

Abstract
Hyperactivity within the ventral hippocampus (vHPC) has been linked to both psychosis in humans and behavioral deficits in animal models of schizophrenia. A local decrease in GABA-mediated inhibition, particularly involving parvalbumin (PV)-expressing GABA neurons, has been proposed as a key mechanism underlying this hyperactive state. However, direct evidence is lacking for a causal role of vHPC GABA neurons in behaviors associated with schizophrenia. Here, we probed the behavioral function of two different but overlapping populations of vHPC GABA neurons that express either PV or GAD65 by selectively inhibiting these neurons with the pharmacogenetic neuromodulator hM4D. We show that acute inhibition of vHPC GABA neurons in adult mice results in behavioral changes relevant to schizophrenia. Inhibiting either PV or GAD65 neurons produced distinct behavioral deficits. Inhibition of PV neurons, affecting ∼80% of the PV neuron population, robustly impaired prepulse inhibition of the acoustic startle reflex (PPI), startle reactivity, and spontaneous alternation, but did not affect locomotor activity. In contrast, inhibiting a heterogeneous population of GAD65 neurons, affecting ∼40% of PV neurons and 65% of cholecystokinin neurons, increased spontaneous and amphetamine-induced locomotor activity and reduced spontaneous alternation, but did not alter PPI. Inhibition of PV or GAD65 neurons also produced distinct changes in network oscillatory activity in the vHPC in vivo. Together, these findings establish a causal role for vHPC GABA neurons in controlling behaviors relevant to schizophrenia and suggest a functional dissociation between the GABAergic mechanisms involved in hippocampal modulation of sensorimotor processes.
Keywords: DREADD, GABA, parvalbumin, schizophrenia, ventral hippocampus
Introduction
GABA dysfunction is a well characterized neuropathology of schizophrenia (Benes and Berretta, 2001; Lisman et al., 2008; Lewis et al., 2012). Alterations in markers of inhibitory GABA interneurons, such as reductions in the GABA-synthesizing enzymes GAD65 and GAD67, have been widely reported in the prefrontal cortex and hippocampus (Akbarian et al., 1995; Todtenkopf and Benes, 1998; Guidotti et al., 2000; Heckers et al., 2002; Benes et al., 2007). These alterations appear to most notably affect the GABA interneuron subtype containing parvalbumin (PV; Benes et al., 1991; Volk et al., 2000; Zhang and Reynolds, 2002; Hashimoto et al., 2003; Gonzalez-Burgos and Lewis, 2008; Fung et al., 2010; Konradi et al., 2011). Robust histochemical changes have been found in PV-positive interneurons in schizophrenia, including decreased PV and GAD67 mRNA and reduced PV immunoreactivity (Beasley and Reynolds, 1997; Zhang and Reynolds, 2002; Hashimoto et al., 2003). Similar to schizophrenia patients, a variety of animal models including neurodevelopmental, pharmacological, and genetic models exhibit reductions in PV expression (Penschuck et al., 2006; Behrens et al., 2007; Harte et al., 2007; Meyer et al., 2008; Shen et al., 2008). Collectively, these molecular alterations suggest impaired functioning in PV-positive interneurons (Nakazawa et al., 2012). The resulting GABA dysfunction in particular brain regions may contribute to specific features of schizophrenia (Grace, 2010; Lewis et al., 2012). Neuroimaging studies have correlated increased activity in the human anterior (ventral in rodents) hippocampus with the emergence and severity of psychosis (Schobel et al., 2009; Schobel et al., 2013). In rodents, manipulations that increase ventral hippocampus (vHPC) activity, including nonselective disruptions in GABAergic transmission, have consistently resulted in hyperlocomotor activity and sensorimotor gating deficits, among other behaviors related to schizophrenia (Bast et al., 2001a; Zhang et al., 2002; Peterschmitt et al., 2008; Taepavarapruk et al., 2008). The prenatal methylazoxymethanol acetate (MAM) rodent model exhibits these behavioral changes, as well as PV neuron loss and increased pyramidal neuron excitability in the vHPC (Lodge and Grace, 2007; Lodge et al., 2009). Because GABA interneurons exert inhibitory control over pyramidal neurons, deficits in vHPC GABA interneuron function may account for local hyperactivity and the associated behavioral changes. Furthermore, specific subtypes of GABA interneurons may subserve distinct behavioral functions and behavior-dependent network activities (Freund and Katona, 2007).
Although chronic developmental and broad GABA manipulations have implicated GABA neuron dysfunction in schizophrenia, to our knowledge, no study has selectively and reversibly examined the behavioral functions of GABA interneuron populations specifically in the vHPC. We therefore investigated the effect of acutely inhibiting two genetically defined and relatively distinct populations of GABA interneurons, those positive for PV or GAD65 (Fukuda et al., 1997; Wierenga et al., 2010; Nagode et al., 2014), on behaviors relevant to schizophrenia. The recent development of DREADDs (Designer Receptors Exclusively Activated by Designer Drugs) provides pharmacogenetic control of neural activity in targeted neuron populations in freely behaving mice (Armbruster et al., 2007). We selectively inhibited either PV-positive neurons, affecting ∼80% of the PV population, or GAD65-positive neurons, a heterogeneous population that included ∼40% of PV neurons and 65% of cholecystokinin (CCK) neurons, in the vHPC using the hM4D DREADD receptor. The effect of this inhibition was determined using multiple behavioral assays and in vivo recording of network oscillatory activity in the vHPC.
Materials and Methods
Animals.
GAD65-Cre (Gad2<tm2(cre)Zjh>/J; JAX #010802) and PV-Cre (B6;129P2-Pvalbtm1(cre)Arbr/J; JAX#008069) mice were obtained from The Jackson Laboratory and bred as homozygotes. Mice were group housed in a temperature-controlled room on a 12 h light/dark cycle with ad libitum access to food and water. Experiments were performed on 2- to 3-month-old mice. Male mice were used for behavioral experiments, whereas both male and female mice were used for electrophysiology experiments. All procedures were performed in accordance with the guidelines of the National Institutes of Health (NIH) and the Canadian Council on Animal Care (CCAC) with approval from the University of Toronto Animal Care Committee.
AAV vector construction.
The recombinant AAV-hSyn-FLEX-hM4D-mCherry plasmid (Krashes et al., 2011) was obtained from Dr. Bryan Roth at the University of North Carolina at Chapel Hill and packaged in serotype 2/8 by the University of Pennsylvania Vector Core service; titers were 1 × 1012 particles/ml.
Surgery.
Mice were anesthetized with isoflurane and mounted onto a stereotaxic frame. The AAV vector containing doubly floxed hM4D-mCherry (AAV2/8-hsyn-FLEX-hM4D-mCherry) was infused into the vHPC (AP: −3.40 mm, DV: −4.75, ML: ±2.75 mm, after Paxinos and Franklin, 2007). A volume of 0.2–0.3 μL was infused via an internal cannula connected by Tygon tubing to a 10 μL Hamilton needle syringe by pressure ejection at a rate of 0.1 μL/min. Following infusion, the internal cannula was left in place for 10 min to prevent solution backflow. After surgery, mice were single-housed.
Drugs.
Clozapine-N-oxide [CNO; obtained from the NIH as part of the Rapid Access to Investigative Drug Program funded by the National Institute of Neurological Disorders and Stroke (NINDS)] was dissolved in DMSO and 0.9% saline. d-amphetamine-sulfate (Amph; Sigma-Aldrich) and SCH-23390 (Sigma-Aldrich) were dissolved in 0.9% saline. Haloperidol (Sigma-Aldrich) was dissolved in 0.025% glacial acetic acid and 0.9% saline and adjusted to pH 5.5 with NaOH. All drugs were administered by intraperitoneal injection at a volume of 10 ml/kg.
Behavioral apparatuses and testing procedures.
Behavioral tests were performed 12–14 d after surgery and conducted 2–3 d apart. Spontaneous locomotor activity and social interaction were tested using a within-subjects design. Mice tested for spontaneous locomotor activity were subsequently tested for social interaction. Amphetamine-induced locomotor activity, prepulse inhibition of the startle reflex (PPI), and spontaneous alternation were tested using a between-subjects design. In these experiments, mice were randomly assigned to receive either vehicle or CNO administration with an equal allocation of littermates to each group. PV-Cre mice tested for amphetamine-induced locomotor activity later underwent PPI and spontaneous alternation testing. All PV-Cre mice expressed hM4D bilaterally. GAD65-Cre mice expressing hM4D unilaterally were tested for both amphetamine-induced locomotor activity and spontaneous alternation. A separate group of GAD65-Cre mice with bilateral hM4D expression were tested for PPI.
Open field.
Locomotor activity was examined in a black Plexiglas box measuring 30 cm L × 30 cm W × 40 cm H under red lighting (3 lux). Mice were habituated to the arena for 2 h on 3 consecutive days before testing and for 1 h immediately before testing. Testing of spontaneous locomotor activity took place over 2 consecutive days with mice receiving a single injection each day. PV-Cre mice were injected with both vehicle and CNO in a counterbalanced order. GAD65-Cre mice were injected with vehicle followed by CNO. For amphetamine-induced locomotor activity, mice were coinjected with either vehicle and a low dose of amphetamine (1 mg/kg) or CNO and amphetamine. In the experiment using haloperidol, mice were pretreated with either vehicle or 0.02 mg/kg haloperidol followed 30 min later with vehicle or CNO. Testing took place over 2 d with mice receiving either haloperidol + vehicle and vehicle + CNO or vehicle + vehicle and haloperidol + CNO in counterbalanced order. To test the effect of SCH-23390 on locomotor activity, mice were injected with 0.05 mg/kg SCH-23390 30 min before vehicle and CNO injection. Mice were administered vehicle, CNO, SCH-23390 + vehicle, and SCH-23390 + CNO over 4 d. After injection, mice were placed into the center of the arena and distance traveled was measured for 1 h using ANY-maze (Stoelting).
