

Abstract
Lung cancer is one of the most deadly human cancers and continues to be a major unsolved health problem worldwide. Here, we evaluate the function of Pbx1 in the proliferation of non-small-cell lung cancer (NSCLC). In contrast with its known proliferative function, we found that Pbx1 inhibits the proliferation of lung cancer cells. In particular, Pbx1-specific RNA interference resulted in increased proliferation in lung cancer cells. In addition, histone H3 phosphorylation was also increased following inhibition of Pbx1 expression. In contrast, Pbx1 overexpression repressed the proliferation of lung cancer cells and inhibited DNA synthesis. Collectively, our data indicate that Pbx1 inhibits proliferation in lung cancer cells, suggesting a complex role for Pbx1 in modulating the proliferation of cancer cells and making this protein a potential new target for lung cancer therapy.
Keywords: Pbx1, non-small-cell lung cancer (NSCLC), proliferation
Introduction
Lung cancer remains one of the most deadly cancers worldwide (1). Compared with small cell lung cancer, non-small-cell lung cancer (NSCLC) exhibits higher resistance to traditional chemotherapy, radiotherapy and surgical resection (2-4). Recently, molecular biological approaches have been used to develop novel therapeutic strategies, although a more detailed understanding of the mechanisms of NSCLC proliferation is required to identify new molecular targets.
As a member of the TALE class homeodomain transcription factor family, Pbx1 forms hetero-oligomeric protein complexes to regulate gene expression (5-8), and it plays a role in morphologic patterning, organogenesis, and hematopoiesis. It has been shown that Pbx1 promotes the proliferation of specific progenitor cells (9). Moreover, Pbx1 potentiates the transcriptional activity of HOX proteins to promote the development of specific cancers, such as leukemia and ovarian cancer (10,11). Therefore, Pbx1 is considered to be a proto-oncogene.
In the present study, we examined the role of Pbx1 in NSCLC cells. Using RNA interference to target Pbx1, we found that NSCLC cell proliferation was enhanced when Pbx1 expression was inhibited. In particular, we show that DNA synthesis was increased and that more cells showed phosphorylation at serine 10 of Histone H3 following Pbx1 downregulation. To confirm an inhibitory role for Pbx1, we examined cell proliferation in Pbx1-overexpressing cells using plasmid transfection. As expected, cell proliferation was indeed impaired in Pbx1-overexpressed cells. These data suggest an inhibitory role for Pbx1 in NSCLC cell proliferation.
Materials and methods
Plasmids, cell culture and the construction of stable cell lines
Cells were purchased from ATCC and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone, UT, USA) containing 10% fetal bovine serum (FBS) (Hyclone, UT, USA) with 50 U/mL penicillin/streptomycin. The full length Pbx1 cDNA was amplified from 293T cells using polymerase chain reaction (PCR) with epitope-tagged primers. The corresponding amplification product was cloned into pLNCX, which was used to transfect confluent 293 cells. The supernatant from these cells was then used to infect A549 cells, which were selected with G418 for 1 month.
RNA interference
The following Pbx1 sequences were used for RNAi: GCAAGCGACAGAAATCCTGAA and TTCAGGATTTCTGTCGCTTCG. RNAi constructs were synthesized by GenePharma (Shanghai, China). A scrambled RNAi was used as a negative control. Cells (5×105) were incubated in 35-mm plates with mixture of RNAi (50 nM) and lipofectamine RNAiMAX reagent (Invitrogen, NY, USA) in serum-free medium for 6 h. Cells were then grown in media with 10% FBS.
Colony formation and MTT assays for the determination of cell proliferation
To perform the colony formation assay, 2,500 cells were plated onto 60-mm dishes containing 2× RPMI 1640 mixed with an equal volume of 1% agar and then incubated at 37 °C. After 8 days, colonies containing greater than 50 cells were counted. Each treatment was carried out in triplicate. For the MTT assay, 8,000 cells were treated as described and then plated into 96-well culture plates. Cells were then incubated with 20 µL MTT (5 mg/mL) for 2 h at 37 °C. The resulting crystals were recovered and dissolved in DMSO, and the optical density (OD) of these solutions was determined by spectrophotometry.
