

Abstract
Cardiovascular disease poses the greatest risk of premature death seen among patients with chronic kidney disease (CKD). Up to 50% of mortality risk in the dialysis population is attributable to cardiovascular disease and the largest relative excess mortality is observed in younger patients. In early CKD, occlusive thrombotic coronary disease is common, but those who survive to reach end-stage renal failure requiring dialysis are more prone to sudden death attributable mostly to sudden arrhythmic events and heart failure related to left ventricular hypertrophy, coronary vascular calcification and electrolyte disturbances. In this review, we discuss the basis of the interaction of traditional risk factors for cardiovascular disease with various pathological processes such as endothelial dysfunction, oxidative stress, low grade chronic inflammation, neurohormonal changes and vascular calcification and stiffness which account for the structural and functional cardiac changes that predispose to excess morbidity and mortality in young people with CKD.
Keywords: Chronic kidney disease, Cardiovascular mortality, Cardiorenal syndrome, Endothelial dysfunction, Vascular calcification and stiffness
Core tip: In this review, we set out to summarise current opinion based on extensive scientific research that might explain the reasons for the disproportionately high death rate in chronic kidney disease and dialysis patients. The cardiovascular “phenotype” that poses increased risk to patients with chronic kidney disease (CKD) changes with progression of kidney dysfunction. Macrovascular disease is more important in early CKD whereas microvascular processes play an increasing role with worsening kidney disease.
EPIDEMIOLOGY
People with chronic kidney disease (CKD) are at higher mortality risk compared with the general population[1,2]. End-stage renal disease is associated with highest mortality despite modern renal replacement therapy and pharmacological interventions. Mortality risk among individuals starting haemodialysis is greatest in the first 120 d, accounting for 27.5 deaths per 100 person-years and, thereafter, the annual mortality rate is around 20%[3,4]. Furthermore, around 9% of deaths in the first 3 years after commencing dialysis are in the 20-54 years age group[5]. Findings from the United Kingdom Renal Registry indicate that a person commencing dialysis aged 25-29 years has a median life expectancy of only 18.5 years, 33 years less than someone in the same age group in the general population without CKD[6]. Similarly, patients aged 65-74 years, which is the commonest age group starting dialysis reported in the European Renal registry, can expect to live 5 years, about 50% less than those in the same age group in the general population[5].
The leading cause for this observed excess morbidity and mortality in end-stage renal disease is cardiovascular disease which accounts for 40%-50% of deaths[5,7-9]. In absolute terms, patients receiving dialysis have a 10-20 fold increased risk of cardiovascular death than do age and sex-matched controls in the general population. For younger dialysis patients, below 45 years of age, cardiac mortality is even higher, exceeding 100 times. After kidney transplantation, the risk is reduced but remains at 3-5 times that in the general population[7,10].
Of the non-cardiovascular causes of death in advanced renal failure, infection and malignancy are most important, accounting for approximately 15% and 8%, respectively, in the first 3 years of dialysis treatment[5,11]. Over this period of time, 10% of mortality due to infection was recorded in younger adults (20-54 years age group) and a similar percentage was attributed to malignancy. Vascular catheter-associated blood-borne infection often due to recurrent Staphylococcus aureus[12-15] and pneumonia[16] are the main causes. The high incidence of infections and inflammation in dialysis patients is related to disturbances in innate and adaptive immune mechanisms[17]. In particular, renal insufficiency is associated with down-regulation of toll-like receptors leading to sub-optimal stimulation of the innate immune system.
Non-adherence to haemodialysis and dietary restriction especially of high potassium-containing food, and excessive interdialytic fluid weight gain leading to congestive cardiomyopathy, are all contributory risk factors of premature death, especially in young dialysis patients[18]. The number refusing treatment or committing suicide is also not insignificant[5]. Treatment by peritoneal dialysis confers survival advantage over haemodialysis, at least in the first few years, before risk equalises[19,20]. Nevertheless, cardiovascular disease still poses the greatest risk in peritoneal dialysis (PD) patients[21,22]. This patient group shares similar cardiovascular risk factors to haemodialysis patients but they typically gain more weight (due to the high glucose load and insulin resistance) and demonstrate higher levels of chronic inflammation in response to exposure to non-physiological peritoneal dialysis fluid and episodes of peritonitis[21]. However, relatively little randomised trial data is available pertaining to cardiovascular risk factors/outcome in peritoneal dialysis patients and so much of the discussion below relates to haemodialysis patients.