Social interaction.
Social interaction was tested in Crawley's 3-chamber sociability test (Moy et al., 2004) under red lighting (3 lux). The apparatus was made of clear Plexiglas and consisted of three chambers, each measuring 40 cm L × 20 cm W × 40 cm H. The two side chambers contained a cylindrical wire cage (10.5 cm D × 11 cm H, 1 cm bar spacing). Subjects were able to freely move between chambers through an opening (5 cm × 40 cm) in the partitioning walls. After 30 min habituation to the test room, subjects were injected with either vehicle or CNO and then placed into the center chamber and allowed to acclimate to the entire apparatus for 10 min. After acclimation, an age-matched male C57BL/6 mouse, unfamiliar to the subject, was placed into a cage in one of the side chambers. The subject's time spent, number of entries, and distance traveled in each chamber was measured for 10 min using ANY-maze. All mice were tested with both vehicle and CNO on two independent trials separated by 48 h. The order of vehicle and CNO administration and the chamber containing the stranger mouse were counterbalanced.
PPI of the acoustic startle reflex.
Mice were placed into a startle apparatus (Med-Associates) consisting of a small metal cage inside a sound-attenuated chamber. Whole-body startle movements were detected by a piezoelectric accelerometer mounted beneath the cage, converted to electrical signals, and digitized and stored by a computer. A loudspeaker mounted to the side of the cage produced white noise acoustic stimuli. A continuous 64 dB background noise was present throughout the test. The test began with 5 min acclimation to the apparatus, followed by 12 pulse-alone trials. To measure PPI, 12 blocks containing 6 trials were presented: 1 pulse-alone trial, 4 prepulse-pulse trials, and 1 no-stimulus trial. Pulse-alone trials consisted of a 40 ms startle pulse at 120 dB; prepulse-pulse trials consisted of a 20 ms prepulse at 66, 68, 72, or 76 dB followed 100 ms after by the pulse; and no-stimulus trials consisted of background noise only. Trials were presented in pseudorandom order, with an intertrial interval of 10–20 s (mean of 15 s). Startle response was determined as the peak amplitude within 100 ms of the startle pulse onset. The entire test lasted ∼30 min. Mice were injected with either vehicle or CNO immediately before acclimation. The percentage PPI for each prepulse intensity level was calculated as follows: [(mean startle amplitude of pulse-alone trials − startle amplitude of prepulse-pulse trial)/mean startle amplitude of pulse-alone trials * 100].
T-maze continuous alternation task.
Spontaneous alternation was tested in a T-shaped maze made of black Plexiglas with one start arm and two goal arms (30 cm L, 10 cm W, 20 cm H each) separated by removable guillotine doors. Mice were injected with either vehicle or CNO and, 5 min later, acclimated to the start arm for 10 min. The test followed procedures described previously (Gerlai, 1998) and began with one forced choice trial followed by 10 free choice trials. During the forced choice trial, the door to the start arm and one of the goal arms was removed, whereas the door to the alternate arm remained lowered, allowing mice to explore the available arm. Both left and right arms served as forced arms and were counterbalanced between vehicle and CNO groups. Upon leaving the arm and reaching the base of the start arm, a free choice trial was initiated. The door to the alternate goal arm was removed and mice were given the choice of either arm. After entering the chosen arm, the door to the other arm was lowered. Mice were considered to have made a choice when the whole body, including the tail tip, was located in the arm. Another free choice trial followed. The percentage alternation [(number of alternations/10) * 100] and direction bias [|(number of right arm entries)/10) − 0.5|] were calculated. Latency to arm entry was measured using ANY-maze.
Immunohistochemistry.
Mice were transcardially perfused with PBS, pH 7.4, followed by 4% paraformaldehyde. Brains were extracted and postfixed overnight in 4% paraformaldehyde at 4°C and then cryoprotected with PBS containing 30% sucrose. Brains were sectioned coronally at 40 μm thickness using a cryostat (CM 1520; Leica). For PV and CCK-8 immunostaining, free-floating brain sections were blocked with 5% normal donkey serum in PBS-T (0.1%) for 1 h. Sections were then incubated with PBS-T containing rabbit polyclonal anti-PV antibody (1:1000; Abcam) or rabbit polyclonal anti-CCK-8 (1:1000; Sigma-Aldrich) for 48 h at 4°C, followed by Alexa Fluor 488–conjugated donkey anti-rabbit secondary antibody (1:500 in PBS-T; Invitrogen) for 2 h at room temperature. For cell-counting experiments, every fourth section between bregma −3.00 mm to −3.64 mm was collected and immunostained for PV or CCK-8. The sections were mounted and imaged on a confocal laser scanning microscope with a 20× objective. mCherry-positive and PV-immunoreactive (PV-ir) or CCK-immunoreactive (CCK-ir) cells were counted in all strata of the ventral CA1 in a 750 × 700 μm area. The percentage transduction selectivity [(total number of double labeled cells/total number of mCherry-positive cells) × 100] and the percentage transduction efficacy [(total number of double labeled cells/total number of PV or CCK-ir cells) × 100] were calculated.
Slice electrophysiology.
Brains were rapidly removed after decapitation and placed into a cutting solution containing the following (in mm): 205 sucrose, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 0.4 ascorbic acid, 1 CaCl2, 2 MgCl2, and 3 sodium pyruvate, pH 7.4, osmolality 300 mOsm/kg. Transverse slices (350 μm) containing the vHPC were prepared from 6- to 8-week-old mice previously infected with AAV containing hM4D-mCherry. Slices recovered at 32°C in a 50:50 mixture composed of cutting saline + artificial CSF (aCSF) for 30 min and then placed in aCSF alone for 30 min. During experimentation slices were perfused at a rate of ∼ 2 ml/min in aCSF. The aCSF solution consisted of the following (in mm): 123 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 2 CaCl2, and 1 MgCl2 in double-distilled water and saturated with 95% O2/5% CO2, pH 7.4, osmolarity ∼300mOsm. Whole-cell patch-clamp recordings were obtained from transduced neurons (identified by mCherry fluorescence in the vHPC) and their untransduced neighbors. Micropipettes were filled with an intracellular fluid containing the following (in mm): 130 potassium gluconate, 10 KCl, 10 HEPES, 0.2 EGTA, 4 ATP, 0.3 GTP, and 10 phosphocreatine, pH 7.4, osmolality 300 mOsm/kg. The resting membrane potential was recorded (in current-clamp mode) in normal aCSF for a minimum of 5 min before the addition of 1 μm CNO for 10 min. Neuronal excitability was determined in current-clamp mode by injecting current in 10 pA steps from −50 pA to 150 pA. Field recordings were made by placing the recording electrode (filled with 1 μm NaCl) in the CA1 pyramidal cell body layer to record the somatic population spike (PSP) and a unipolar stimulating electrode in the Schaffer collaterals. PSPs were evoked at a frequency of 0.06 Hz; stimulation intensity was increased at stepwise intervals. PSP amplitude was normalized by dividing each amplitude by the PSP amplitude evoked by the lowest stimulation intensity.
In vivo electrophysiology.
In a separate group of GAD65-Cre (n = 8) and PV-Cre (n = 7) mice expressing hM4D unilaterally in the vHPC, local field potential (LFP) activity was measured from area CA1. The LFP activity was recorded bilaterally to use the hemisphere without hM4D expression as an internal control for factors unrelated to pharmacogenetic manipulations, such as body temperature and anesthesia depth. Mice were initially anesthetized with isoflurane (1–1.5% by volume in oxygen at a flow rate of 1.5 L/min; Halocarbon Laboratories) and then maintained with intraperitoneal injection of urethane (1.7 g/kg). Additional doses of urethane (at 0.05 g/ml in 0.1 ml amounts) were given if necessary to maintain a surgical plane of anesthesia verified by the absence of the hindpaw withdraw reflex. Mice were then mounted onto a stereotaxic instrument with the skull surface in the horizontal plane. Body temperature was maintained with a heating pad at 36°C. Bipolar Teflon-coated stainless steel electrodes (0.4 mm vertical stagger; A-M Systems) secured to a 26 ga stainless steel cannula were then slowly lowered bilaterally into the vHPC at the following coordinates relative to bregma: −3.15 mm posterior, ± 3.00 mm lateral, and −4.6 mm ventral (Paxinos and Franklin, 2007). Stainless steel screws were implanted into the fontal cortex and the cerebellum for ground and reference, respectively. All wires were connected centrally to an interface board (EIC-36-PTB; Neuralynx). LFP activity was recorded with the RZ-5 recording system (Tucker-Davis Technologies). The signal was amplified 1000 times, filtered between 1 and 400 Hz, and digitized at 2 kHz. After electrode implantation, mice were given 30 min to stabilize and the recordings were made with the mice positioned on the stereotaxic instrument in a quiet artificially lit room. LFP activity was continuously recorded from both the hM4D-expressing hemisphere and the control hemisphere. All recordings began with a 10 min baseline period, after which mice received an intraperitoneal injection of saline followed by an injection of CNO (2 mg/kg, i.p.) 35 min after the saline injection. Thirty-five minutes after CNO injection, the recording was stopped and electrode locations were marked with electrolytic lesion. Mice were then killed with an overdose of urethane, perfused intracardially with 10% formalin, and brain tissue was collected for histological analysis. All analyses were performed offline using custom codes written in MATLAB (The MathWorks). Power spectral density estimates were computed using Welch's method (MATLAB function PWelch) in series of 10 s bins covering the entire time course of the recording for each channel. Only mice in which 1 electrode in each hemisphere was positioned in area CA1 of the vHPC were used for further analyses (GAD65-Cre, n = 5; PV-Cre, n = 4) and these channels were selected for statistical analyses. In each animal, the power during the last 10 min block of each injection period (25–35 min postinjection) was then normalized to the 10 min baseline period and averaged across animals. In a subsequent analysis, the normalized power was averaged within 5 frequency bands (delta, 1–4 Hz; theta, 4–12 Hz; alpha, 12–20 Hz; beta, 20–40 Hz; gamma, 40–100 Hz) and averaged across animals.