Real-time RT-qPCR
RNA was isolated using TRIzol (Invitrogen), according to the manufacturer’s protocol. First-strand cDNAs were generated using SuperScript III Reverse Transcriptase (Invitrogen). Real-time RT-qPCR was performed using a StepOne/StepOnePlus™ Real-time PCR System (Applied Biosystems) with SYBR Green PCR Master mix, according to the manufacturer’s instructions.
Western blotting analysis
Cells were lysed in lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.25% Triton-X 100, phosphatase inhibitors cocktail and protein inhibitor cocktail] for 30 min and then centrifuged (13,000 g, 10 min, 4 °C). Anti-Pbx1, anti-p21 and anti-p27 antibodies were purchased from Santa Cruz. Anti-GAPDH was purchased from Vazyme Biotech (Nanjing, China).
Immunofluorescence analysis
Cells were fixed with 4% paraformaldehyde for 1 h and then permeabilized with 0.2% Triton X-100 for 1 h. Blocking buffer (3% BSA) was then added into cells for 1 h at room temperature. Primary antibodies were used as indicated. Species-specific fluorescent secondary IgG (Invitrogen) antibodies were diluted in blocking solution and then incubated with the samples. The slides were washed and counterstained with DAPI. All of the images were captured using florescence microscopy (Leica).
EdU
Cells were plated in 6-well plates and treated as described. EdU was added to the cells (10 µM final concentration) 8 h prior to collection. Cells were then fixed. EdU detection was performed using the Click-iT EdU cell proliferation kit (Invitrogen), according to the manufacturer’s instructions. Nuclei were visualized with DAPI.
Statistical analysis
All experiments were performed with 3 to 5 replicates. The data are presented as mean and standard error of the mean (SEM) values. Comparisons within groups were performed with t-tests; the P values indicated in the figures are <0.05 (*), <0.01 (**) and <0.001 (***).
Results
Knockdown of Pbx1 in NSCLC cells
To explore the role of Pbx1 in lung cancer cells, we first identified a sequence for RNA interference that specifically targets human Pbx1. Pbx1-specific RNAi treatment resulted in greatly decreased mRNA levels in A549 cells (Figure 1A). Protein levels were also strongly downregulated (>70%) (Figure 1B).
Figure 1.
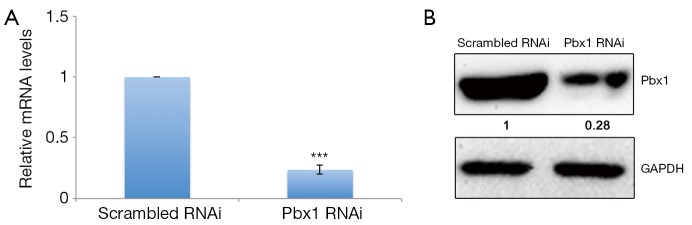
Efficiency of Pbx1 RNAi. (A) A549 cells were transfected with Pbx1 or scrambled RNAi for 72 h. mRNA levels were then analyzed (mean SD, n=3, ***P<0.001); (B) cells were treated as in (A) and subjected to western blotting analysis. Experiments were performed in triplicate and representative images are shown.
Pbx1 regulates the proliferation of NSCLC cells
Pbx1 has been classified as a proto-oncogene, as it promotes the proliferation of a variety of cancer cells. However, its role in NSCLC cells was unknown. Therefore, we examined its role in A549 cell proliferation. Interestingly, colony formation was induced (Figure 2A,B) and cell proliferation was increased (Figure 2C, left panel) when Pbx1 was inhibited in A549 cells. Similarly, knockdown of Pbx1 promoted SPC-A1 proliferation (Figure 2C, right panel). These results indicate an inhibitory role for Pbx1 in NSCLC cell proliferation.
Figure 2.

