, spatial memory in APP transgenic (TG) animals (Chen et al., 2012; Piro et al., 2012), suggesting that MAGL is a new therapeutic target for prevention and treatment of AD. However, the molecular mechanisms responsible for these beneficial effects produced by inhibition of 2-AG metabolism in AD remain unclear. In particular, the anti-inflammatory and neuroprotective effects produced by MAGL inactivation are apparently mediated neither via the CB1 nor CB2 receptors (Nomura et al., 2011; Chen et al., 2012; Piro et al., 2012).
MicroRNAs (miRNAs) are small noncoding RNAs that repress expression and function of target proteins at translational levels by binding to 3′-UTR of target mRNAs (Bartel, 2009; Wang et al., 2012). Several studies show that miRNA dysfunction in the brain contributes to neurodegenerative diseases (Junn and Mouradian, 2010; Im and Kenny, 2012; Abe and Bonini, 2013). Indeed, it has been shown that several miRNAs are upregulated or downregulated both in AD patients and animal models, suggesting important roles of miRNAs in pathogenesis and neuropathology of AD (Delay et al., 2012; Satoh, 2012; Schonrock and Götz, 2012). However, little is known about whether restoring or reversal of deregulated miRNAs is capable of counteracting neuropathology of AD. β-Site amyloid precursor protein cleaving enzyme 1 (BACE1) is a key enzyme responsible for Aβ formation. It is possible that elevated expression of BACE1 in AD may arise from misregulated expression of miRNAs that directly or indirectly regulate BACE1 expression, resulting in aberrant Aβ formation. Decreases in Aβ by MAGL inhibition in TG animals are likely associated with the suppression BACE1 expression (Chen et al., 2012). Here we show that suppression of BACE1 by inhibition of 2-AG metabolism is likely associated with upregulating miR-188-3p targeting BACE1 in TG mice. Overexpression of miR-188-3p in the hippocampus resulted in decreases in BACE1 expression, Aβ synthesis, and neuroinflammation and improvements in LTP and spatial memory in TG animals. miR-188-3p expression was upregulated by 2-AG signaling through increasing PPARγ activity and expression and suppressing NF-κB activity, resulting in repression of BACE1 expression and Aβ formation. Our study provides a novel signaling pathway that mediates the beneficial effects produced by inhibition of 2-AG metabolism in AD.
Materials and Methods
Animals and chemicals.
5XFAD TG mice, expressing both mutant human APP(695) with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) Familial Alzheimer's Disease (FAD) mutations and human PS1 harboring two FAD mutations, M146L and L286V (Oakley et al., 2006), were obtained from The Jackson Laboratory (stock #006554). Breeder mice homozygous for retinal degeneration 1 (Pde6b) mutation were removed from the colonies. Therefore, mice used in the present studies do not have Pde6b mutants. A total of 298 female TG and age-matched wild-type (WT) littermates 4–6 months of age were used in the present study. All animal studies were performed in compliance with the Department of Health and Human Services Guide for the Care and Use of Laboratory Animals, and the care and use of the animals reported in this study were approved by the Institutional Animal Care and Use Committee of Louisiana State University Health Sciences Center.
4-Nitrophenyl-4-[bis(1,3-benzodioxol-5-yl)(hydroxy)methyl]piperidine-1-carboxylate (JZL184), a potent and selective inhibitor of MAGL, was dissolved in the vehicle containing Tween 80 (10%), DMSO (10%), and saline (80%). TG mice were treated with vehicle or JZL184 (12 mg/kg) three times per week by intraperitoneal injections starting at 4 months of ages for 8 weeks as described previously (Chen et al., 2012, 2013). In some experiments, mice received JZL184 plus GW9662 (GW, 5 mg/kg) three times/week starting at 4 months of age for 8 weeks.
RNA samples from human brain tissues.
RNA samples from human brain tissues used in this study were provided by Dr. W. Lukiw of Louisiana State University Health Sciences Center. The use of human tissues and RNA samples was in accordance with and approved by the Institutional Review Board at the Louisiana State University Health Sciences Center. These human brain tissues were from the temporal lobe of normal controls at ages from 83 to 97 years (91 ± 2 years) with the mean postmortem interval 3.7 ± 0.5 h, and AD patients at ages from 87 to 96 years (90 ± 1 years) with postmortem interval 3.6 ± 0.5 h.
Cell culture.
Primary hippocampal neurons (astroglial cells ∼5%), HEK 293, HEK293T, and NG108-15 cells were cultured as described previously (Sang et al., 2005; Zhang and Chen, 2008; Chen et al., 2013).
miRNA microarray analysis.
Expression of miRNAs in the hippocampus was profiled using GenoExplorer microRNA microarray analysis (GenoSensor) in APP TG and WT mice that received vehicle or JZL184 three times/week starting at 2 months of age for 8 weeks. RNAs were sampled and isolated 1 d after cessation of the last injection.
Plasmid and lentiviral constructs.
The TargetScan (http://www.targetscan.org), microRNA.org (http://www.microrna.org/microrna/home.do), and mirbase.org (http://www.mirbase.org) webs were used for computational prediction of miRNA targets. Two computationally predicted binding sites (BSs) of miR-188-3p in the 3′UTR of BACE1 (BS1, nt 1511-1517; BS2, nt 1597-1603) were identified. The sequences of pre-miR-188-3p were cloned in the miRNASelectTM pEGP expression vector (Cell Biolab), and the sequences of the complete 3′UTR of mouse BACE1 (NCBI accession number BC048189) were amplified by PCR and cloned in the psiCHECK vector (Promega). Mutations of the BS sequences were made in the 3′UTR of BACE1 recognized by the seed region of miR-188-3p. All the constructs were verified by sequencing. The FUGW2.1 lentiviral vector (LV, provided by Dr. B. Luikart of Dartmouth Medical School) was used to insert mature miR-188-3p driven by U6 promoter and GFP reporter gene driven by ubiquitin promoter. pCMVΔ8.9 and VSVg vectors were used for viral envelope and production. FUGW lentiviruses were generated and packaged in HEK293T cells and titered by FACS analysis using flow cytometry (Chen et al., 2013). The titer of the LV at least 1.0 × 109 infection functional units/ml was used for in vivo injections.
The FUGW LV was also used for PPARγ and NF-κB p65 shRNA in primary cultured hippocampal neurons (Chen et al., 2013). The oligos for the PPARγ (AB644275.1) shRNA construct (5′TGACCAAGTGACTCTGCTCAA3′) and scramble (5′ AGTTAGCAACCTAGCCGATCT3′) were used for knockdown of PPARγ; and the oligos for the NF-κB p65 (NM_009045.4) shRNA construct (5′AGGACCTATGAGACCTTCAAG3′) and scramble: (5′GAATCATTACGCGAGACTGCA3′) were used for knockdown of NF-κB p65.
To knock down miR-188-3p, we used the lentiviral-based miRNA “sponge” technique, which renders an efficient and permanent miRNA loss of function by complementary binding to an miRNA of interest (Ebert et al., 2007; Edbauer et al., 2010). The LIB01 lentiviral vector was used for the construct of miR-188-3p sponge, which contained six concatenated miR-188-3p BSs with a central bulge/mismatch to prevent mRNA cleavage by Argonaute 2 (Edbauer et al., 2010). The sponge senses (6×) forward: CTAGCTGCAAACCCACGTTGTGGGAGaattTGCAAACCCACGTTGTGGGAGaattTGCAAACCCACGTTGTGGGAGaattTGCAAACCCACGTTGTGGGAGaattTGCAAACCCACGTTGTGGGAGaattTGCAAACCCACGTTGTGGGAGaattGG; and reverse: CGCGCC AATTCTCCCACAACGTGGGTTTGCAAATTCTCCCACAACGTGGGTTTGCAAATTCTCCCACAACGTGGGTTTGCAAATTCTCCCACAACGTGGGTTTGCAAATTCTCCCACAACGTGGGTTTGCAAATTCTCCCACAACGTGGGTTTGCAG. Control and miR-188-3p sponge LV were expressed in cultured hippocampal neurons.
Functional validation.
The putative miR-188-3p recognition in the 3′UTR of BACE1 was functionally validated by the luciferase reporter vector expressing 3′UTR of BACE1, or mutated BS1, BS2, and BS1 + BS2 in the 3′UTR of BACE1 cotransfected with the pEGP vector expressing pre-miR-188 in HEK293 cells. The reporter activity (light units) was detected using a microplate luminometer.
Stereotaxic injection.
WT or TG mice at 4 months of age were anesthetized with ketamine/xylazine (200/10 mg/kg) and placed in a stereotaxic frame. LV-scramble control or LV-miR-188-3p (2 μl at 0.2 μl /min) was injected into each side of the hippocampus, primarily in the dentate gyrus area, at the coordinate: anteroposterior, −2; mediolateral, ±1.8; and dorsoventral, −2. Immunobot, qPCR, immunostaining, electrophysiological recordings, and behavioral assays were performed 8 weeks after LV injections.
Reverse transcription and real-time PCR.
Total RNA was prepared from harvested tissue or cells with the RNeasy Mini Kit (QIAGEN) and treated with RNase-free DNase (QIAGEN) according to the manufacturer's instructions. The iScript cDNA synthesis kit (Bio-Rad) was used for the reverse transcription reaction. We used 1 μg total RNA, with 4 μl 5× iScript reaction mix and 1 μl iScript reverse transcriptase. The total volume was 20 μl. Samples were incubated for 5 min at 25°C. All samples were then heated to 42°C for 30 min, and reactions were stopped by heating to 85°C for 5 min. The primers for pre- and miR-188-3p were obtained from QIAGEN. Real-time RT-PCR-specific primers for PPARγ and GAPDH were selected using Beacon Designer Software (Bio-Rad) and synthesized by IDT. They are listed as follows: name: forward primer, reverse primer (amplicon size), genebank number: PPARγ: 5′TATGACCTGAAGCTCCAAGAATACC3′, 5′CCACAGACTCGGCACTCAATG3′ (145 bp), NM_11146; GAPDH: 5′ACCACAGTCCATGCCATCAC3′, 5′ACCTTGCCCACAGCCTTG3′ (134 bp), M32599. All the PCR products were verified by sequencing. Samples were compared using the relative CT method. The fold increase or decrease was determined relative to a vehicle-treated control after normalizing to a housekeeping gene using 2−ΔΔCT, where ΔCT is (gene of interest CT) − (GAPDH CT), and ΔΔCT is (ΔCT treated) − (ΔCT control), as described previously (Sang et al., 2005; Zhang and Chen, 2008; Chen et al., 2012, 2013).