Statistical analysis.
Data were analyzed using two-way repeated-measures ANOVA, two-way mixed ANOVA, three-way mixed ANOVA, and Student's t test. Where appropriate, ANOVAs were followed by planned pairwise comparisons. Statistical analyses were performed using IBM SPSS software version 21.
Results
hM4D-mediated inhibition of PV neurons in the vHPC
Cre-dependent rAAV and PV-Cre mice were used to target the pharmacogenetic activity silencer hM4D to PV neurons of the vHPC (Fig. 1A,B) (Hippenmeyer et al., 2005; Atasoy et al., 2008; Krashes et al., 2011). Targeting of hM4D was selective to PV neurons since 83% of hM4D expressing neurons were positive for PV immunoreactivity (PV-ir) (Fig. 1C,D). Furthermore, 80% of the total PV-ir neuron population in the ventral CA1 expressed hM4D, demonstrating high efficacy of PV neuron transduction (Fig. 1C,D). Next, we confirmed that the synthetic ligand for hM4D, CNO, effectively inhibited PV neurons by performing whole-cell patch-clamp recordings on vHPC slices. Application of CNO significantly hyperpolarized membrane potential from −67.3 ± 1.7 mV to −72.6 ± 1.2 mV (Fig. 1Ei, paired t test p = 0.009) and significantly suppressed current-induced spiking activity in hM4D-positive neurons from 7.8 ± 3.7 to 2.1 ± 2.1 action potentials (induced by a 500 ms, 120 pA current injection; p = 0.008; Fig. 1Eii). No significant changes in membrane potential or current-induced spiking were observed in hM4D-negative neurons (Fig. 1Eiii,Eiv).
Figure 1.

Inhibition of PV neurons in the vHPC using hM4D. A, Construct design for AAV-hsyn-FLEX-hM4D-mCherry. hM4D-mCherry sequences are inverted between two pairs of heterotypic, antiparallel loxP sites. In the presence of Cre recombinase, the sequences are inverted and thus transgene expression is activated. CAG, CMV enhancer/beta-globin chimeric promoter; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element; ITR, inverted terminal repeat. B, Representative image of hM4D-mCherry expression in the vHPC of PV-Cre mice. Inset, High-magnification confocal image of boxed area. Red, hM4D-mCherry; blue, DAPI. C, Representative confocal image of immunostaining for PV in the ventral CA1 of PV-Cre mice expressing hM4D. Red, hM4D-mCherry; Green, PV-ir. D, Bar graph showing the percentage of double-labeled cells (hM4D+/PV+) within the total number of hM4D+ and PV+ cells in PV-Cre mice (n = 6). Data are presented as mean ± SEM. E, Representative voltage traces of CNO-induced hyperpolarization (i) and CNO-induced suppression of evoked action potential firing (ii) in hM4D-positive neurons (n = 8) and of no significant effects of CNO on hyperpolarization (iii) or evoked action potential firing (iv) in hM4D-negative neurons of PV-Cre mice.
Inhibition of PV neurons in the vHPC disrupts PPI and spatial working memory
To examine the effect of vHPC PV neuron inhibition on behaviors relevant to positive symptoms of schizophrenia, we tested locomotor activity in the open field (Geyer and Moghaddam, 2002; Arguello and Gogos, 2006). Total spontaneous locomotor activity after CNO administration was not significantly different from vehicle administration at doses of 2 or 5 mg/kg CNO (Fig. 2A; mixed ANOVA, no main effect of treatment, F < 1). Examination of locomotor activity in 10 min intervals revealed no significant differences between vehicle and CNO administration at any time point for both 2 mg/kg CNO (Fig. 2B; 2-way repeated-measures ANOVA, no main effect of treatment, F < 1; no treatment × time interaction, F < 1) and 5 mg/kg CNO (Fig. 2C; 2-way repeated-measures ANOVA, no main effect of treatment, F < 1; no treatment × time interaction, F < 1). Similarly, no significant differences in locomotor activity after administration of a low dose of amphetamine (1 mg/kg) was observed between vehicle- and CNO-treated mice (Fig. 2D; mixed ANOVA, no main effect of treatment, F < 1).
Figure 2.

hM4D-mediated inhibition of vHPC PV neurons did not affect spontaneous or amphetamine-induced locomotor activity. PV-Cre mice expressing hM4D in PV neurons of the vHPC were administered vehicle or CNO intraperitoneally. A, CNO activation of hM4D did not significantly change total spontaneous locomotor activity after either 2 mg/kg CNO (n = 8) or 5 mg/kg CNO (n = 8) compared with vehicle treatment. There were no significant differences in distance traveled across 10 min bins between vehicle and 2 mg/kg CNO (B) or 5 mg/kg CNO (C). D, Amphetamine (1 mg/kg)-induced locomotor activity was not significantly different between CNO-treated (5 mg/kg, n = 10) and vehicle-treated mice (n = 10). Data are presented as mean ± SEM.
Next, we determined whether vHPC PV neuron inhibition affects sensorimotor gating by measuring PPI, a process disrupted in schizophrenia (Braff et al., 2001). As expected, percentage PPI increased as a function of prepulse intensity (Fig. 3A; mixed ANOVA, F(3,54) = 16.09, p < 0.0001; linear trend F(1,18) = 29.07, p < 0.0001). Interestingly, CNO treatment significantly reduced the percentage PPI compared with vehicle treatment (Fig. 3A; mixed ANOVA, main effect of treatment, F(3,54) = 8.44, p = 0.009). This effect was dependent on prepulse intensity level (Fig. 3A; mixed ANOVA, treatment × prepulse intensity interaction, F(5,40) = 3.03, p = 0.037) and was significant at 66, 68, and 72 dB but not at 76 dB (Fig. 3A; unpaired t tests, 66 dB: t(18) = 2.19, p = 0.042; 68 dB: t(18) = 3.08, p = 0.006; 72 dB: t(18) = 2.91, p = 0.009; 76 dB: t(18) = 1.82, p = 0.09). To determine whether vHPC PV neuron inhibition affected the startle response, we also measured startle amplitude during trials in which the 120 dB pulse was presented alone. CNO-treated mice exhibited a significant reduction in startle response compared with vehicle-treated mice (Fig. 3B; unpaired t test, t(18) = 3.29, p = 0.004). Therefore, vHPC PV neuron inhibition decreased both startle response and PPI.
Figure 3.

hM4D-mediated inhibition of vHPC PV neurons reduced percentage PPI, startle amplitude, and spontaneous alternation, but did not affect social interaction. PV-Cre mice expressing hM4D in PV neurons of the vHPC were administered vehicle or CNO intraperitoneally. A, CNO treatment (5 mg/kg, n = 10) reduced percentage PPI compared with vehicle treatment (n = 10). *p < 0.05; **p < 0.01. B, Startle amplitude during pulse-alone (120 dB) trials was significantly reduced in CNO-treated compared with vehicle-treated mice. **p < 0.01. C, CNO-treated mice (5 mg/kg, n = 7) alternated significantly less than vehicle-treated mice (n = 8). **p = 0.01. Di, Dii, Percentage time spent in the stranger and empty chambers (i) and distance traveled during the social interaction test (ii) was not significantly different between vehicle and CNO treatment (5 mg/kg, n = 12). Data are presented as mean ± SEM.
We then examined the effect of vHPC PV neuron inhibition on spatial working memory by conducting the T-maze continuous spontaneous alternation task (Gerlai, 1998; Dudchenko, 2004; Hughes, 2004). Although vehicle-treated mice showed alternation above 50% of chance (Fig. 3C; 1-sample t test, t(8) = 4.264, p = 0.003), alternation in CNO-treated mice was lower than in vehicle-treated mice (Fig. 3C; unpaired t test, t(15) = 2.96, p = 0.01) and was not significantly different from chance level (Fig. 3C; 1-sample t test, t(7) = 1.0, p = 0.351). No significant differences in direction bias or latency to arm entry were observed between groups (unpaired t tests, t(15) = 0.33, p = 0.74; t(15) = 0.10, p = 0.99).
Last, to determine whether vHPC PV neuron inhibition affects behaviors associated with negative symptoms of schizophrenia, we measured social interaction in Crawley's three-chamber sociability test (Moy et al., 2004; Arguello and Gogos, 2006). After both vehicle and CNO treatment, mice exhibited significant preference for the chamber containing a stranger mouse compared with the empty chamber (Fig. 3Di; 2-way repeated-measures ANOVA, main effect of chamber, F(1,11) = 81.93, p < 0.0001). No significant differences in chamber preference (Fig. 3Di; 2-way repeated-measures ANOVA, no main effect of treatment, F(1,11) = 2.17, p = 0.18; no treatment × chamber interaction, F(1,11) = 2.80, p = 0.12) or locomotor activity (Fig. 3Dii; paired t test, t(11) = 1.096, p = 0.30) were observed between treatments.