Pbx RNAi inhibits NSCLC cell proliferation. (A) A549 cells were transfected for 24 h and then colony formation assays were performed. Representative images are shown; (B) quantification of clones in (A) (mean SD, n=3, *P<0.05); (C) scrambled or Pbx1 RNAi constructs were transfected into A549 (left panel) and SPC-A1 (right panel) cells for 48 h. Cells were subjected to MTT analysis (mean SD, n=3, **P<0.01).
Pbx1 regulates cell cycle progression in NSCLC cells
To further characterize the effects of Pbx1 on NSCLC cell proliferation, we next examined cell cycle progression. Cell cycle distribution analysis revealed that Pbx1 knockdown resulted in more cells distributed throughout the cell cycle, indicating that Pbx1 represses cell cycle entry in A549 cells (Figure 3A). Phosphorylation of Histone H3 at serine10 is an established and consistent marker of mitosis. As shown in Figure 3B,C, the number of cells with phosphorylated histone H3 was increased following Pbx1 RNAi. Collectively, these data indicate that Pbx1 inhibits cell cycle progression in A549 cells.
Figure 3.

Pbx1 regulates cell cycle progression in NSCLC cells. (A) Cell cycle analysis was performed after RNAi transfection in A549 cells for 72 h. M1 indicates G0/G1 phase, M2 indicates S phase and M3 indicates M phase; (B) cells were transfected as in (A) and then fixed for immunofluorescence to detect histone H3 phosphorylation. Representative images from three parallel experiments are shown; (C) quantification of clones in (B) (mean SD, n=3, ***P<0.001).
Pbx1 regulates CDK inhibitors
It has been shown that Pbx1 modulates CDK inhibitors, such as p21 and p27. Considering that Pbx1 represses cell cycle entry in A549 cells, we hypothesized that Pbx1 might regulate p21 and p27 expression. To examine this, we measured p21 and p27 transcript levels, and as expected, both p21 and p27 were downregulated when Pbx1 expression was compromised (Figure 4A). The protein levels of p21 and p27 were also consistently reduced (Figure 4B) in these cells.
Figure 4.
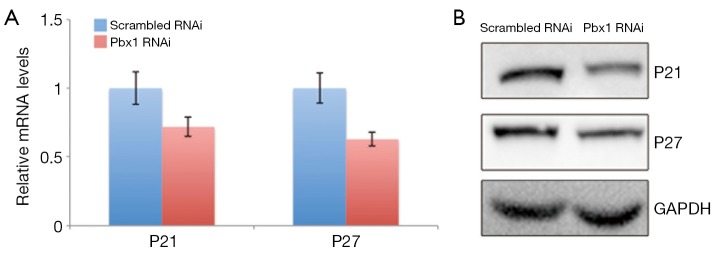
(A) A549 cells were transfected for 72 h and then subjected to RT-PCR analysis; (B) A549 cells were transfected with RNAi for 72 h and then lysed for western blotting analysis. Specific antibodies were used as indicated. Experiments were performed in triplicate, and representative images are shown.
Pbx1 overexpression inhibits NSCLC cell proliferation
Next, we were interested in whether Pbx1 overexpression could inhibit cell proliferation. To test this, we established an A549 cell line that stably expressed Pbx1 to eliminate artifacts due to transient transfection. Pbx1 expression was increased ~2-fold compared with a control cell line transfected with an empty vector (Figure 5A). We then measured proliferation in this cell line. Consistent with the RNAi experiments, cell proliferation was impaired when Pbx1 was overexpressed (Figure 5B). Furthermore, EdU incorporation was also compromised in Pbx1-overexpressing A549 cells, suggesting that DNA synthesis was repressed (Figure 5C), and the number of cells with histone H3 phosphorylation was also decreased. These data confirm that Pbx1 inhibits proliferation in NSCLC cells.
Figure 5.