Chromatin immunoprecipitation (ChIP) analysis.
ChIP analysis was performed as described previously (Chen et al., 2013) to determine the binding of NF-κBp65 in the promoter of the miR-188-3p gene according to the manufacturer's instruction (EMD Millipore). The potential NF-κB BS(s) in the promoter region was identified using the TFSEARCH (http://www.cbrc.jp/research/db/TFSEARCH.html). The primers for CHIP are as follows: forward primer, 5′CAGGGAGACAGAAGCCAGAC3′; and reverse primer, 5′ CTCCTGTACCTTCCCTGCAT3′ for the BS1 and 5′TCTCATGTGCTCTCTGTGTGG3′, and 5′ ACTGATGGTGGGTAAGGCAG3′ for the BS2.
Luciferase activity assay.
PPARγ activity stimulated by 2-AG was assessed in NG108–15 cells transfected with pCMX-Gal-LBD-mPPARγ and TK-MH100x4-luc vectors (provided by Dr. Ronald Evans of the Salk Institute). Luciferase values were normalized to the level of β-galactosidase activity. The effect of 2-AG and miR-188-3p on BACE1 promoter activity was detected in HEK293 cells transfected with pGL3 luciferase reporter vector (Promega) and pB1-A BACE1 promoter luciferase construct (provided by Dr. Weihong Song of the University of British Columbia) as described previously (Ly et al., 2013). The miRNASelect pEGP-miR Cloning and Expression Vector (Cell Biolabs) was used for expressing miR-188-3p and the pEGP-miR-null vector for the control. The NF-κB luciferase reporter assay kit (QIAGEN) was used to determine the effect of 2-AG and PPARγ on NF-κB promoter activity according to the procedures described by the manufacturer.
Immunoblot.
Western blot assay was conducted to determine production of Aβ42 and expression of BACE1, NF-κB phosphorylation, and glutamate receptor subunits in the brain from TG mice that received LV-scramble control or -miR-188-3p. Hippocampal tissue was extracted and immediately homogenized in RIPA lysis buffer and protease inhibitors and incubated on ice for 30 min, then centrifuged for 10 min at 10,000 rpm at 4°C. Supernatants were fractionated on 4%–15% SDS-PAGE gels and transferred onto PVDF membranes (Bio-Rad). A special 16.5% Criterion Tris-Tricine Gelforgel (Bio-Rad) was used for detecting Aβ42 immunoreactivity. The membrane was incubated with the following: anti-Aβ42 (1:1000, Invitrogen, catalog #44-344, RRID:AB_2313572), anti-BACE1 (1:1000, Covance, catalog #PRB-617C-100 RRID:AB_10063987), anti-p-NF-κB (1:1000, Cell Signaling Technology, catalog #3039S RRID:AB_330579), anti-GluA1 (1:1000, EMD Millipore, catalog #AB1504 RRID:AB_2113602), GluA2 (1:1000, EMD Millipore, catalog #MAB397 RRID:AB_2113875), GluN1 (1:500, EMD Millipore, catalog #MAB363 RRID:AB_94946), GluN2A (1:1000, EMD Millipore, catalog #AB1555P RRID:AB_90770), and GluN2B (1:1000, Abcam, catalog #ab73001 RRID:AB_1269571) at 4°C overnight. The blots were washed and incubated with a secondary antibody (goat anti-rabbit 1:2000, Cell Signaling Technology, catalog #7074 RRID: AB_2099233) at room temperature for 1 h. Proteins were visualized by enhanced chemiluminescence (GE Healthcare). The densities of specific bands were quantified by densitometry using FUJIFILM Multi Gauge software (version 3.0). Band densities were quantified and converted to the total amount of protein loaded in each well as determined by mouse anti β-actin (1:4000, Sigma-Aldrich, catalog #A2228 RRID:AB_476697) as described previously (Zhang and Chen, 2008; Du et al., 2011; Chen et al., 2012, 2013).
Aβ42 ELISA.
Levels of Aβ42 in hippocampal tissues of TG mice that received scramble control and miR-188-3p lentiviruses were detected using a colorimetric Aβ42 ELISA kit (AnaSpec) according to the instructions provided by the manufacturer.
Immunocytochemistry and immunohistochemistry.
Immunostaining analyses were performed to determine total Aβ, Aβ42, astrocytic, and microglial markers (GFAP and CD11b) in coronal sectioned brain slices and BACE1 expression in cultured neurons as described previously (Sang et al., 2005; Chen et al., 2012, 2013). Mice were anesthetized with ketamine/xylazine (200/10 mg/kg) and subsequently transcardially perfused with PBS followed by 4% PFA in phosphate buffer. The brains were quickly removed from the skulls and fixed in 4% PFA overnight, and then transferred into the PBS containing 30% sucrose until sinking to the bottom of the small glass jars. Cryostat sectioning was made on a freezing Vibratome at 40 μm, and series sections (10–12 slices) were collected in 0.1 m phosphate buffer. Free floating sections or cultured neurons were immunostained using antibodies specific for total Aβ (4G8, 1:2000, Covance, catalog #SIG-39220-200 RRID: AB_10174824), Aβ42 (1:1000, Invitrogen, catalog #44-344, RRID:AB_2313572), GFAP (1:200, Sigma-Aldrich, catalog #G3893 RRID:AB_477010), and OX42/CD11b (1:200, Abcam, catalog #ab1211 RRID:AB_442947) followed by incubation with the corresponding fluorescent-labeled secondary antibody. DAPI, a fluorescent stain that binds strongly to DNA, was used to detect cell nuclei in the sections. The sections were then mounted on slides for immunofluorescence detection using a Zeiss deconvolution microscope with SlideBook 5.5 software. Every 10th section from 6 animals per group was analyzed. The numbers and the area of total Aβ and Aβ42 plaques, and the fluorescence intensity (in arbitrary densitometric units) of GFAP and OX42/CD11b in the hippocampal area in each image were analyzed and quantified using SlideBook 5.0 and NIH ImageJ software as described previously (Chen et al., 2012, 2013).
Hippocampal slice preparation.
Mouse hippocampal slices were prepared as described previously (Chen et al., 2002, 2012, 2013; Fan et al., 2010). Briefly, after decapitation, brains were rapidly removed and placed in cold oxygenated (95% O2/5% CO2) ACSF. Slices were cut at a thickness of 350–400 μm and transferred to a holding chamber in an incubator containing oxygenated ACSF containing 3 pyruvic acid and 1 ascorbic acid at 36°C for 0.5–1 h, and then maintained in an incubator containing oxygenated ACSF at room temperature (∼22°C–24°C) for >1.5 h before recordings. Slices were then transferred to a recording chamber where they were continuously perfused with 95% O2/5% CO2-saturated standard ACSF at ∼32°C–34°C.
Electrophysiological recordings.
Field EPSP (fEPSP) recordings were made in response to stimulation at perforant path synapses in the dentate gyrus of the hippocampus at a frequency of 0.05 Hz using an Axoclamp-2B patch-clamp amplifier in bridge mode. Hippocampal LTP was induced by a theta burst stimulation as described previously (Fan et al., 2010; Chen et al., 2012, 2013). The input–output function was tested before recording of LTP, and the baseline stimulation strength was set to provide fEPSP with an amplitude of ∼30% from the subthreshold maximum derived from the input–output function.
Morris water maze.
The classic Morris water maze test was used to determine spatial learning and memory as described previously (Chen et al., 2012, 2013). A circular water tank (diameter 120 and 75 cm in high) was filled with water, and a round platform (diameter 15 cm) was hidden 1 cm beneath the surface of the water at the center of a given quadrant of the water tank. Before hidden platform training, the mice were given 3 d of nonspatial training (4 trials per day) to find the platform above the water. Animals that failed to find the platform were gently guided to the platform and allowed to stay on it for 10 s. Invisible platform training was performed continuously for 7 d (7 sessions), and each session consisted of 4 trials. For each trial, the mouse was released from the wall of the tank and allowed to search, find, and stay on the platform for 10 s within the 60 s trial period. For each training session, the starting quadrant and sequence of the four quadrants from where the mouse was released into the water tank were randomly chosen so that it was different among the separate sessions for each animal and was different for individual animals. The mice in the water pool were recorded by a video-camera, and the task performances, including swimming paths, speed, and time spent in each quadrant, were recorded by using an EthoVision video tracking system (Noldus). A probe test was conducted 24 h after the completion of the training. During the probe test, the platform was removed from the pool, and the task performances were recorded for 60 s. The time spent in each quadrant was analyzed.
Open field.
The “open field” test was conducted to determine the effects of miR-188-3p overexpression on motor function and anxiety using an automatic-recording open-field working station (MED Associates) as described previously (Chen et al., 2013). During the test, mice were individually released into the center of the box and allowed to explore the field for 30 min. The animal behaviors were recorded by a photobeam-scanning system, and the data were analyzed automatically by the computer-sampling system.
Rotarod.