These results demonstrate that inhibition of PV neurons in the vHPC disrupted PPI and spontaneous alternation, but had no effect on locomotor activity or social interaction. Together, these findings suggest that PV neurons have a specialized function in regulating sensorimotor gating and spatial working memory. The vHPC has a well established role in mediating locomotor activity (Bast and Feldon, 2003), so a subset of vHPC GABA neurons separate from PV neurons may contribute to this behavior. In addition, the deficits observed in PV-Cre mice may be particularly sensitive to general GABA system dysregulation and may be observed when a certain number of vHPC GABA neurons are inhibited regardless of subtype identity. To investigate these possibilities, we inhibited the population of vHPC GABA neurons expressing GAD65 using GAD65-Cre knock-in mice (Taniguchi et al., 2011).
hM4D expression in GAD65 neurons in the vHPC
When combined with doubly floxed reporter strains, the GAD65-Cre knock-in line (Taniguchi et al., 2011) has proven useful for pan-GABA neuron targeting since the reporter expression pattern reflects the entire history of GAD65 expression up to the time of analysis (Taniguchi et al., 2011; Ledri et al., 2014). However, when combined with Cre-dependent AAV infusion into adult brains, thus excluding embryonic and early postnatal GAD65 expression, the line has been used to study CCK GABA neurons due to its biased expression of Cre recombinase in non-PV GABA neurons (Losonczy et al., 2010; Nagode et al., 2014). In the hippocampus, CCK GABA neurons have been shown to be largely nonoverlapping with PV GABA neurons (Gulyás et al., 1991; Pawelzik et al., 2002).
We used an identical Cre-dependent AAV vector (AAV-hsyn-FLEX-hM4D-mCherry) in GAD65-Cre mice to target hM4D to GAD65 neurons (Fig. 4A). hM4D was expressed in ∼3 times as many neurons in GAD65-Cre mice compared with PV-Cre mice (Fig. 4B; unpaired t test, t(5) = 7.23, p = 0.0008). Immunohistochemical staining revealed that hM4D-expressing neurons in GAD65-Cre mice are heterogeneous in their cell type identity, including more CCK-GABA neurons than PV-GABA neurons; only 20% of hM4D-expressing neurons were PV-ir (Fig. 4C,E; 19.7 ± 4.55%), whereas 55% were CCK-ir (Fig. 4D,E; 55.5 ± 4.37%). In addition, 42% of total PV-ir neurons (Fig. 4C; 41.9 ± 3.82%) and 64% of total CCK-ir neurons were hM4D-positive (Fig. 4D; 64.4 ± 1.48%). These findings confirm previous reports that the GAD65-Cre line shows biased Cre expression in CCK-GABA neurons and demonstrate that hM4D-expressing neurons in GAD65-Cre and PV-Cre mice represent two relatively distinct, yet partially overlapping, subpopulations of GABA neurons.
Figure 4.

Inhibition of GAD65 neurons in the vHPC using hM4D. A, Representative image of hM4D-mCherry expression in the vHPC of GAD65-Cre mice. Inset, High-magnification confocal image of boxed area. Red, hM4D-mCherry; blue, DAPI. B, Mean number of hM4D-mCherry positive cells in the ventral CA1 was significantly higher in GAD65-Cre (n = 4) compared with PV-Cre (n = 3) mice. ***p = 0.0008. C, Representative confocal image of immunostaining for PV in the ventral CA1 of GAD65-Cre mice expressing hM4D. Red, hM4D-mCherry; Green, PV-ir. D, Representative confocal image of immunostaining for CCK in the ventral CA1 of GAD65-Cre mice expressing hM4D. Red, hM4D-mCherry; Green, CCK-ir. E, Bar graph showing the percentage of cells positive for the respective marker out of the total number of hM4D-positive cells in GAD65-Cre mice (n = 9). Data are presented as mean ± SEM.
GAD65 neuron inhibition in the vHPC enhances locomotor activity and disrupts spatial working memory
In contrast to PV-Cre mice, CNO administration in GAD65-Cre mice significantly increased spontaneous locomotor activity compared with vehicle administration (Fig. 5A; mixed ANOVA, main effect of treatment, F(1,20) = 36.05, p < 0.0001). This enhancement was observed for all doses of CNO tested (Fig. 5A; paired t tests, 0.5 mg/kg: t(3) = 3.30, p = 0.046; 1 mg/kg: t(9) = 3.46, p = 0.007; 2 mg/kg: t(8) = 5.69, p = 0.0005). No significant main effect of dose (Fig. 5A; mixed ANOVA, F(2,20) = 2.38, p = 0.12) or dose by treatment interaction was found (Fig. 5A; mixed ANOVA, F < 1). The increased locomotor activity induced by CNO significantly changed across time (Fig. 5B–D; 2-way repeated-measures ANOVA, main effect of treatment, 0.5 mg/kg: F(1,3) = 10.86, p = 0.046, 1 mg/kg: F(1,9) = 11.94, p = 0.007, 2 mg/kg: F(1,8) = 32.34, p = 0.0004; treatment × time interaction, 0.5 mg/kg: F(5,15) = 9.67, p < 0.0001, 1 mg/kg: F(5,45) = 3.10, p = 0.017, 2 mg/kg: F(5,40) = 9.96, p < 0.0001), with peak activity occurring at 30 min after 0.5 mg/kg CNO (Fig. 5B; paired t test, t(3) = 3.61, p = 0.037), at 10–20 min after 1 mg/kg CNO (Fig. 5C; paired t test, 10 min: t(9) = 3.82, p = 0.004, 20 min: t(9) = 2.23, p = 0.053), and at 20 min after 2 mg/kg CNO (Fig. 5D; paired t test, t(8) = 4.87, p = 0.001).
Figure 5.

hM4D-mediated inhibition of vHPC GAD65 neurons enhanced spontaneous and amphetamine-induced locomotor activity. GAD65-Cre mice expressing hM4D in GAD65 neurons of the vHPC were administered vehicle or CNO intraperitoneally. A, CNO significantly increased total distance traveled at 0.5 mg/kg (n = 4), 1 mg/kg (n = 10), and 2 mg/kg (n = 9) compared with vehicle treatment. *p < 0.05; **p < 0.01; ***p < 0.001. B–D, Distance traveled across 10 min bins was enhanced after 0.5 mg/kg CNO (p = 0.05; B); 1 mg/kg CNO (p = 0.007; C); or 2 mg/kg CNO (p < 0.001; D). E, Distance traveled after 1 mg/kg CNO was reduced by 0.02 mg/kg haloperidol pretreatment (n = 6) compared with vehicle pretreatment (n = 6). *p < 0.05. There were no significant differences between vehicle + vehicle–treated (n = 6) and haloperidol + vehicle–treated mice (n = 6). F, Distance traveled after vehicle and CNO was reduced by SCH23390 (n = 6). *p < 0.05. G, Amphetamine (1 mg/kg)-induced locomotor activity was significantly enhanced in CNO-treated (0.5 mg/kg, n = 8) compared with vehicle-treated mice (n = 8). p < 0.0001.
To determine whether the enhanced locomotor activity was mediated by dopamine receptor binding, we pretreated mice with either the D2 receptor antagonist and typical antipsychotic haloperidol or with the selective D1 receptor antagonist SCH-23390. Haloperidol (0.02 mg/kg) pretreatment had no significant effect on locomotor activity after vehicle administration (Fig. 5E; unpaired t test, t(10) = 0.86, p = 0.41), but significantly attenuated locomotor activity induced by 1 mg/kg CNO administration (Fig. 5E; unpaired t test, t(10) = 3.049, p = 0.012). However, this attenuation was partial because locomotor activity of mice treated with haloperidol + CNO was significantly higher than of vehicle + vehicle controls (Fig. 5E; paired t test, t(5) = 2.674, p = 0.044). SCH-23390 (0.05 mg/kg) pretreatment significantly reduced, but did not completely abolish, locomotor activity after both vehicle and 2 mg/kg CNO administration (Fig. 5F; paired t tests, vehicle, t(5) = 3.97, p = 0.011, CNO: t(5) = 3.05, p = 0.028).
In addition, amphetamine-induced locomotor activity was significantly enhanced by CNO treatment (Fig. 5G). CNO-treated mice exhibited increased amphetamine-induced locomotor activity compared with vehicle-treated mice (Fig. 5G; mixed ANOVA, main effect of treatment, F(1,14) = 38.17, p < 0.0001), with peak activity occurring 30 min after CNO and amphetamine coinjection (Fig. 5G; mixed ANOVA, time × treatment interaction, F(5,70) = 9.96, p < 0.0001; unpaired t test, t(14) = 6.61, p < 0.0001).
In contrast to PV-Cre mice, PPI in GAD65-Cre mice was not affected by CNO treatment (Fig. 6A). No significant differences in PPI were observed between vehicle and CNO treatment at any prepulse intensity (Fig. 6A; mixed ANOVA, no main effect of treatment, F < 1; no treatment × prepulse intensity interaction, F(3,30) = 1.57, p = 0.22). As expected, percentage PPI increased gradually with prepulse intensity level (Fig. 6A; mixed ANOVA, main effect of prepulse intensity, F(3,30) = 23.52, p < 0.0001; linear trend, F(1,10) = 41.27, p < 0.0001). Furthermore, startle response was not significantly different between CNO- and vehicle-treated mice (Fig. 6B; unpaired t test, t(13) = 1.77, p = 0.1).