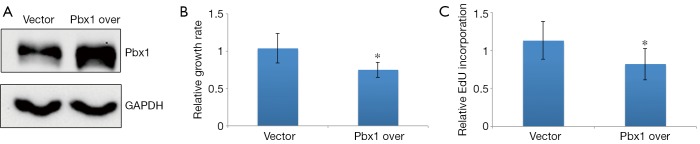
Pbx1 overexpression represses NSCLC cell proliferation. (A) Determination of Pbx1 protein levels by western blotting analysis. GAPDH was used as a loading control. Representative images from three independent experiments are shown; (B) MTT analysis of Pbx1-overexpressing A549 cells (mean SD, n=5, *P<0.05); (C) EdU was added to control or Pbx1-overexpressing A549 cells; cells were cultured for 8 h and then collected for EdU analysis (mean SD, n=5, *P<0.05).
Discussion
A positive role for Pbx1 in the proliferation of certain tumors has been well established. Previous studies have indicated that Pbx1 promotes leukemogenesis (12,13), and high expression levels of Pbx1 have been observed in melanoma cells (14). Furthermore, Pbx1 controls the progression of ovarian tumors (11). In the current study, we report the interesting finding that Pbx1 inhibits NSCLC cell proliferation, suggesting a noncanonical function for Pbx1 in the regulation of cell proliferation. Recently, it was shown that Pbx1-deficient long-term hematopoietic stem cells exhibited increased cycling and reduced quiescence, indicating that Pbx1 represses cell cycle entry in long-term hematopoietic stem cells, in turn promoting the self-renewal of hematopoietic stem cells (15). Furthermore, Pbx1 also limits the competitive ability of HOXB4-transduced hematopoietic stem cells by repressing proliferation (16). Although hematopoietic stem cells possess distinct cellular contexts from NSCLC cells, these studies do indicate a complex regulatory role for Pbx1 in cell proliferation.
As a transcription factor, Pbx1 cooperates with other transcription factors to modulate gene expression. A previous study showed that TGF-β signaling was impaired in Pbx1-deficient hematopoietic stem cells (15). TGF-β signaling has been shown to inhibit proliferation in a variety of normal and cancer cells (17), including NSCLC cells (18). These findings suggest that Pbx1 limits proliferation by modulating TGF-β signaling in NSCLC cells, a hypothesis that requires further study.
Here, we also found that Pbx1 can modulate cell cycle progression in NSCLC cells. In particular, an increase in the number of cycling cells was observed when Pbx1 was repressed. These observations indicate that Pbx1 can regulate cell cycle entry in NSCLC cells, suggesting a novel therapeutic role for Pbx1 in treating NSCLC. Therefore, specific cell cycle inhibitors that modulate the G1-S transition, such as p21 or p27, may be targets of Pbx1-mediated transcription. Consistent with this hypothesis, we showed that the expression of p21 and p27 was downregulated when the Pbx1 was repressed by RNAi in NSCLC cells. Taken together, our results suggest that Pbx1 could regulate the cell cycle machinery by modulating a downstream cell cycle-associated signaling pathway in NSCLC cells. It has been also shown that TGF-β signaling promotes cell quiescence by regulating the expression of cell cycle inhibitors (18). Therefore, it is interesting to hypothesize that Pbx1 might be a downstream effector of TGF-β signaling, suggesting a close relationship between Pbx1 and TGF-β signaling in the regulation of cell proliferation. To test this, further experiments will be required to examine the interplay between Pbx1 and TGF-β signaling in NSCLC cells.
Conclusions
In conclusion, our results reveal that Pbx1 inhibits cell proliferation in NSCLC, suggesting a complex role for Pbx1 in modulating the proliferation of cancer cells. Therefore, Pbx1 is also a potential new target for lung cancer therapy.
Acknowledgements
Author contributions: Weihao Li conceived and designed the project, performed the experiments, collected and analyzed the data and co-wrote the manuscript. Kai Huang performed the experiments, and collected and analyzed the data. Haizhou Guo and Guanghui Cui performed the experiments and analyzed the data. Song Zhao supervised the project and wrote the manuscript.
Disclosure: The authors declare no conflict of interest.