The rotarod test was used to determine sensorimotor function in animals that received control and miR-188-3p lentiviruses as described previously (Jiao et al., 2008). The test was performed on an accelerating rotarod apparatus (MED Associates) starting at an initial rotation of 4 RPM accelerating to 40 RPM over 5 min. Mice were given three trials every day with a 30 min intertrial interval, and each trial was ended by either falling down from the rod or staying on it over 300 s. The time for the mouse keeping on the rod was recorded and averaged from three trials in each training session.
Data analysis.
Data are presented as mean ± SEM. Unless stated otherwise, ANOVA with post hoc tests were used for statistical comparison when appropriate. Differences were considered significant when p < 0.05.
Results
Inhibition of 2-AG metabolism restores the downregulated miR-188-3p expression in TG animals
Aβ is the initiator in the etiology and pathogenesis of AD, and BACE1 is the key enzyme responsible for Aβ formation. We demonstrated previously that MAGL inactivation by JZL184, a highly selective and potent MAGL inhibitor (Long et al., 2009a, b), robustly reduces BACE1 expression and Aβ plaques in TG mice (Chen et al., 2012). To determine epigenetic mechanisms by which inhibition of 2-AG metabolism reduces BACE1 and Aβ, we profiled expression of miRNAs in hippocampal tissue of 4-month-old TG mice using the GenoExplorer microRNA microarray analysis. Animals at 2 months of age received vehicle or JZL184 (12 mg/kg, i.p.) three times per week for 8 weeks, as described previously (Chen et al., 2012). As shown in Figure 1A, a significant number of miRNAs was upregulated or downregulated in TG animals compared with the WT control. It appears that the altered expressions of miRNAs in TG mice were reversed in animals that received JZL184. Figure 1B lists the most significantly upregulated or downregulated miRNAs in TG animals that received vehicle or JZL184 when normalized to that of the WT control. We noticed that expression of miR-188-3p, which has computationally predicted BSs in the 3′UTR of BACE1 (Fig. 1D), was significantly decreased to 0.25 ± 0.02 in vehicle-treated TG and increased to 1.30 ± 0.04 in JZL184-treated TG animals when normalized to the WT controls. miR-188-3p is a previously unrecognized miRNA that targets BACE1. As demonstrated previously, 5XFAD mice exhibit deficits in synaptic and cognitive function starting at ages of 5–6 months (Oakley et al., 2006; Chen et al., 2012). To confirm whether expression of miR-188-3p is still altered in 6-month-old TG animals, we assessed the expression in TG mice-treated with JZL184 for 8 weeks starting at 4 months of age. As seen in Figure 1C, expression of miR-188-3p was still significantly downregulated in 6-month-old TG animals, and this decrease was rescued by JZL184. These results indicate that inhibition of 2-AG metabolism counteracts deregulation of miRNA expressions in TG animals. To determine whether expression of miR-188-3p is also downregulated in the brains of AD patients, we assessed expression of miR-188-3p in RNAs extracted from a cohort of 8 AD patients and 8 age-matched normal subjects. As shown in Figure 1C, expression of miR-188-3p was also significantly reduced in the brains of AD patients. Our results indicate that expression of miR-188–3p is downregulated in the brains of both AD humans and TG animals, suggesting that downregulated expression of miR-188-3p may contribute to aberrant BACE1 expression and Aβ formation in AD.
Figure 1.

Reduced expression of miR-188-3p targeting BACE1 is rescued by inhibition of 2-AG metabolism in 5XFAD APP TG mice. A, Expression profiling of miRNAs that are robustly altered in TG mice and reversed by JZL184 in the hippocampus using the miRNA microarray analysis (n = 3 animals/group). B, List of the most significantly upregulated or downregulated miRNAs in TG animals that received vehicle or JZL184 compared with the WT control using the miRNA microarray analysis. C, Expression of miR-188-3p is significantly reduced in the brains of both AD humans (n = 8) and TG animals, and the decrease is restored by inhibition of 2-AG metabolism in TG mice (n = 6). D, Two BSs of miR-188-3p in the 3′UTR of BACE1. E, Luciferase reporter construct (psiCHECK vector) of WT (normal) and mutated BSs in the 3′UTR of BACE1 recognized by the seed region of miR-188-3p. F, Expression of pre-miR-188-3p using the pEGP vector decreases the luciferase reporter activity in 293 cells, (G) but fails to inhibit the reporter activity in cotransfection of BS1 or 2 mutated reporter vectors (n = 6). Data are mean ± SEM. **p < 0.01, compared with the vehicle or normal controls. ##p < 0.01, compared with TG vehicle (one-way ANOVA with Fisher's PLSD test).
Functional validation of the predicted binding of miR-188-3p in the 3′UTR of BACE1
Based on computationally predicted BSs, miR-188-3p has at least two BSs in the 3′UTRs of BACE1 (Fig. 1D,E). To functionally validate these putative BSs, we used the luciferase reporter construct (psiCHECK vector, Promega) inserted with the sequences of the complete 3′UTR of mouse BACE1. As seen in Figure 1E, F, cotransfection of the reporter vector with the pEGP vector expressing pre-miR-188 in 293 cells resulted in inhibition of the reporter activity compared with the control vector. Then, we mutated the BS sequences in the 3′UTR of BACE1 recognized by the seed region of miR-188-3p and observed that cotransfection of the BS1 or BS2 mutated reporter vectors with the pEGP vector expressing pre-miR-188-3p led to a loss of inhibition of the reporter activity when compared with the control (WT; Fig. 1E,G). Mutations in both BS1 and BS2 further augmented this effect (Fig. 1G). These results indicate that miR-188-3p functionally binds to the 3′UTR of BACE1 but fails to inhibit the reporter activity when its BS sequences in the 3′UTR of BACE1 are mutated.
miR-188-3p suppresses BACE1 expression
If miR-188-3p is capable of functional binding to the 3′UTR of BACE1, then overexpression of it should suppress BACE1 expression. To test this prediction, we first detected BACE1 expression in cultured hippocampal neurons overexpressing miR-188-3p using the FUGW lentiviral vector (LV) (Luikart et al., 2011; Chen et al., 2013). As shown in Figure 2A, overexpression of miR-188-3p led to a suppression of BACE1, as detected by immunostaining in cultured hippocampal neurons. Suppression of BACE1 by miR-188-3p was further confirmed by immunoblot analysis (Fig. 2B). These results from in vitro preparations indicate that overexpression of miR-188-3p represses BACE1 expression. To determine whether miR-188-3p inhibits BACE1 in vivo, we stereotaxically injected LV into the hippocampus (Fig. 2C) to overexpress miR-188-3p in TG mice, which exhibit elevated expression of BACE1 (Zhao et al., 2007; Chen et al., 2012). The injected LV was well localized in the hippocampal area indicated by GFP fluorescence (Fig. 2C). Overexpression of miR-1883p significantly reduced BACE1 expression in the hippocampus compared with the LV scramble control in TG mice (Fig. 2D). We used GFP expression as a marker to indicate the viral infection in the immunoblot assay (Fig. 2D). The results from in vitro and in vivo studies show that miR-188-3p functionally represses BACE1 expression.
Figure 2.

Overexpression of miR-188-3p reduces BACE1 expression both in vitro and in vivo. A, Immunostaining analysis of BACE1 expression in cultured hippocampal neurons treated with LVs expressing scramble control or miR-188-3p. Imaging was taken 5 d after transfection. Scale bars, 20 μm. B, Immunoblot analysis of BACE1 expression in hippocampal neurons transfected with LV expressing miR-188-3p. Data are mean ± SEM. **p < 0.01, compared with the scramble control (ANOVA with Fisher's PLSD test, n = 3). C, Stereotaxic injection sites of LV, hippocampal expression of GFP after LV injection, and enlarged GFP expression in dentate granule neurons and CA1 pyramidal neurons. Scale bars, 20 and 200 μm. D, BACE1 expression in hippocampal tissue of WT and TG mice at 6 months of age injected with LV-scramble control or LV-miR-188-3p for 8 weeks. Expression of GFP was used as the control. **p < 0.01, compared with the WT-LV control. ##p < 0.01, compared with the TG-LV control (n = 3). E, 2-AG (3 μm) reduces BACE1 expression in cultured hippocampal neurons. **p < 0.01, compared with the vehicle control (n = 3). F, miR-188-3p loss of function blocks the 2-AG (3 μm)-induced decrease in BACE1 expression in culture treated with LV-control (LV-Con) or LV-miR-188-3p sponge (LV-188-s). **p < 0.01, compared with the LV-control (n = 4).
As we reported previously (Chen et al., 2012), inhibition of MAGL by JZL184 reduces BACE1 expression in TG mice. To confirm whether this effect is mediated via 2-AG, we detected BACE1 expression in cultured hippocampal neurons in the presence of 2-AG. As shown in Figure 2E, 2-AG (3 μm) significantly reduced BACE1 expression. To further determine whether the effect of 2-AG on BACE1 expression is mediated through miR-188-3p, we used the miRNA “sponge” technique, which renders a continuous miRNA loss of function by complementary binding to a miRNA of interest (Ebert et al., 2007; Edbauer et al., 2010). As shown in Figure 2F, knockdown of miR-188-3p in cells by miR-188–30 sponge eliminated 2-AG-induced suppression of BACE1, whereas 2-AG still suppressed BACE1 expression in cells treated with the scramble controls. These results indicate that the suppression of BACE1 by 2-AG is mediated through miR-188-3p.
Overexpression of miR-188-3p decreases Aβ in TG animals
We demonstrated previously that inhibition of 2-AG hydrolysis by JZL184 reduces BACE1 expression and Aβ formation (Chen et al., 2012). Also, inhibition of 2-AG metabolism reversed downregulated expression of miR-188-3p, as shown in Figure 1. We next asked whether overexpression of miR-188-3p is able to reduce Aβ production in TG animals by suppressing BACE1. To test this prediction, we measured total Aβ and Aβ42 in TG animals that received LV-scramble control and LV-miR-188-3p at 4 months of age. Total Aβ and Aβ42 were detected 2 months after LV injections. As illustrated in Figure 3, total Aβ and Aβ42 were significantly reduced in the hippocampus by miR-188-3p overexpression using immunoblot, immunostaining, and ELISA. This information suggests that suppression of Aβ production by inhibition of 2-AG metabolism likely results from upregulating miR-188-3p, which suppresses BACE1, thereby decreasing Aβ formation and accumulation in TG animals.