Figure 6.

hM4D-mediated inhibition of vHPC GAD65 neurons impaired spontaneous alternation but did not affect percentage PPI, startle amplitude, or social interaction. GAD65-Cre mice expressing hM4D in GAD65 neurons of the vHPC were administered vehicle or CNO intraperitoneally. A, B, No significant differences in percentage PPI (A) or startle amplitude (B) during pulse-alone trials were found between mice treated with CNO (5 mg/kg, n = 8) and vehicle (n = 7). C, CNO (0.5 mg/kg, n = 8) reduced percentage alternation compared with vehicle (n = 8). **p = 0.001. Di, Dii, In the social interaction test, percentage time spent in the stranger and empty chambers (i) and distance traveled (ii) were not significantly different between vehicle and CNO treatment (0.5 mg/kg, n = 8). Data are presented as mean ± SEM.
Similar to PV-Cre mice, CNO-treated GAD65-Cre mice exhibited impaired spontaneous alternation (Fig. 6C). Spontaneous alternation in CNO-treated mice was significantly lower than vehicle-treated mice (Fig. 6C; unpaired t test, t(14) = 4.13, p = 0.001) and was below chance level (Fig. 6C; 1-sample t test, CNO: t(7) = 3.64, p = 0.008; vehicle: t(7) = 2.31, p = 0.054). Although no differences in latency to arm entry were found (unpaired t test, t(14) = 1.85, p = 0.086), direction bias was significantly higher in CNO-treated mice (unpaired t test, t(14) = 2.29, p = 0.038). The increased direction bias may account for the below-chance alternation observed and may indicate a stereotyped or perseverative tendency to enter one arm of the maze.
In the test for social interaction, no significant differences in preference for the stranger chamber compared with the empty chamber (Fig. 6Di; two-way repeated-measures ANOVA, main effect of chamber, F(1,7) = 53.44, p = 0.0001; no main effect of treatment, F < 1; no treatment × chamber interaction, F(1,7) = 1.39, p = 0.28) or in locomotor activity (Fig. 6Dii; paired t test, t(7) = 0.425, p = 0.684) were observed between vehicle and CNO treatment. The lack of enhancement in locomotor activity may be due to the structure of the three-chambered apparatus, which is likely less conducive for locomotor activity compared with the open field.
Together, inhibition of GAD65 neurons in the vHPC increased spontaneous locomotor activity through both D2 and D1 receptor binding, potentiated amphetamine-induced locomotor activity, and impaired spontaneous alternation, but had no effect on PPI or social interaction.
Inhibition of PV and GAD65 neurons alters vHPC excitability and network activity
The differences in behavioral changes observed upon inhibiting PV and GAD65 neurons in the vHPC may be associated with distinct changes in network activity. To explore this possibility, we first compared the effects of PV and GAD65 neuron inhibition on network excitability by performing extracellular recordings from ventral hippocampal slices. Specifically, we determined the input-output relationship in the CA1 (stimulus intensity vs PSP) in slices obtained from mice transduced with hM4D in either the PV or GAD65 neuron populations. As expected, we found that application of CNO significantly increased the PSP amplitude of the input-output curves (Fig. 7A; 3-way mixed ANOVA, main effect of treatment, F(1,28) = 9.99, p = 0.004), reflecting an increase in network excitability when either population of interneurons was inhibited (Fig. 7A; 2-way repeated-measures ANOVA, main effect of treatment, GAD65-Cre: F(1,14) = 7.03, p = 0.019; PV-Cre: F(1,14) = 5.02, p = 0.042). In addition, the CNO-induced increase in PSP amplitude was significantly larger for GAD65-Cre mice compared with PV-Cre mice (Fig. 7A; 2-way mixed ANOVA, genotype × stimulus intensity interaction, F(2,56) = 3.66, p = 0.032), with a significant difference observed at the high-intensity range (Fig. 7A,B; unpaired t test, t(28) = 2.07, p = 0.048).
Figure 7.
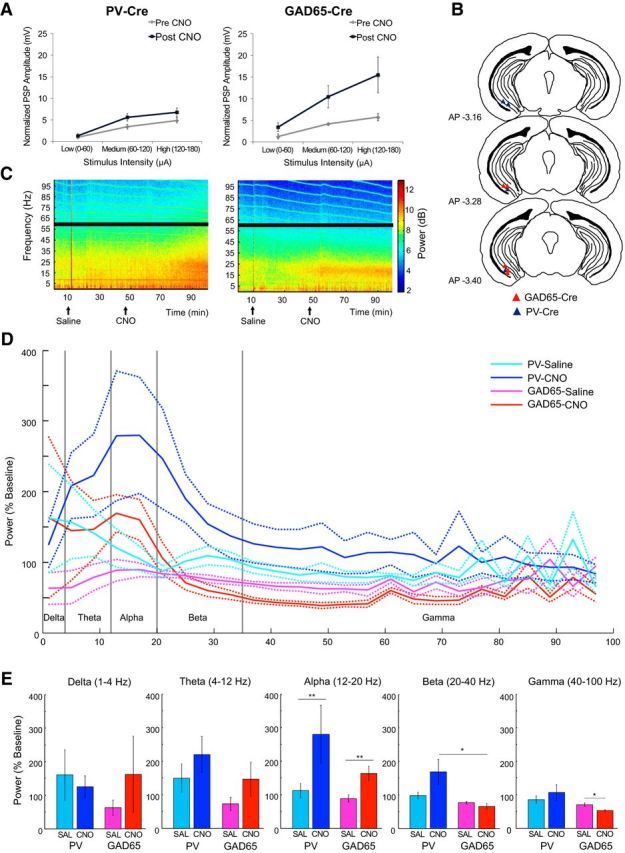
Effect of hM4D-mediated inhibition of vHPC GAD65 and PV neurons on network excitability and oscillations. Summary curves showing the relationship between stimulus intensity and PSP measured by in vitro extracellular recordings in the vHPC CA1 subfield of GAD65-Cre (A, left, n = 6) and PV-Cre mice (A, right, n = 5) before and after CNO application. B–E, Examining changes in the power of oscillations in the vHPC after saline and CNO injections intraperitoneally using in vivo electrophysiology in anesthetized mice. B, Electrode locations for recordings of LFP activity in the vHPC of the hM4D-expressing hemisphere of PV-Cre (blue triangle) and GAD65-Cre (red triangle) mice. Images are adapted from Paxinos and Franklin (2007). C, vHPC LFP power (color coded) for different frequencies (y-axis) across time (x-axis) depicting changes in power after saline (arrow) and CNO (arrow) injections. Black bars mask the power line noise. The source of horizontal lines in the high-frequency band is unknown. D, Power normalized to baseline after saline or CNO injection in PV-Cre (n = 4, cyan and blue lines) and GAD65-Cre (n = 5, pink and red lines, respectively) mice. Dotted lines show the SEM. E, Averaged power in delta, theta, alpha, beta, and gamma bands after saline or CNO injection in PV-Cre (cyan and blue bars) and GAD65-Cre (pink and red bars) mice. The error bars show the SEM. A significant main effect of treatment (**p = 0.008) in the alpha band, a significant difference between PV-Cre + CNO and GAD65-Cre + CNO in the beta band (*p = 0.020), and a significant difference between GAD65-Cre + saline and GAD65-Cre + CNO in the gamma band (*p = 0.016) were observed.
Next, we investigated whether inhibition of PV or GAD65 neurons affects network oscillatory activity in the vHPC in vivo by comparing the effects of systemic injection of saline or CNO on LFP power recorded bilaterally in the vHPC under urethane anesthesia (Fig. 7B–E). We chose this preparation over recording in behaving mice to isolate the effect of CNO on LFP activity from potential LFP changes induced by behavior. Mice expressed hM4D unilaterally in the vHPC, with the nonexpressing side serving as an internal control for factors other than drug injection, such as the depth of anesthesia and body temperature (Fig. 7B). In both PV-Cre and GAD65-Cre mice, CNO injection gradually increased the power of oscillations mainly in the bands <∼35 Hz, whereas saline injection had no effect (Fig. 7C). This change was not observed in the control hemisphere (data not shown). To determine whether CNO had different effects on the power of oscillation between PV-Cre and GAD65-Cre mice, we compared the normalized oscillation power (up to 100 Hz) during the last 10 min block of each injection period (25–35 min postinjection; Fig. 7D). The effect of CNO significantly differed depending on the groups and frequency of oscillatory activity (Fig. 7D; 3-way repeated-measures ANOVA, an interaction of treatment, group, and frequency, F(24,336) = 1.56, p = 0.047). To identify which frequency band showed the group-dependent effect of CNO, the normalized power was compared across the treatment types and groups in five different frequency bands (Fig. 7E). CNO did not have any effect on the power of delta (1–4 Hz) or theta (4–12 Hz) oscillation in either group (Fig. 7E; 2-way repeated-measures ANOVA, delta: no interaction of treatment and group, F(1,7) = 1.16, p = 0.32, no main effect of treatment, F < 1, no main effect of group, F < 1; theta: no interaction of treatment and group, F < 1, no main effect of treatment, F(1,7) = 4.67, p = 0.068, no main effect of group, F(1,7) = 2.16, p = 0.19). In contrast, CNO increased the power of alpha oscillation (12–20 Hz) in both GAD65-Cre and PV-Cre mice (Fig. 7E; main effect of treatment, F(1,7) = 13.7, p = 0.008). In the beta band (20–40 Hz), CNO had a differential effect on the power depending on the groups (Fig. 7E; interaction of treatment and group, F(1,7) = 7.56, p = 0.029; a follow-up t test, PV-Cre + CNO versus GAD65-Cre + CNO, p = 0.020). In addition, CNO decreased the power of gamma oscillation (40–100 Hz) in GAD65-Cre, but not PV-Cre, mice (Fig. 7E; interaction of treatment and group, F(1,7) = 10.56, p = 0.014; a follow-up t test, GAD65-Cre + saline vs GAD65-Cre + CNO, p = 0.016, PV-Cre + saline vs PV-Cre + CNO, p = 0.456). Together, inhibition of PV and GAD65 neurons in the vHPC both increased network excitability in the vHPC, but produced qualitatively distinct changes in spontaneous network oscillatory activity.