References
- 1.Landis SH, Murray T, Bolden S, et al. Cancer statistics, 1999. CA Cancer J Clin 1999;49:8-31,1. [DOI] [PubMed]
- 2.Mountain CF. Prognostic implications of the International Staging System for Lung Cancer. Semin Oncol 1988;15:236-45. [PubMed] [Google Scholar]
- 3.Perez CA, Pajak TF, Rubin P, et al. Long-term observations of the patterns of failure in patients with unresectable non-oat cell carcinoma of the lung treated with definitive radiotherapy. Report by the Radiation Therapy Oncology Group. Cancer 1987;59:1874-81. [DOI] [PubMed] [Google Scholar]
- 4.Johnson DH, Einhorn LH, Bartolucci A, et al. Thoracic radiotherapy does not prolong survival in patients with locally advanced, unresectable non-small cell lung cancer. Ann Intern Med 1990;113:33-8. [DOI] [PubMed] [Google Scholar]
- 5.DiMartino JF, Selleri L, Traver D, et al. The Hox cofactor and proto-oncogene Pbx1 is required for maintenance of definitive hematopoiesis in the fetal liver. Blood 2001;98:618-26. [DOI] [PubMed] [Google Scholar]
- 6.Kim SK, Selleri L, Lee JS, et al. Pbx1 inactivation disrupts pancreas development and in Ipf1-deficient mice promotes diabetes mellitus. Nat Genet 2002;30:430-5. [DOI] [PubMed] [Google Scholar]
- 7.Manley NR, Selleri L, Brendolan A, et al. Abnormalities of caudal pharyngeal pouch development in Pbx1 knockout mice mimic loss of Hox3 paralogs. Dev Biol 2004;276:301-12. [DOI] [PubMed] [Google Scholar]
- 8.Selleri L, Depew MJ, Jacobs Y, et al. Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development 2001;128:3543-57. [DOI] [PubMed] [Google Scholar]
- 9.Moens CB, Selleri L. Hox cofactors in vertebrate development. Dev Biol 2006;291:193-206. [DOI] [PubMed] [Google Scholar]
- 10.Schnabel CA, Jacobs Y, Cleary ML. HoxA9-mediated immortalization of myeloid progenitors requires functional interactions with TALE cofactors Pbx and Meis. Oncogene 2000;19:608-16. [DOI] [PubMed] [Google Scholar]
- 11.Naora H, Montz FJ, Chai CY, et al. Aberrant expression of homeobox gene HOXA7 is associated with müllerian-like differentiation of epithelial ovarian tumors and the generation of a specific autologous antibody response. Proc Natl Acad Sci U S A 2001;98:15209-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nourse J, Mellentin JD, Galili N, et al. Chromosomal translocation t(1;19) results in synthesis of a homeobox fusion mRNA that codes for a potential chimeric transcription factor. Cell 1990;60:535-45. [DOI] [PubMed] [Google Scholar]
- 13.Kamps MP, Look AT, Baltimore D. The human t(1;19) translocation in pre-B ALL produces multiple nuclear E2A-Pbx1 fusion proteins with differing transforming potentials. Genes Dev 1991;5:358-68. [DOI] [PubMed] [Google Scholar]
- 14.Matsuzaki H, Dong S, Loi H, et al. Genotyping over 100,000 SNPs on a pair of oligonucleotide arrays. Nat Methods 2004;1:109-11. [DOI] [PubMed] [Google Scholar]
- 15.Ficara F, Murphy MJ, Lin M, et al. Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell 2008;2:484-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krosl J, Beslu N, Mayotte N, et al. The competitive nature of HOXB4-transduced HSC is limited by PBX1: the generation of ultra-competitive stem cells retaining full differentiation potential. Immunity 2003;18:561-71. [DOI] [PubMed] [Google Scholar]
- 17.Gold LI. The role for transforming growth factor-beta (TGF-beta) in human cancer. Crit Rev Oncog 1999;10:303-60. [PubMed] [Google Scholar]
- 18.Liu ZY, Zhang GL, Wang MM, et al. MicroRNA-663 targets TGFB1 and regulates lung cancer proliferation. Asian Pac J Cancer Prev 2011;12:2819-23. [PubMed] [Google Scholar]