Figure 3.
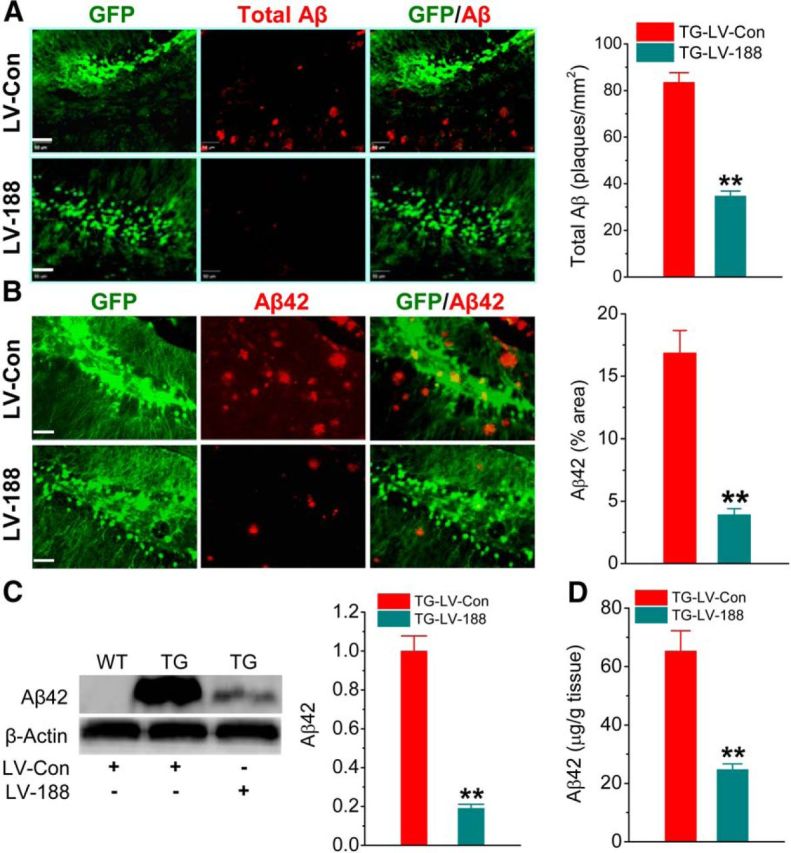
Overexpression of miR-188-3p reduces Aβ formation. A, Total Aβ (all forms) detected by anti-4G8 antibody in the hippocampus of 6-month-old TG mice that received LV-scramble control or LV-miR-188-3p at 4 months of age. B, C, D, Aβ42 is reduced in the hippocampus of TG mice that received LV-miR-188-3p using immunostaining, Western blot, and ELISA analyses. **p < 0.01, compared with the TG-LV scramble control (ANOVA with Bonferroni post hoc or Fisher's PLSD test); n = 4–6 mice per group. Scale bar, 50 μm.
miR-188-3p suppresses neuroinflammation
Neuroinflammation is one of the major pathogenic mechanisms that lead to synaptic and cognitive deficits in AD. Previous studies have shown that MAGL inactivation reduces gliosis in terms of activated astrocytes and microglia (Nomura et al., 2011; Chen et al., 2012; Piro et al., 2012). Because neuroinflammation is closely associated with increased production of Aβ in AD, we asked whether an increase in miR-188-3p expression decreases neuroinflammation in TG animals. Reactive astrocytes and microglia were determined using specific markers (GFAP and OX42/CD11b) in TG animals. As shown in Figure 4A, B, an increase in miR-188-3p expression significantly reduced expression of GFAP and CD11b in the hippocampus, suggesting that overexpression of miR-188-3p reduces neuroinflammation in TG mice.
Figure 4.

Overexpression of miR-188-3p decreases gliosis in TG animals. A, Hippocampal GFAP (astrocytic marker) expression is suppressed by miR-188-3p. LV-control or miR-188-3p was injected into the hippocampi at 4 months of age, and immunostaining was conducted at 6 months of age. B, Expression of CD11b (microglial marker) is suppressed by miR-188-3p overexpression. Data are mean ± SEM. **p < 0.01, compared with the TG-LV scramble control (n = 6 mice/group).
Overexpression of miR-188-3p prevents synaptic dysfunction
It is well established that increased Aβ and neuroinflammation are detrimental to synaptic and cognitive function in AD. Because miR-188-3p significantly reduces Aβ and gliosis, we expected that increase in miR1–88-3p expression would be able to improve synaptic function in TG animals. As shown in Figure 5A, B, hippocampal basal synaptic transmission (in terms of input–output function) and LTP were significantly improved by overexpression of miR-188-3p in TG animals. The results indicate that overexpression of miR-188-3p improves hippocampal basal synaptic transmission and long-term synaptic plasticity in AD animals.
Figure 5.

Overexpression of miR-188-3p improves basal synaptic transmission and LTP and rescues expression of glutamate receptor subunits in TG mice. A, Input–output function recorded at hippocampal perforant synapses in 6-month-old TG injected with LV-scramble or -miR-188-3p for 8 weeks. Stimulus intensity was normalized to the maximum intensity. Data are mean ± SEM averaged from 9–12 recordings/group and 6–8 mice/group. B, LTP curves and mean ± SEM of fEPSP slope averaged from 56 to 60 min after theta burst stimulation (TBS) in 6-month-old TG mice injected with LV scramble or -miR-188-3p for 8 weeks. **p < 0.01, compared with the TG-LV scramble control (n = 8–10 recordings/group and 5–7 mice/group). C, Immunoblot analysis of hippocampal expression of AMPA (GluA1 and GluA2) and NMDA (GluN1, GluN2A, and GluN2B) receptor subunits in 6-month-old TG mice injected with LV-scramble or -miR-188-3p for 8 weeks. Data are mean ± SEM. *p < 0.05, compared with the TG-LV-scramble control (n = 3 mice/group). **p < 0.01, compared with the TG-LV-scramble control (n = 3 mice/group).
The functional integrity of excitatory synapses is largely dependent on expression of glutamate receptors. We demonstrated previously that expressions of glutamate receptor subunits are significantly reduced in TG animals and rescued by inhibition of 2-AG metabolism (Chen et al., 2012). To determine whether improved basal synaptic transmission and LTP by miR-188-3p is associated with upregulation of glutamate receptor expression in TG mice, we assessed expression of glutamate receptor subunits GluA1, GluA2, GluN1, GluN2A, and GluN2B in TG mice that received LV-miR-188-3p. As shown in Figure 5C, expression of these subunits in the hippocampus was significantly increased in TG animals overexpressed with miR-188-3p compared with the LV-scramble control. This information suggests that improvements in synaptic function by miR-188-3p overexpression are likely associated with the restoration of glutamate receptor subunit expressions in TG animals.
Improvements in cognitive function by miR-188-3p overexpression
The most important characteristic in AD is the impairments in learning and memory. To determine whether overexpression of miR-188-3p is able to improve spatial learning and memory, we used the classic Morris water maze test. As shown in Figure 6A, TG mice that received LV-miR-188-3p exhibited improved behavioral performance in the water maze test, suggesting that miR-188-3p may have potential for therapeutic intervention to improve cognitive function in AD.
Figure 6.
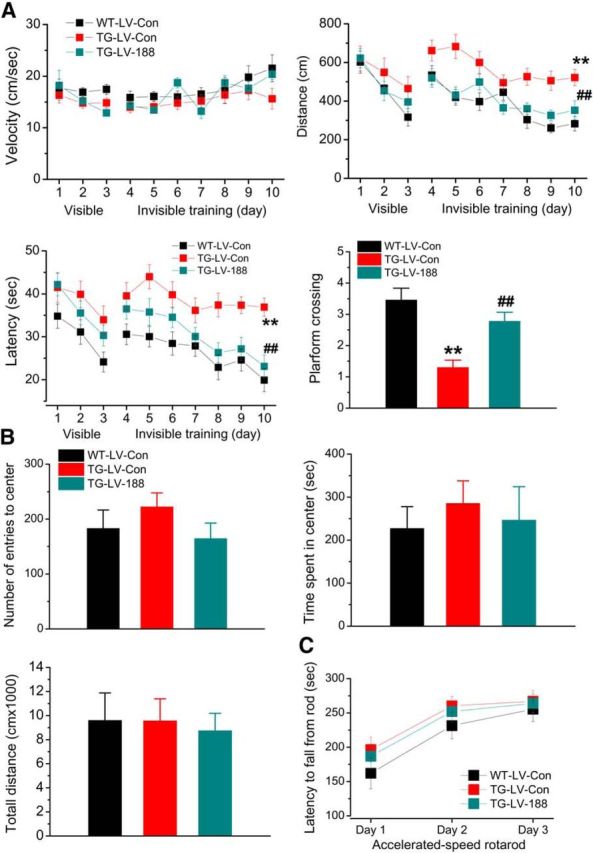
Overexpression of miR-188-3p prevents deterioration in cognitive function in TG mice. A, Spatial learning and memory in TG mice are improved by overexpression of miR-188-3p. The Morris water maze test was performed in 6-month-old TG mice that received LV-scramble or -miR-188-3p for 8 weeks. The probe test was conducted 24 h after 7 d of invisible training. Data are mean ± SEM. **p < 0.01, compared with the WT-LV-scramble control. ##p < 0.01, compared with TG-LV-scramble control (n = 17–23 mice/group). B, The open field test was conducted in TG 6-month-old TG mice that received LV-scramble or -miR-188-3p for 8 weeks. Overexpression of miR-188-3p does not significantly affect behavioral performance. C, Sensorimotor function is not affected by miR-188-3p overexpression in TG mice in an accelerated-speed rotarod test (n = 9–11 mice/group).