Discussion
GABAergic dysfunction has been proposed as a key process underlying the vHPC hyperactivity observed in schizophrenia (Heckers et al., 2002; Lodge and Grace, 2008; Schobel et al., 2013). A diverse population of GABAergic interneurons participates in inhibitory mechanisms controlling pyramidal cell activity in the hippocampus (Klausberger et al., 2003; Klausberger and Somogyi, 2008). This raises the important question of whether all subtypes of vHPC GABA interneurons are equally involved in schizophrenia pathophysiology or if specific interneuron subtypes contribute differentially.
Electrophysiology experiments in the present study demonstrate that inhibiting either PV neurons or GAD65 neurons is sufficient to produce a hyperactive state in the vHPC. In vitro extracellular recordings suggest that GAD65 neuron inhibition results in a greater change in network excitability than PV neuron inhibition. Moreover, inhibiting these two subpopulations of GABA neurons produced distinct changes in network oscillatory activity during in vivo LFP recordings. Inhibition of GAD65 neurons increased LFP power of the alpha oscillation (12–20 Hz), whereas PV neuron inhibition increased LFP power of both alpha (12–20 Hz) and beta (20–40 Hz) oscillations. A caveat of the present study, however, is that the LFP power was measured in anesthetized animals, in which the peak frequencies of LFP bands likely differ from those measured in freely moving animals. Therefore, changes in the LFP frequency bands reported in the present study cannot be linked directly to the behavioral changes observed upon inhibiting GAD65 or PV neurons.
Consistent with their functional dissociation in controlling vHPC network activity, our study shows that inhibition of PV neurons and GAD65 neurons produces distinct behavioral deficits. We found that acute inhibition of PV neurons in the vHPC, but not GAD65 neurons, disrupted PPI, an operational measure of sensorimotor gating, as well as reduced startle reactivity. Although GAD65 neurons in the vHPC far exceed PV neurons in number, inhibiting GAD65 neurons did not impair PPI. Therefore, PPI impairment elicited by PV neuron inhibition is not simply a result of a nonspecific and overall decrease in GABA transmission; rather, it suggests a specific role of vHPC PV neurons in PPI regulation. GAD65 neurons represent a heterogeneous population of GABA neurons including CCK, PV, and likely other GABA neuron subtypes. In GAD65-Cre mice, 42% of vHPC PV-ir cells expressed hM4D and were recruited for inhibition as a part of GAD65 neurons. This suggests that there may be a threshold of vHPC PV neurons that must be inhibited before PPI impairment is observed.
Although the hyperactive state of the vHPC has consistently been associated with PPI deficits, the underlying mechanism has not yet been determined (Wan et al., 1996; Bast and Feldon, 2003). The vHPC-evoked PPI deficit may be mediated by a dopamine-dependent pathway similar to vHPC-evoked hyperlocomotor activity, in which case, local blockade of glutamatergic transmission in the nucleus accumbens (NAc) or systemically inhibiting dopamine activity would likely reverse this deficit. However, Wan et al. (1996) found that neither local NAc infusion of the AMPA receptor blocker CNQX nor systemic injection of the dopamine antagonist haloperidol reversed the vHPC-evoked PPI deficit. In contrast, vHPC-evoked hyperlocomotor activity was completely abolished by haloperidol or the D1 receptor antagonist SCH23390 (Bardgett and Henry, 1999; Bast et al., 2001c). These findings suggest that the vHPC modulates PPI and locomotor activity through different mechanisms. vHPC-evoked hyperlocomotion appears to be initiated by vHPC-NAc efferent activity and to arise from excess mesolimbic dopamine activity, whereas vHPC-evoked PPI disruption is mediated by pathways independent of the mesolimbic dopamine system (Bast and Feldon, 2003). It is possible that hippocampal targets other than the NAc are involved in the PPI disruption observed upon inhibiting vHPC PV neurons. Inhibiting PV neurons, but not GAD65 neurons, could evoke changes along efferent pathways relevant for PPI, such as vHPC projections to the mPFC or the amygdala (Bakshi and Geyer, 1998; Shoemaker et al., 2005). Consistent with this idea, PV neurons in the vHPC have been found to preferentially innervate and inhibit pyramidal neurons projecting to the amygdala (Lee et al., 2014).
Furthermore, the vHPC-evoked PPI impairment may reflect a disruption in synchronized network activity within the vHPC relevant to sensorimotor gating. Consistent with this idea, systemic administration of the NMDA receptor antagonist phencyclidine results in PPI impairment and an abnormally high amplitude of gamma-frequency oscillation in the hippocampus, suggesting that hippocampal modulation of PPI may be associated with gamma rhythms (Ma et al., 2004). Convergent evidence has implicated the synchronous activity of fast-spiking PV neurons in gamma-frequency oscillation (Cardin et al., 2009; Sohal et al., 2009; Carlén et al., 2012). Therefore, the PPI impairment induced by vHPC PV neuron inhibition may be mediated by changes in local hippocampal gamma activity. This possibility will have to be examined further by measuring evoked LFP activity in the vHPC during PPI testing upon inhibiting PV neurons.
We also found that acute inhibition of GAD65 neurons, but not PV neurons, in the vHPC is sufficient to enhance both spontaneous and amphetamine-induced locomotor activity. Consistent with our findings, pharmacological stimulation of the vHPC by NMDA, picrotoxin, or carbachol has been shown to increase locomotor activity, whereas pharmacological inhibition of the vHPC by TTX or muscimol produces the opposite effect (Yang and Mogenson, 1987; Brenner and Bardgett et al., 1998; Bast et al., 2001a; Bast et al., 2001b). Locomotor activity evoked by GAD65 neuron inhibition is likely mediated by excess DA activity, because it was attenuated by systemic administration of the D2 receptor antagonist haloperidol. GAD65 neuron inhibition in the vHPC may increase dopamine transmission through a well characterized pathway involving the NAc and ventral pallidum (Floresco et al., 2001, 2003). vHPC stimulation has been shown to increase the number of spontaneously firing dopamine neurons in the ventral tegmental area, reflecting increased dopamine neuron population activity (West and Grace, 2000; Lodge and Grace, 2006). In contrast to GAD65 neuron inhibition, selective inhibition of PV neurons in the vHPC failed to enhance either spontaneous or amphetamine-induced locomotor activity. This finding suggests that the hyperactive state of the vHPC achieved upon PV neuron inhibition may be insufficient to increase dopamine neuron population activity in the VTA, although this possibility should be examined in future studies.
A functional dissociation between GAD65 and PV neurons has been described previously by Nagode et al. (2014). Using AAV-mediated expression of halorhodopsin (NpHR) in GAD65-Cre and PV-Cre mice, optogenetic silencing of GAD65 neurons suppressed carbachol-induced theta oscillations in acute hippocampal slices, whereas silencing of PV neurons did not. The study found that ∼90% of CCK-expressing cells were labeled with NpHR in GAD65-Cre mice, suggesting that CCK-expressing GABA neurons contribute to generating carbachol-induced theta oscillations. In the present study, we found that 64% of hM4D-expressing neurons in GAD65-Cre mice express CCK. The inhibition of CCK-GABA neurons may largely account for the increase in amphetamine-induced locomotion observed upon inhibiting GAD65 neurons, so one may expect that inhibition of CCK-GABA neurons alone should be sufficient to recapitulate the hyperlocomotor activity. Therefore, it would be of great interest to examine the effect of selectively inhibiting vHPC CCK-GABA neurons on theta-frequency oscillations during locomotor activity and compare the effects with those of inhibiting other GABA neuron subtypes, including somatostatin, VIP, calbindin, or calretinin-expressing GABA neurons (Klausberger and Somogyi, 2008).
The importance of PV neurons in spatial working memory is supported by studies using genetic strategies to impair PV neuron function both globally and specifically in the dorsal hippocampus (Fuchs et al., 2007; Belforte et al., 2010; Korotkova et al., 2010; Murray et al., 2011; Carlén et al., 2012). Upon inhibiting either PV or GAD65 neurons in the vHPC, we observed deficits in T-maze spontaneous alternation, which is used to measure spatial working memory. Unlike PV neuron inhibition, GAD65 neuron inhibition reduced alternation to below chance level and increased direction bias. Although these behaviors likely reflected stereotypy or increased perseveration typical of hippocampal disturbances (Stevens and Cowey, 1973; Dalland, 1976; Gerlai, 1998), intact working memory of the previously visited arm cannot be excluded. These findings suggest that spatial working memory is sensitive to disruptions in the activity of a broad set of vHPC GABA neurons that include PV neurons. However, more extensive testing of working memory using well validated assays such as the delayed match/nonmatch to sample task (Olton, 1987; Dudchenko, 2004) are required to demonstrate conclusively the role of vHPC PV and Gad65 neurons in spatial working memory.