An important issue for overexpressing miR-188-3p in the hippocampus is whether this intervention would affect sensorimotor function and/or anxiety. To answer this question, we conducted open field and rotarod tests. As shown in Figure 6B, C, there were no differences in number of entry to the center area, time stayed in the center, total traveled distance, and accelerated-speed rotarod between the animals treated with LV-miR-188-3p and LV-scramble control, suggesting that miR-188-3p overexpression does not affect sensorimotor function and anxiety.
Regulation of BACE1 by 2-AG signaling is mediated through PPARγ
The anti-inflammatory and neuroprotective effects produced by inhibition of 2-AG metabolism appear independently of CB1 or CB2 receptors (Nomura et al., 2011; Chen et al., 2012; Piro et al., 2012; Naydenov et al., 2014). Therefore, we hypothesized that regulation of BACE1 expression by inhibition of 2-AG metabolism may be through previously unidentified signaling pathways. PPARγ functions as a transcription factor that regulates expression of the genes involved in metabolism and inflammation (Bensinger and Tontonoz, 2008; Bright et al., 2008). Available information indicates that PPARγ is a target of 2-AG (O'Sullivan, 2007; Du et al., 2011) and can be activated by 2-AG independently of CB1R or CB2R (Rockwell et al., 2006). For this reason, we targeted PPARγ and determined whether PPARγ contributes to the beneficial effects produced by inhibition of 2-AG metabolism. As shown in Figure 7A, 2-AG was able to directly stimulate PPARγ activity using the luciferase reporter assay in the absence and presence of 2-AG. 15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), an endogenous PPARγ agonist, was used as a positive control. Dose-dependent increase in PPARγ luciferase reporter activity by 2-AG suggests that 2-AG likely is an endogenous PPARγ agonist. Interestingly, the reduced expression of PPARγ in TG animals was rescued by MAGL inhibition, which boosts the level of brain 2-AG (Fig. 7B).
Figure 7.
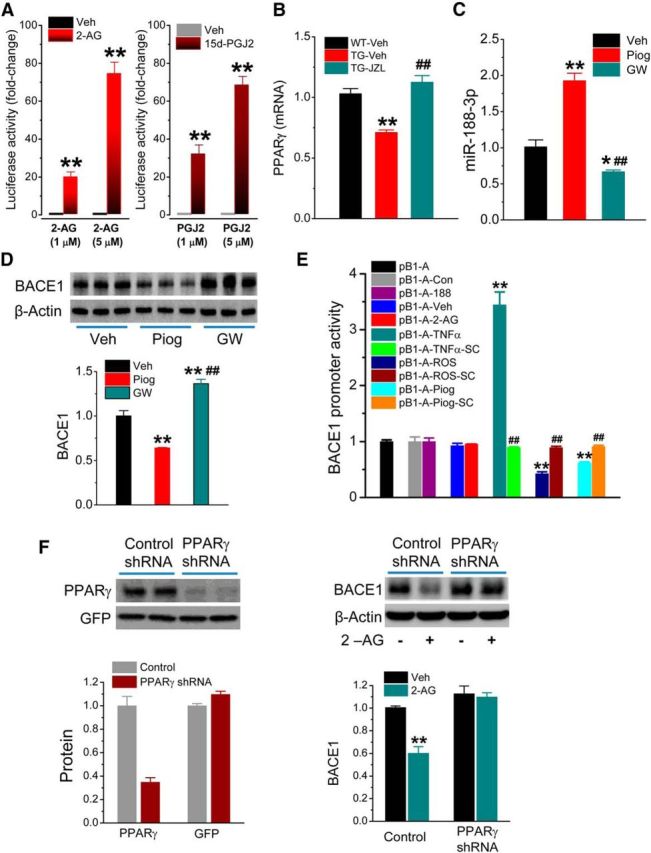
PPARγ mediates 2-AG-induced suppression of BACE1. A, 2-AG increases PPARγ activity by detecting luciferase reporter activity in NG108–15 cells transfected with pCMX-Gal-LBD-mPPARγ and TK-MH100x4-luc vectors. Luciferase activity values were normalized to the level of β-galactosidase activity. **p < 0.01, compared with the vehicle control (n = 6). B, Expression of PPARγ in the hippocampus of TG mice is reduced and restored by JZL184. PPARγ expression was analyzed by qPCR in 6-month-old WT and TG animals that received JZL184 (12 mg/kg) three times/week starting at 4 months of age for 8 weeks. **p < 0.01, compared with the WT vehicle control. ##p < 0.01, compared with the TG vehicle (n = 3 animals/group). C, Expression of miR-188-3p is increased by PPARγ activation and suppressed by PPARγ antagonism in the hippocampus. Piog (5 mg/kg, i.p.) or GW (5 mg/kg) was injected into animals once a day for 7 d. Expression of miR-188-3p in hippocampal tissue was assessed by qPCR analysis. *p < 0.05, compared with the vehicle control. **p < 0.01, compared with the vehicle control. ##p < 0.01, compared with Piog (n = 3 animals/group). D, Immunoblot analysis of BACE1 expression in animals that received Piog or GW. **p < 0.01, compared with the vehicle control. ##p < 0.01, compared with Piog (n = 3 animals/group). E, 2-AG and miR-188-3p do not affect BACE1 promoter activity. BACE1 promoter luciferase activity was detected in HEK293 cells transfected with pB1-A or pGL3 vectors in the presence an absence of 2-AG (3 μm), pEGP-miR null control or pEGP- miR-188-3p vector, TNFα (2.5 pg/ml), TNFα + SC (100 μm), ROS (1 μm), ROS + SC, Piog (1 μm) + SC. **p < 0.01, compared with pB1-A. ##p < 0.01, compared with TNFα, ROS, or Piog alone (n = 3). F, Knockdown of PPARγ blocks 2-AG-reduced BACE1 expression. Hippocampal neurons in culture were treated with FUGW lentiviral vectors expressing PPARγ shRNA or scramble control. BACE1 expression was detected in the presence and absence of 2-AG (3 μm). **p < 0.01, compared with the vehicle-scramble control (n = 3).
We next asked the question of whether PPARγ activation augments and its antagonism suppresses miR-188-3p expression. As shown in Figure 7C, pioglitazone (Piog), a PPARγ agonist, significantly increased miR-188-3p expression, whereas GW, a PPARγ antagonist, suppressed the expression in the hippocampus. Meanwhile, we observed that PPARγ activation reduced BACE1 expression, whereas PPARγ antagonism increased the expression (Fig. 7D). These results are consistent with the reports by others (Sastre et al., 2003, 2006; Wang et al., 2013). To further confirm that inhibition of BACE1 expression by 2-AG is through regulation of miR-188-3p at the post-transcriptional levels, we detected the effect of 2-AG on BACE1 promoter activity. As illustrated in Figure 7E, 2-AG did not induce changes in BACE1 reporter activity, suggesting that 2-AG does not affect BACE1 transcription. As expected, miR-188-3p also did not change BACE1 reporter activity (Fig. 7E). It has been shown previously that PPARγ activation suppresses BACE1 promoter activity (Sastre et al., 2006). We observed that Piog or rosiglitazone (ROS), another PPARγ agonist, reduced BACE1 promoter activity, whereas TNFα increased it (He et al., 2007). The changes in the BACE1 promoter activity were blocked by SC-510 (SC), an IKK-2 inhibitor, indicating that reduction of the BACE1 promoter activity by PPARγ agonists is mediated through NF-κB. These results also suggest that there might be currently undefined mechanisms involved in this process.
If PPARγ mediates 2-AG-produced suppression of BACE1, then knockdown of PPARγ should eliminate this effect. To test this prediction, we used the lentiviral-based shRNA technique as described previously (Chen et al., 2013) to silence PPARγ. As expected, 2-AG still suppressed BACE1 in cells treated with LV-scramble control but failed to suppress BACE1 in cells treated with LV-PPARγ shRNA (Fig. 7F), suggesting that 2-AG-induced suppression of BACE1 is mediated via PPARγ.
To further define the role of PPARγ in mediating 2-AG-induced upregulation of miR-188-3p and suppression of BACE1, we determined Aβ production and astrocytic reactivation in TG animals that received JZL184 or JZL184 plus GW three times/week starting at 4 months of age for 8 weeks. As shown in Figure 8A, the Aβ reducing effect by JZL184 was eliminated by GW. Similarly, the suppressive effect of JZL184 on GFAP expression was diminished by GW in TG animals (Fig. 8B). These data indicate that PPARγ is an important mediator in suppressing BACE1 expression, Aβ formation, and neuroinflammation produced by MAGL inhibition.
Figure 8.
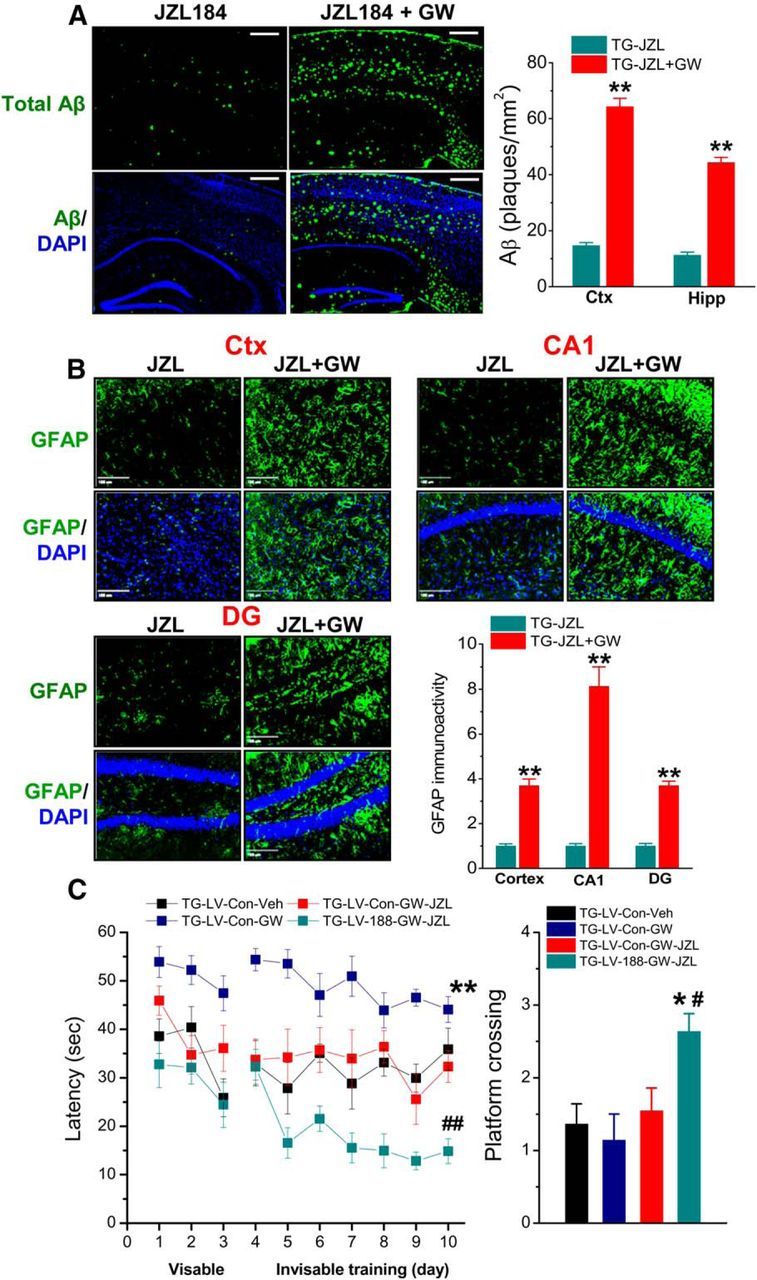
Aβ reducing and anti-inflammatory effects and improving cognitive function by inhibition of 2-AG metabolism are occluded by PPARγ antagonism. TG mice received JZL184 (JZL, 12 mg/kg) or JZL184 plus GW (5 mg/kg) three times/week starting at 4 months of age for 8 weeks. A, JZL184 decreases Aβ, but the decrease is eliminated by GW. B, Hippocampal and cortical GFAP (astrocytic marker) expression is suppressed by JZL184 but reversed by GW. **p < 0.01, compared with JZL184 alone (n = 5 mice/group). C, miR-188-3p overexpression rescues PPARγ antagonism-disrupted memory improvement by MAGL inhibition. TG animals were injected with LV-scramble control or LV-miR-188-3p. One week after injection, the animals received vehicle, GW, JZL184 (12 mg/kg), or JZL184 plus GW three times/week starting at 4 months of age for 8 weeks. The behavioral performance in the Morris water maze was tested 1 d after cessation of the last treatments. Data are mean ± SEM. *p < 0.01, compared with LV-scramble and vehicle control. #p < 0.05, compared with LV-scramble and JZL-GW (n = 11 to 13 mice/group). ##p < 0.01, compared with LV-scramble and JZL-GW (n = 11 to 13 mice/group).
We demonstrated previously that MAGL inhibition by JZL184 significantly promotes learning and memory in TG animals (Chen et al., 2012). To determine whether PPARγ blockade disrupts learning and memory improvement produced by MAGL inhibition and whether this disruption could be rescued by overexpression of miR-188-3p, LV-scramble control or LV-miR-188-3p was stereotaxically injected into the hippocampi of TG animals that received vehicle, JZL184, GW, or JZL184 plus GW three times a week for 8 weeks. As shown in Figure 8C, PPARγ antagonism by GW disrupted the improvement of spatial learning induced by JZL184. However, this disruptive effect on spatial learning was reversed by overexpression of miR-188-3p. It appeared that TG mice that received GW alone took much longer time to reach the hidden platform compared with the vehicle controls, suggesting that PPARγ antagonism further deteriorates spatial learning in TG mice (Fig. 8C). These results suggest that improved cognitive function by inhibition of 2-AG metabolism is mediated through PPARγ signaling.
Regulation of miR-188-3p expression by 2-AG is mediated by the PPARγ and NF-κB signaling pathway
Inhibition of 2-AG metabolism by JZL184 robustly raises brain 2-AG levels and reduces AA and its derivatives (Blankman et al., 2007; Long et al., 2009a,b; Nomura et al., 2011; Piro et al., 2012). To determine whether the reduced expression of miR-188-3p in AD is associated with increased Aβ and neuroinflammation and whether upregulated miR-188-3p expression by inhibition of 2-AG metabolism in TG animals, as shown in Figure 1, is mediated via 2-AG signaling, we assessed miR-188-3p expression in hippocampal neurons in cultures treated with Aβ, IL-1β, or TNFα in the absence or presence of 2-AG. As seen in Figure 9A, miR-188-3p expression was significantly decreased by Aβ42, IL-1β, or TNFα, suggesting that Aβ and proinflammatory insults may lead to downregulation of miR-188-3p expression. The decrease in miR-188-3p expression induced by Aβ42, IL-1β, or TNFα was prevented by 2-AG, ROS, and SC. These results indicate that Aβ- and proinflammatory cytokine-suppressed miR-188-3p can be rescued not only by 2-AG and activation of PPARγ, but also by inhibition of NF-κB, an important transcription factor. This information suggests that NF-κB is involved in the regulation of miR-188-3p by 2-AG and PPARγ. Available information suggests that anti-inflammatory and neuroprotective effects exerted by PPARγ are largely through interaction with NF-κB (Bensinger and Tontonoz, 2008; Bright et al., 2008). We observed that Aβ42 or TNFα increased NF-κB p65 phosphorylation, and the phosphorylation was inhibited by 2-AG, ROS, and SC (Fig. 9B). These results suggest that 2-AG and PPARγ agonist are able to inhibit NF-κB p65 activity.
Figure 9.

Aβ42 and proinflammatory cytokines suppress miR-188-3p expression, and the suppression is blocked by 2-AG, PPARγ activation, and NF-κB inhibition. A, Real-time qPCR analysis of miR-188-3p expression in hippocampal neurons in culture treated with vehicle, Aβ42 (10 μm), IL-1β (10 ng/ml), TNFα (10 ng/ml), 2-AG (3 μm), ROS (1 μm), and SC (100 μm). **p < 0.01, compared with the vehicle control. ##p < 0.01 compared with Aβ, IL-1β, or TNFα (n = 3). B, NF-κB phosphorylation induced by Aβ42 and TNFα is attenuated in the presence of 2-AG, ROS, and SC. **P < 0.01, compared with the vehicle control. #p < 0.05, compared with Aβ42 or TNFα (n = 3). ##p < 0.01, compared with Aβ42 or TNFα (n = 3).
Next, we asked whether regulation of miR-188-3p expression by 2-AG and PPARγ signaling is via NF-κB transcription. To this end, we searched BSs of NF-κB p65 in the promoter region of the miR-188-3p gene through TFSEARCH (http://www.cbrc.jp/research/db/TFSEARCH.html) and found that there are at least two NF-κB BSs. These BSs were confirmed by the ChIP analysis (Fig. 10A) and validated by qPCR analysis (data not shown). The NF-κB reporter assay further provided the evidence that 2-AG and PPARγ activation inhibited NF-κB reporter activity (Fig. 10B), suggesting that upregulation of miR-188-3p by 2-AG and PPARγ is likely through suppressing NF-κB, relieving the inhibitory effect of NF-κB on miR-188-3p expression (Fig. 11). The results from the NF-κB p65 shRNA analysis, which show that knockdown of NF-κB p65 blocked the suppressive effect of 2-AG and PPARγ activation on BACE1 (Fig. 10C), further support our hypothesis.
Figure 10.

Regulation of BACE1 by 2-AG and PPARγ is mediated through NF-κB. A, Binding of the NF-κB p65 subunit in the promoter of the miR-188-3p gene. ChIP analysis of promoter binding activity of NF-κB was analyzed. A 100 bp DNA ladder was used for the marker. B, 2-AG and PPARγ agonist reduce NF-κB reporter activity using a luciferase analysis. The concentrations used for 2-AG and ROS were 3 and 1 μm, respectively. **p < 0.01, compared with the vehicle control (n = 4). C, Knockdown of NF-κB p65 using the shRNA silencing technique eliminates reductions in BACE1 expression induced by 2-AG and ROS. Hippocampal neurons in culture were transfected with lentiviral vectors expressing scramble control or NF-κB p65 shRNA. BACE1 expression was assessed in the presence and absence of 2-AG (3 μm) or ROS (1 μm). **p < 0.01, compared with the vehicle control (n = 3).
Figure 11.

Signaling pathways mediating the beneficial effects produced by inhibition of 2-AG metabolism. Inhibition of MAGL increases 2-AG and decreases arachidonic acid (AA) and its metabolite prostaglandins through cyclooxygenase 1 and 2 (COX-1/2) and leukotrienes (LT4s) through 5-lipoxygenase (LOX). Increases in PPARγ activity and expression by 2-AG inhibit NF-κB, relieving the inhibitory effect of NF-κB on miR-188-3p transcription. Increased miR188–3p expression represses BACE1 and leads to a decrease in Aβ formation, which in turn suppresses neuroinflammation and improves synaptic and cognitive function.