It is not well understood whether the vHPC plays a critical role in social interaction. Ibotenic acid lesions in the vHPC in adult rats did not affect social behavior (Becker et al., 1999), but a recent optogenetic study showed that amygdala-mediated vHPC activation robustly reduced social interaction (Felix-Ortiz and Tye, 2014). The present study found that vHPC activation achieved by inhibition of vHPC GABA neurons did not significantly alter social interaction. The hippocampal CA1 area innervated by the amygdala in Felix-Ortiz and Tye (2014) is more dorsal to the region included in the present study. This suggests that there may be a functional dissociation within the vHPC in controlling social behaviors.
It is important to note that the behavioral changes observed in our study are the result of the acute loss of GABA neuron activity restricted to the vHPC in adult mice, which is in contrast to more broad and chronic changes reported in developmental models of schizophrenia. A prevailing hypothesis proposes that GABA dysfunction in forebrain regions arises from alterations in development during specific periods of prenatal, postnatal, or postpubertal life and later becomes a core feature of the disease in adulthood (Gonzalez-Burgos et al., 2011; Powell et al., 2012). The plausibility of this neurodevelopmental hypothesis has been explored in various animal models of schizophrenia, including the MAM model (Flagstad et al., 2004; Moore et al., 2006; Lodge et al., 2009). Notably, acute pharmacological inhibition of adult vHPC activity, via local infusion of an allosteric modulator for GABAA α5 subunit, was sufficient to normalize both aberrant dopamine activity and hyperlocomotor response to amphetamine in the MAM model (Gill et al., 2011). Furthermore, Perez and Lodge (2013) showed that transplanting GABAergic precursor cells of the medial ganglionic origin into the vHPC can reverse both aberrant dopamine activity and hyperlocomotor response to amphetamine in the MAM model. Together with ours, these findings suggest that, in schizophrenia, a hyperactive state of the vHPC in the adult brain may be a direct cause of behavioral deficits and the associated pathological increase in DA activity.
In summary, our results support the idea that GABA dysfunction in the vHPC could account, at least in part, for the specific behavioral deficits found in schizophrenia and suggest a functional dissociation among the hippocampal GABAergic mechanisms underlying schizophrenia pathophysiology. Impaired functioning of PV neurons may underlie sensorimotor gating deficits, whereas GAD65 neurons may be involved in hyperlocomotor activity arising from excess dopamine activity.
Footnotes
CNO was obtained from the NIH as part of the Rapid Access to Investigative Drug Program funded by the NINDS. This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC Discovery Grant MOP 491009 to J.C.K. and 29319 to M.A.W.) and the Canadian Institutes of Health Research (Grant MOP 496401 to J.C.K.). We thank Elena Soukhov and Michael McPhail for technical assistance and members of the Kim laboratory for their input.
The authors declare no competing financial interests.
References
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE, Jr, Jones EG. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- Arguello PA, Gogos JA. Modeling madness in mice: one piece at a time. Neuron. 2006;52:179–196. doi: 10.1016/j.neuron.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Aponte Y, Su HH, Sternson SM. A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci. 2008;28:7025–7030. doi: 10.1523/JNEUROSCI.1954-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi VP, Geyer MA. Multiple limbic regions mediate the disruption of prepulse inhibition produced in rats by the noncompetitive NMDA antagonist dizocilpine. J Neurosci. 1998;18:8394–8401. doi: 10.1523/JNEUROSCI.18-20-08394.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardgett ME, Henry JD. Locomotor activity and accumbens Fos expression driven by ventral hippocampal stimulation require D1 and D2 receptors. Neuroscience. 1999;94:59–70. doi: 10.1016/S0306-4522(99)00303-6. [DOI] [PubMed] [Google Scholar]
- Bast T, Feldon J. Hippocampal modulation of sensorimotor processes. Prog Neurobiol. 2003;70:319–345. doi: 10.1016/S0301-0082(03)00112-6. [DOI] [PubMed] [Google Scholar]
- Bast T, Zhang W-N, Feldon J. Hyperactivity, decreased startle reactivity, and disrupted prepulse inhibition following disinhibition of the rat ventral hippocampus by the GABA(A) receptor antagonist picrotoxin. Psychopharmacology (Berl) 2001a;156:225–233. doi: 10.1007/s002130100775. [DOI] [PubMed] [Google Scholar]
- Bast T, Zhang WN, Feldon J. The ventral hippocampus and fear conditioning in rats. Exp Brain Res. 2001b;139:39–52. doi: 10.1007/s002210100746. [DOI] [PubMed] [Google Scholar]
- Bast T, Zhang WN, Heidbreder C, Feldon J. Hyperactivity and disruption of prepulse inhibition induced by N-methyl-D-aspartate stimulation of the ventral hippocampus and the effects of pretreatment with haloperidol and clozapine. Neuroscience. 2001c;103:325–335. doi: 10.1016/S0306-4522(00)00589-3. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Reynolds GP. Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophr Res. 1997;24:349–355. doi: 10.1016/S0920-9964(96)00122-3. [DOI] [PubMed] [Google Scholar]
- Becker A, Grecksch G, Bernstein H-G, Höllt V, Bogerts B. Social behaviour in rats lesioned with ibotenic acid in the hippocampus: quantitative and qualitative analysis. Psychopharmacology (Berl) 1999;144:333–338. doi: 10.1007/s002130051015. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Benes FM, McSparren J, Bird ED, SanGiovanni JP, Vincent SL. Deficits in small interneurons in prefrontal and cingulate cortices of schizophrenic and schizoaffective patients. Arch Gen Psychiatry. 1991;48:996–1001. doi: 10.1001/archpsyc.1991.01810350036005. [DOI] [PubMed] [Google Scholar]
- Benes FM, Lim B, Matzilevich D, Walsh JP, Subburaju S, Minns M. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc Natl Acad Sci U S A. 2007;104:10164–10169. doi: 10.1073/pnas.0703806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner DM, Bardgett ME. Haloperidol blocks increased locomotor activity elicited by carbachol infusion into the ventral hippocampal formation. Pharmacol Biochem Behav. 1998;60:759–764. doi: 10.1016/S0091-3057(98)00053-7. [DOI] [PubMed] [Google Scholar]
- Braff D, Geyer M, Swerdlow N. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlén M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, Rühlmann C, Jones SR, Deisseroth K, Sheng M, Moore CI, Tsai LH. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17:537–548. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalland T. Response perseveration of rats with dorsal hippocampal lesions. Behav Biol. 1976;17:473–484. doi: 10.1016/S0091-6773(76)90865-8. [DOI] [PubMed] [Google Scholar]
- Dudchenko PA. An overview of the tasks used to test working memory in rodents. Neurosci Biobehav Rev. 2004;28:699–709. doi: 10.1016/j.neubiorev.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Felix-Ortiz AC, Tye KM. Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. J Neurosci. 2014;34:586–595. doi: 10.1523/JNEUROSCI.4257-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagstad P, Mørk A, Glenthøj BY, van Beek J, Michael-Titus AT, Didriksen M. Disruption of neurogenesis on gestational day 17 in the rat causes behavioral changes relevant to positive and negative schizophrenia symptoms and alters amphetamine-induced dopamine release in nucleus accumbens. Neuropsychopharmacology. 2004;29:2052–2064. doi: 10.1038/sj.npp.1300516. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Blaha CD, Yang CR, Phillips AG. Modulation of hippocampal and amygdalar-evoked activity of nucleus accumbens neurons by dopamine: cellular mechanisms of input selection. J Neurosci. 2001;21:2851–2860. doi: 10.1523/JNEUROSCI.21-08-02851.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, Rozov A, Whittington MA, Traub RD, Rawlins JN, Monyer H. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53:591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Heizmann CW, Kosaka T. Quantitative analysis of GAD65 and GAD67 immunoreactivities in somata of GABAergic neurons in the mouse hippocampus proper (CA1 and CA3 regions), with special reference to parvalbumin-containing neurons. Brain Res. 1997;764:237–243. doi: 10.1016/S0006-8993(97)00683-5. [DOI] [PubMed] [Google Scholar]
- Fung SJ, Webster MJ, Sivagnanasundaram S, Duncan C, Elashoff M, Weickert CS. Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am J Psychiatry. 2010;167:1479–1488. doi: 10.1176/appi.ajp.2010.09060784. [DOI] [PubMed] [Google Scholar]
- Gerlai R. A new continuous alternation task in T-maze detects hippocampal dysfunction in mice. A strain comparison and lesion study. Behav Brain Res. 1998;95:91–101. doi: 10.1016/S0166-4328(97)00214-3. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Moghaddam B. Animal models relevant to schizophrenia disorder. In: Davis KL, Charney C, Coyle JT, Nemeroff C, editors. Psychopharmacology: the fifth generation of progress. Philadelphia: Lippincott; 2002. pp. 689–701. [Google Scholar]