Discussion
In the present study, we provided experimental evidence that counteracting neuropathology and improving synaptic and cognitive function in AD animals by inhibition of MAGL are primarily mediated by 2-AG signaling, which upregulates expression of miR-188-3p through the PPARγ and NF-κB signaling pathway, resulting in suppression of BACE1 expression and Aβ formation. We demonstrated that expression of miR-188-3p was significantly reduced both in the brains of AD humans and TG animals and that overexpression of miR-188-3p robustly reduced BACE1 expression, Aβ formation, and neuroinflammation and prevented deterioration in long-term synaptic plasticity, spatial learning, and memory in TG animals. Expression of miR-188-3p was upregulated by MAGL inhibition, 2-AG and PPARγ activation, or NF-κB inhibition and suppressed by PPARγ antagonism or NF-κB activation. In addition, reducing Aβ and neuroinflammation by inhibition of 2-AG metabolism were occluded by PPARγ antagonism. Moreover, improved spatial learning by MAGL inhibition was disrupted by PPARγ antagonism, but this disruptive effect was rescued by overexpression of miR-188-3p. Our results provide a novel epigenetic mechanism responsible for the improved synaptic and cognitive function by inhibition of 2-AG metabolism in an animal model of AD.
Previous studies show that inhibition of 2-AG metabolism reduces neuroinflammation in response to proinflammatory stimuli and prevents neurons from degenerating in an experimental animal model of Parkinson's disease (Nomura et al., 2011). We and others demonstrated recently that inhibition of MAGL robustly reduces production and accumulation of Aβ, reactive astroglial cells, and neurodegeneration, improves synaptic and cognitive function in TG animals (Chen et al., 2012; Piro et al., 2012). The results from these previous studies indicate that MAGL is a novel therapeutic target for inflammatory and neurodegenerative disorders. However, the molecular mechanisms underlying the beneficial effects produced by inhibition of 2-AG metabolism are largely unknown. MAGL is the primary enzyme that degrades 85% of 2-AG in the brain, resulting in the release of glycerol and AA, a precursor of proinflammatory prostaglandins and leukotrienes (Blankman et al., 2007; Long et al., 2009a,b; Nomura et al., 2011). Although inactivation of MAGL robustly elevates brain 2-AG and reduces AA and its derivatives, the anti-inflammatory and neuroprotective effects appear to be mediated neither by CB1R nor by CB2R (Nomura et al., 2011; Chen et al., 2012; Piro et al., 2012). It is possible that the anti-neuroinflammatory and neuroprotective effects of MAGL inactivation might be associated with reduced AA and its derivatives, such as prostaglandins (Nomura et al., 2011). 2-AG has been shown to possess significant anti-inflammatory and neuroprotective effects in response to harmful insults (Panikashvili et al., 2001, 2005; Marsicano and Lutz, 2006; Centonze et al., 2007; Zhang and Chen, 2008; Arevalo-Martin et al., 2010; Chen et al., 2011; Du et al., 2011). We hypothesized that enhanced 2-AG signaling by inhibition of 2-AG metabolism in the brain contributes to amelioration of AD neuropathology (Chen et al., 2012). Because Aβ is an initiator of AD etiology and pathogenesis and accumulated Aβ deposition is a well-recognized neuropathological hallmark of AD (Walsh and Selkoe, 2004; Ashe and Zahs, 2010; Huang and Mucke, 2012), we thus primarily focused on whether and how Aβ formation are regulated by 2-AG signaling. We observed previously that MAGL inhibition-reduced Aβ accumulation and deposition are accompanied with a decreased expression of BACE1, a key enzyme responsible for Aβ synthesis (Chen et al., 2012). We believed that understanding of how BACE1 is regulated would be the key to elucidating the underlying mechanisms mediating the beneficial events induced by inhibition of 2-AG metabolism. Our results show that 2-AG is capable of suppressing BACE1 expression.
To address the question of how expression of BACE1 is regulated by 2-AG signaling, we focused on the epigenetic regulation of BACE1 expression. Growing information suggests that miRNAs are likely involved in Aβ production and AD neuropathology (Junn and Mouradian, 2010; Delay et al., 2012; Satoh, 2012; Abe and Bonini, 2013; Lukiw et al., 2013). It has been shown that expressions of miRNAs, including those targeting BACE1, are altered both in AD humans and/or animal models (Boissonneault et al., 2009; Hébert et al., 2008; Wang et al., 2008; Schonrock and Götz, 2012). At least five miRNAs (e.g., miR-9, miR-29, miR-107, miR-298, and miR-328) have been reported previously to target BACE1. However, little is known about whether restoring or reversal of these deregulated miRNAs in AD animal models is capable of ameliorating neuropathology of AD by reducing BACE1 expression and Aβ formation. In our miRNA microarray screening analysis, we observed that expression of miR-188-3p, a previously unrecognized miRNA that targets BACE1, was robustly downregulated both in AD humans and TG animals and upregulated by inhibition of 2-AG metabolism in TG mice. We demonstrate that MAGL inhibition, which elevates 2-AG in the brain, is able to rescue downregulated miR-188-3p expression induced by Aβ and proinflammatory cytokines and suppress BACE1 expression, suggesting that upregulation of miR-188-3p and decrease in BACE1 by MAGL inhibition are likely a result of enhanced 2-AG signaling. We observed that 2-AG suppressed BACE1, and this effect was not mediated through inhibiting BACE1 transcription. Our results showing that overexpression of miR-188-3p resulted in suppression of BACE1 in both in vitro and in vivo preparations support this speculation. Importantly, miR-188-3p overexpression in TG animals significantly reduced Aβ formation and neuroinflammation, reversed the reduced expression of glutamate receptor subunits, and improved hippocampal basal synaptic transmission, LTP, spatial learning, and memory. These effects produced by miR-188-3p are similar to those produced by MAGL inhibition, suggesting that the beneficial effects resulting from MAGL inactivation in TG animals are associated with upregulation of miR-188-3p through 2-AG signaling.
As demonstrated previously, pharmacological or genetic inhibition of CB1R or CB2R fails to block neuroprotective and anti-inflammatory and antiepileptic effects induced by inhibition of 2-AG metabolism (Nomura et al., 2011; Chen et al., 2012; Piro et al., 2012; Naydenov et al., 2014). This suggests that there may be other mechanisms involved in regulation of miR-188-3p expression by 2-AG signaling. It has been shown that treatments with PPARγ agonists exhibit beneficial effects both in AD patients and animal models (Heneka et al., 2005; Jiang et al., 2008; Kummer and Heneka, 2008; Sato et al., 2011). Interestingly, 2-AG is able to exert an action through activation of PPARγ independently of CB1R or CB2R (Rockwell et al., 2006; O'Sullivan, 2007), suggesting that 2-AG is able to cross the plasma membrane and go nuclear directly to activate nuclear receptors (O'Sullivan, 2007). We observed that expression of PPARγ is significantly reduced in TG animals and that the decrease is reversed by inhibition of 2-AG metabolism. Also, 2-AG is able to directly activate PPARγ activity. Elimination of the improvements in spatial memory by PPARγ antagonism and anti-inflammatory and Aβ-reducing effects produced by MAGL inhibition suggest that 2-AG functions as an endogenous agonist for PPARγ activation and expression. Indeed, PPARγ activation increased miR-188-3p expression and decreased BACE1 expression, mimicking 2-AG-induced upregulation of miR-188-3p and suppression of BACE1. In contrast, PPARγ antagonism reduced miR-188-3p and increased BACE1. Knockdown of PPARγ blocked 2-AG-produced suppression of BACE1. Our results provide compelling evidence that regulation of miR-188-3p expression and BACE1 by 2-AG signaling is likely through PPARγ. Anti-inflammatory and neuroprotective effects produced by PPARγ are primarily through limiting NF-κB activity (Bensinger and Tontonoz, 2008; Bright et al., 2008). Our ChIP analysis shows that NF-κB p65 has BSs in the promoter region of the miR-188-3p gene. Also, 2-AG and PPARγ activation suppressed NF-κB reporter activity, suggesting that inhibition of NF-κB by 2-AG would relieve the inhibitory effect of NF-κB on miR-188-3p transcription. This assumption is supported by the results showing that expression of miR-188-3p was suppressed by Aβ and proinflammatory cytokines that increase phosphorylation of NF-κB, and the suppression was reversed by 2-AG, PPARγ activation, or NF-κB inhibition. In addition, silencing NF-κB p65 prevented BACE1 suppression by 2-AG and PPARγ activation. Our results suggest that neuroinflammation (e.g., increased release of proinflammatory cytokines, such as IL-1β and TNFα) and Aβ per se facilitate Aβ formation by augmenting NF-κB activity, which suppresses miR-188-3p expression. A single miRNA may have multiple targets. Thus, we cannot exclude the possibility that the effects we observed may be associated with other targets of miR-188-3p.
Collectively, the results presented in this report provide evidence that reduced neuroinflammation and Aβ and improved synaptic and cognitive function by MAGL inhibition are mediated through 2-AG signaling, which upregulates expression of miR-188-3p through the PPARγ and NF-κB signaling pathway, resulting in suppression of BACE1 expression and Aβ formation (Fig. 11). Growing evidence suggests that miRNA-based therapies, either restoring or reversal of deregulated miRNAs expression and activity, hold great promise for clinical applications (van Rooij et al., 2012; Hydbring and Badalian-Very, 2013; Zhang et al., 2013). The results from the present study suggest that restoration of miR-188-3p expression by inhibition of 2-AG metabolism or direct delivery of the vector expressing miR-188-3p would potentially be novel interventions for an AD therapy.
Footnotes
This work was supported by National Institutes of Health Grants NS076815 and AG039669 to C.C., M.H. was supported in part by the State Scholarship Fund from the China Scholarship Council No. 201207060033. We thank Dr. Ronald Evans of Salk Institute for providing PPARγ luciferase vectors, Dr. Bryan W. Luikart of Dartmouth Medical School for providing FUGW lentiviral vectors, Dr. Walter Lukiw and Mr. Surjyadipta Bhattacharjee of Louisiana State University Health Sciences Center for providing human RNA samples, Dr. Weihong Song of the University of British Columbia for providing the BACE1 promoter luciferase construct, and National Institutes of Health Mental Health Institute Chemical Synthesis and Drug Supply Program for providing JZL184.
The authors declare no competing financial interests.
References
- Abe M, Bonini NM. MicroRNAs and neurodegeneration: role and impact. Trends Cell Biol. 2013;23:30–36. doi: 10.1016/j.tcb.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arevalo-Martin A, Garcia-Ovejero D, Molina-Holgado E. The endocannabinoid 2-arachidonoylglycerol reduces lesion expansion and white matter damage after spinal cord injury. Neurobiol Dis. 2010;38:304–312. doi: 10.1016/j.nbd.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. (2009) MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Di Marzo V. Cannabinoid receptors and endocannabinoids: role in neuroinflammatory and neurodegenerative disorders. CNS Neurol Disord Drug Targets. 2010;9:564–573. doi: 10.2174/187152710793361568. [DOI] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissonneault V, Plante I, Rivest S, Provost P. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J Biol Chem. 2009;284:1971–1981. doi: 10.1074/jbc.M807530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright JJ, Kanakasabai S, Chearwae W, Chakraborty S. PPAR regulation of inflammatory signaling in CNS diseases. PPAR Res. 2008;2008:658520. doi: 10.1155/2008/658520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Finazzi-Agrò A, Bernardi G, Maccarrone M. The endocannabinoid system in targeting inflammatory neurodegenerative diseases. Trends Pharmacol Sci. 2007;28:180–187. doi: 10.1016/j.tips.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Chen C, Magee JC, Bazan NG. Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J Neurophysiol. 2002;87:2851–2857. doi: 10.1152/jn.2002.87.6.2851. [DOI] [PubMed] [Google Scholar]
- Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang YP, Teng Z, Chen C. Monoacylglycerol lipase is a therapeutic target for Alzheimer's disease. Cell Rep. 2012;2:1329–1339. doi: 10.1016/j.celrep.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zhang J, Fan N, Teng Z, Wu Y, Yang H, Tang Y, Sun H, Song Y, Chen C. Δ9-THC-caused synaptic and memory impairments are mediated through COX-2 signaling. Cell. 2013;155:1554–1565. doi: 10.1016/j.cell.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang J, Chen C. Endocannabinoid 2-arachidonoylglycerol protects neurons against β-amyloid insults. Neuroscience. 2011;178:159–168. doi: 10.1016/j.neuroscience.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delay C, Mandemakers W, Hébert SS. MicroRNAs in Alzheimer's disease. Neurobiol Dis. 2012;46:285–290. doi: 10.1016/j.nbd.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Du H, Chen X, Zhang J, Chen C. Inhibition of COX-2 expression by endocannabinoid 2-arachidonoylglycerol is mediated via PPAR-γ. Br J Pharmacol. 2011;163:1533–1549. doi: 10.1111/j.1476-5381.2011.01444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Neilson JR, Foster KA, Wang CF, Seeburg DP, Batterton MN, Tada T, Dolan BM, Sharp PA, Sheng M. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron. 2010;65:373–384. doi: 10.1016/j.neuron.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan N, Yang H, Zhang J, Chen C. Reduced expression of glutamate receptors and phosphorylation of CREB are responsible for Δ9-THC-impaired hippocampal synaptic plasticity. J Neurochem. 2010;112:691–702. doi: 10.1111/j.1471-4159.2009.06489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein AM, O'Banion MK. Neuroinflammation and memory: the role of prostaglandins. Mol Neurobiol. 2009;40:15–32. doi: 10.1007/s12035-009-8066-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O'Banion K, Klockgether T, Van Leuven F, Landreth GE. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- Hensley K. Neuroinflammation in Alzheimer's disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis. 2010;21:1–14. doi: 10.3233/JAD-2010-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hydbring P, Badalian-Very G. Clinical applications of microRNAs. F1000 Res. 2013;2:136. doi: 10.12688/f1000research.2-136.v3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im HI, Kenny PJ. MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 2012;35:325–334. doi: 10.1016/j.tins.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Heneka M, Landreth GE. The role of peroxisome proliferator-activated receptor-gamma (PPARgamma) in Alzheimer's disease: therapeutic implications. CNS Drugs. 2008;22:1–14. doi: 10.2165/00023210-200822010-00001. [DOI] [PubMed] [Google Scholar]
- Jiao J, Nakajima A, Janssen WG, Bindokas VP, Xiong X, Morrison JH, Brorson JR, Tang YP. Expression of NR2B in cerebellar granule cells specifically facilitates effect of motor training on motor learning. PLoS One. 2008;3:e1684. doi: 10.1371/journal.pone.0001684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junn E, Mouradian MM. MicroRNAs in neurodegenerative disorders. Cell Cycle. 2010;9:1717–1721. doi: 10.4161/cc.9.9.11296. [DOI] [PubMed] [Google Scholar]
- Kummer MP, Heneka MT. PPARs in Alzheimer's disease. PPAR Res. 2008;2008:403896. doi: 10.1155/2008/403896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Normura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem. Biol. 2009a;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009b;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Bensen AL, Washburn EK, Perederiy JV, Su KG, Li Y, Kernie SG, Parada LF, Westbrook GL. miR-132 mediates the integration of newborn neurons into the adult dentate gyrus. PLoS One. 2011;6:e19077. doi: 10.1371/journal.pone.0019077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Andreeva TV, Grigorenko AP, Rogaev EI. Studying micro RNA function and dysfunction in Alzheimer's disease. Front Genet. 2013;3:327. doi: 10.3389/fgene.2012.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, Song W. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest. 2013;123:224–235. doi: 10.1172/JCI64516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Neuromodulatory functions of the endocannabinoid system. J Endocrinol Invest. 2006;29(Suppl 3):27–46. [PubMed] [Google Scholar]
- Mulvihill MM, Nomura DK. Therapeutic potential of monoacylglycerol lipase inhibitors. Life Sci. 2013;92:492–497. doi: 10.1016/j.lfs.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naydenov AV, Horne EA, Cheah CS, Swinney K, Hsu KL, Cao JK, Marrs WR, Blankman JL, Tu S, Cherry AE, Fung S, Wen A, Li W, Saporito MS, Selley DE, Cravatt BF, Oakley JC, Stella N. ABHD6 blockade exerts antiepileptic activity in PTZ-induced seizures and in spontaneous seizures in R6/2 mice. Neuron. 2014;83:361–371. doi: 10.1016/j.neuron.2014.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- Panikashvili D, Mechoulam R, Beni SM, Alexandrovich A, Shohami E. CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J Cereb Blood Flow Metab. 2005;25:477–484. doi: 10.1038/sj.jcbfm.9600047. [DOI] [PubMed] [Google Scholar]
- Piro JR, Benjamin DI, Duerr JM, Pi Y, Gonzales C, Wood KM, Schwartz JW, Nomura DK, Samad TA. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer's disease. Cell Rep. 2012;1:617–623. doi: 10.1016/j.celrep.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwell CE, Snider NT, Thompson JT, Vanden Heuvel JP, Kaminski NE. Interleukin-2 suppression by 2-arachidonyl glycerol is mediated through peroxisome proliferator-activated receptor gamma independently of cannabinoid receptors 1 and 2. Mol Pharmacol. 2006;70:101–111. doi: 10.1124/mol.105.019117. [DOI] [PubMed] [Google Scholar]
- Salmon JA, Higgs GA. Prostaglandins and leukotrienes as inflammatory mediators. Br Med Bull. 1987;43:285–296. doi: 10.1093/oxfordjournals.bmb.a072183. [DOI] [PubMed] [Google Scholar]
- Sang N, Zhang J, Marcheselli V, Bazan NG, Chen C. Postsynaptically synthesized prostaglandin E2 modulates hippocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J Neurosci. 2005;25:9858–9870. doi: 10.1523/JNEUROSCI.2392-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003;23:9796–9804. doi: 10.1523/JNEUROSCI.23-30-09796.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci U S A. 2006;103:443–448. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging. 2011;32:1626–1633. doi: 10.1016/j.neurobiolaging.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Satoh J. Molecular network of microRNA targets in Alzheimer's disease brains. Exp Neurol. 2012;235:436–446. doi: 10.1016/j.expneurol.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Schonrock N, Götz J. Decoding the non-coding RNAs in Alzheimer's disease. Cell Mol Life Sci. 2012;69:3543–3559. doi: 10.1007/s00018-012-1125-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T. Physiological roles of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Biofactors. 2009;35:88–97. doi: 10.1002/biof.18. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Purcell AL, Levin AA. Developing microRNA therapeutics. Circ Res. 2012;110:496–507. doi: 10.1161/CIRCRESAHA.111.247916. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Wang R, Li JJ, Diao S, Kwak YD, Liu L, Zhi L, Büeler H, Bhat NR, Williams RW, Park EA, Liao FF. Metabolic stress modulates Alzheimer's β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab. 2013;17:685–694. doi: 10.1016/j.cmet.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer's disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Kwon EJ, Tsai LH. MicroRNAs in learning, memory, and neurological diseases. Learn Mem. 2012;19:359–368. doi: 10.1101/lm.026492.112. [DOI] [PubMed] [Google Scholar]
- Xu J, Chen C. Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist. 2014 doi: 10.1177/1073858414524632. doi: 10.1177/1073858414524632. Advance online publication. Retrieved Feb. 25, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen C. Endocannabinoid 2-arachidonoylglycerol protects neurons by limiting COX-2 elevation. J Biol Chem. 2008;283:22601–22611. doi: 10.1074/jbc.M800524200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang Z, Gemeinhart RA. Progress in microRNA delivery. J Control Release. 2013;172:962–974. doi: 10.1016/j.jconrel.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. β-Site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]