- Gill KM, Lodge DJ, Cook JM, Aras S, Grace AA. R-positive allosteric modulator reverses hyperactivation of the dopamine system in the MAM model of schizophrenia. Neuropsychopharmacology. 2011;36:1903–1911. doi: 10.1038/npp.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Lewis DA. GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia. Schizophr Bull. 2008;34:944–961. doi: 10.1093/schbul/sbn070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Fish KN, Lewis DA. GABA neuron alterations, cortical circuit dysfunction and cognitive deficits in schizophrenia. Neural Plast. 2011;2011:723184. doi: 10.1155/2011/723184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA. Ventral hippocampus, interneurons, and schizophrenia: a new understanding of the pathophysiology of schizophrenia and its implications for treatment and prevention. Current Directions in Psychology. 2010;19:232–237. doi: 10.1177/0963721410378032. [DOI] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E, DiGiorgi Gerevini V. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- Gulyás AI, Tóth K, Dános P, Freund TF. Subpopulations of GABAergic neurons containing parvalbumin, calbindin D28k, and cholecystokinin in the rat hippocampus. J Comp Neurol. 1991;312:371–378. doi: 10.1002/cne.903120305. [DOI] [PubMed] [Google Scholar]
- Harte MK, Powell SB, Swerdlow NR, Geyer MA, Reynolds GP. Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J Neural Transm. 2007;114:893–898. doi: 10.1007/s00702-007-0627-6. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23:6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM. Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry. 2002;59:521–529. doi: 10.1001/archpsyc.59.6.521. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RN. The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci Biobehav Rev. 2004;28:497–505. doi: 10.1016/j.neubiorev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Magill PJ, Márton LF, Roberts JD, Cobden PM, Buzsáki G, Somogyi P. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature. 2003;421:844–848. doi: 10.1038/nature01374. [DOI] [PubMed] [Google Scholar]
- Konradi C, Yang CK, Zimmerman EI, Lohmann KM, Gresch P, Pantazopoulos H, Berretta S, Heckers S. Hippocampal interneurons are abnormal in schizophrenia. Schizophr Res. 2011;131:165–173. doi: 10.1016/j.schres.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68:557–569. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, Lowell BB. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 2011;121:1424–1428. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledri M, Madsen MG, Nikitidou L, Kirik D, Kokaia M. Global optogenetic activation of inhibitory interneurons during epileptiform activity. J Neurosci. 2014;34:3364–3377. doi: 10.1523/JNEUROSCI.2734-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Marchionni I, Bezaire M, Varga C, Danielson N, Lovett-Barron M, Losonczy A, Soltesz I. Parvalbumin-positive basket cells differentiate among hippocampal pyramidal cells. Neuron. 2014;82:1129–1144. doi: 10.1016/j.neuron.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31:234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. The hippocampus modulates dopamine neuron responsivity by regulating the intensity of phasic neuron activation. Neuropsychopharmacology. 2006;31:1356–1361. doi: 10.1038/sj.npp.1300963. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci. 2007;27:11424–11430. doi: 10.1523/JNEUROSCI.2847-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Hippocampal dysfunction and disruption of dopamine system regulation in an animal model of schizophrenia. Neurotox Res. 2008;14:97–104. doi: 10.1007/BF03033801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci. 2009;29:2344–2354. doi: 10.1523/JNEUROSCI.5419-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczy A, Zemelman BV, Vaziri A, Magee JC. Network mechanisms of theta related neuronal activity in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2010;13:967–972. doi: 10.1038/nn.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Shen B, Rajakumar N, Leung LS. The medial septum mediates impairment of prepulse inhibition of acoustic startle induced by a hippocampal seizure or phencyclidine. Behav Brain Res. 2004;155:153–166. doi: 10.1016/j.bbr.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008;22:469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry. 2006;60:253–264. doi: 10.1016/j.biopsych.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L, Bartos M, Wisden W, Wulff P. Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat Neurosci. 2011;14:297–299. doi: 10.1038/nn.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagode DA, Tang AH, Yang K, Alger BE. Optogenetic identification of an intrinsic cholinergically driven inhibitory oscillator sensitive to cannabinoids and opioids in hippocampal CA1. J Physiol. 2014;592:103–123. doi: 10.1113/jphysiol.2013.257428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, Belforte JE. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology. 2012;62:1574–1583. doi: 10.1016/j.neuropharm.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olton DS. The radial arm maze as a tool in behavioral pharmacology. Physiol Behav. 1987;40:793–797. doi: 10.1016/0031-9384(87)90286-1. [DOI] [PubMed] [Google Scholar]
- Pawelzik H, Hughes DI, Thomson AM. Physiological and morphological diversity of immunocytochemically defined parvalbumin- and cholecystokinin-positive interneurones in CA1 of the adult rat hippocampus. J Comp Neurol. 2002;443:346–367. doi: 10.1002/cne.10118. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB. The Mouse Brain in Stereotaxic Coordinates. (Fourth Edition) 2007 [Google Scholar]
- Penschuck S, Flagstad P, Didriksen M, Leist M, Michael-Titus AT. Decrease in parvalbumin-expressing neurons in the hippocampus and increased phencyclidine-induced locomotor activity in the rat methylazoxymethanol (MAM) model of schizophrenia. Eur J Neurosci. 2006;23:279–284. doi: 10.1111/j.1460-9568.2005.04536.x. [DOI] [PubMed] [Google Scholar]
- Perez SM, Lodge DJ. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Mol Psychiatry. 2013;18:1193–1198. doi: 10.1038/mp.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterschmitt Y, Meyer F, Louilot A. Differential influence of the ventral subiculum on dopaminergic responses observed in core and dorsomedial shell subregions of the nucleus accumbens in latent inhibition. Neuroscience. 2008;154:898–910. doi: 10.1016/j.neuroscience.2008.03.073. [DOI] [PubMed] [Google Scholar]
- Powell SB, Sejnowski TJ, Behrens MM. Behavioral and neurochemical consequences of cortical oxidative stress on parvalbumin-interneuron maturation in rodent models of schizophrenia. Neuropharmacology. 2012;62:1322–1331. doi: 10.1016/j.neuropharm.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, Small SA. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66:938–946. doi: 10.1001/archgenpsychiatry.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, Inbar BP, Corcoran CM, Lieberman JA, Moore H, Small SA. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78:81–93. doi: 10.1016/j.neuron.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Lang B, Nakamoto C, Zhang F, Pu J, Kuan SL, Chatzi C, He S, Mackie I, Brandon NJ, Marquis KL, Day M, Hurko O, McCaig CD, Riedel G, St Clair D. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J Neurosci. 2008;28:10893–10904. doi: 10.1523/JNEUROSCI.3299-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker JM, Saint Marie RL, Bongiovanni MJ, Neary AC, Tochen LS, Swerdlow NR. Prefrontal D1 and ventral hippocampal N-methyl-D-aspartate regulation of startle gating in rats. Neuroscience. 2005;135:385–394. doi: 10.1016/j.neuroscience.2005.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens R, Cowey A. Effects of dorsal and ventral hippocampal lesions on spontaneous alternation, learned alternation and probability learning in rats. Brain Res. 1973 doi: 10.1016/0006-8993(73)90659-8. [DOI] [PubMed] [Google Scholar]
- Taepavarapruk P, Howland JG, Ahn S, Phillips AG. Neural circuits engaged in ventral hippocampal modulation of dopamine function in medial prefrontal cortex and ventral striatum. Brain Struct Funct. 2008;213:183–195. doi: 10.1007/s00429-008-0177-1. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB, Huang ZJ. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todtenkopf MS, Benes FM. Distribution of glutamate decarboxylase65 immunoreactive puncta on pyramidal and nonpyramidal neurons in hippocampus of schizophrenic brain. Synapse. 1998;29:323–332. doi: 10.1002/(SICI)1098-2396(199808)29:4<323::AID-SYN4>3.0.CO%3B2-7. [DOI] [PubMed] [Google Scholar]
- Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry. 2000;57:237–245. doi: 10.1001/archpsyc.57.3.237. [DOI] [PubMed] [Google Scholar]
- Wan FJ, Caine SB, Swerdlow NR. The ventral subiculum modulation of prepulse inhibition is not mediated via dopamine D2 or nucleus accumbens non-NMDA glutamate receptor activity. Eur J Pharmacol. 1996;314:9–18. doi: 10.1016/S0014-2999(96)00535-3. [DOI] [PubMed] [Google Scholar]
- West AR, Grace AA. Striatal nitric oxide signaling regulates the neuronal activity of midbrain dopamine neurons in vivo. J Neurophysiol. 2000;83:1796–1808. doi: 10.1152/jn.2000.83.4.1796. [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Müllner FE, Rinke I, Keck T, Stein V, Bonhoeffer T. Molecular and electrophysiological characterization of GFP-expressing CA1 interneurons in GAD65-GFP mice. PLoS One. 2010;5:e15915. doi: 10.1371/journal.pone.0015915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CRC, Mogenson GJG. Hippocampal signal transmission to the pedunculopontine nucleus and its regulation by dopamine D2 receptors in the nucleus accumbens: an electrophysiological and behavioural study. Neuroscience. 1987;23:1041–1055. doi: 10.1016/0306-4522(87)90179-5. [DOI] [PubMed] [Google Scholar]
- Zhang WN, Bast T, Feldon J. Effects of hippocampal N-methyl-[D]-aspartate infusion on locomotor activity and prepulse inhibition: Differences between the dorsal and ventral hippocampus. Behav Neurosci. 2002;116:72–84. doi: 10.1037/0735-7044.116.1.72. [DOI] [PubMed] [Google Scholar]
- Zhang ZJ, Reynolds GP. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res. 2002;55:1–10. doi: 10.1016/S0920-9964(01)00188-8. [DOI] [PubMed] [Google Scholar]