CARDIORENAL SYNDROME
Large population studies have indicated that all stages of CKD predispose to premature death from cardiovascular and other causes, and is not restricted to those on dialysis[1]. Go and colleagues’ landmark study demonstrated that the age standardised mortality rate from any cause increased in a step-wise manner, independent of known risk factors (Figure 1). Even in cases of moderate renal insufficiency (eGFR 45-59 mL/min), the risk of death was 8% higher per 100 person years than in the general population without kidney impairment. Thereafter, this study indicated that the risk increased exponentially with declining eGFR, exceeding 11 times risk in CKD stage 4. Similarly, when considering age-standardised rate of cardiovascular events, there was a significant increase in events with progressive renal insufficiency (Figure 2). With mild degrees of renal impairment (eGFR > 60 mL/min), the rate of coronary events was 2.11 per 100 person years greater than the general population with no kidney impairment, rising to 21.8 times the risk with CKD stage 4.
Figure 1.
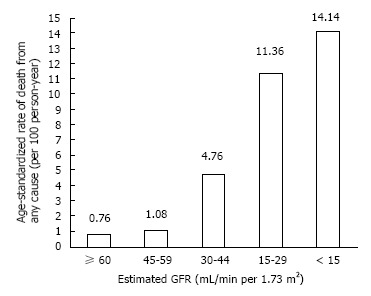
All-cause mortality and its relationship to worsening chronic kidney disease. After Go AS 2004[1].
Figure 2.
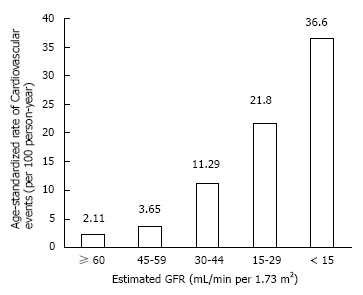
Cardiovascular event rates according to chronic kidney disease stage. After Go AS 2004[1].
Most striking was the adjusted hazard ratio for death from any cause found to be 20% greater in CKD stage 3a and a 40% increase of having a coronary event, rising to 590% and 340% at CKD stage 5, respectively (Table 1). This study confirmed the findings of many longitudinal studies since the 1970s that had shown that patients with advanced CKD died from cardiovascular causes[23,24], and has been substantiated by a recent study[20]. Recognition that cardiovascular disease is a major threat to patients with CKD and, conversely, renal dysfunction is prevalent in patients with cardiac disease, indicating a poorer prognosis, led to adoption of the term, cardio-renal syndrome. Ronco et al[25] published their widely adopted classification to highlight this crucial interaction.
Table 1.
Adjusted hazard ratio for death from any cause and cardiovascular events in 1120295 adults according to estimated glomerular filtration rate
| Estimated GFR (mL/min per 1.73 m2) | Death from any cause | Any Cardiovascular event |
| ≥ 60 | 1 | 1 |
| 45-59 | 1.2 (1.1-1.2) | 1.4 (1.4-1.5) |
| 30-44 | 1.8 (1.7-1.9) | 2.0 (1.9-2.1) |
| 15-29 | 3.2 (3.1-3.4) | 2.8 (2.6-2.9) |
| < 15 | 5.9 (5.4-6.5) | 3.4 (3.1-3.8) |
Adjusted hazard ratio with 95%confidence intervals given in parentheses. Data adjusted for age, gender, presence or absence of prior coronary heart disease, prior chronic heart failure, prior ischaemic stroke or ischaemic attack, prior peripheral arterial disease, diabetes mellitus, hypertension, dyslipidaemia, cancer, liver disease, chronic lung disease. After Go et al[1], 2004.
The bidirectional effect of heart failure and kidney failure is a key concept in the cardiorenal syndrome[26]. In the context of a failing heart due to pump failure, pressor systems (the sympathetic nervous system and renin-angiotensin-aldosterone axis) are activated to maintain the haemodynamic status quo[26,27]. Increased glomerular filtration pressure, achieved by efferent vasoconstriction helps to maintain glomerular filtration rate in low-output states, but the increased vascular resistance decreases kidney perfusion. Over time, this causes tubular hypoxic damage, renal cell apoptosis and replacement fibrosis, which leads to loss of nephron mass and to progressive renal dysfunction. Conversely, as chronic kidney disease progresses, the sympathetic nervous system is overactivated as a result of renal ischaemia, raised angiotensin II levels and suppression of nitric oxide, causing hypertension, left ventricular hypertrophy and progressive left ventricular dilatation. Cardiac myocyte dysfunction and fibrosis, so-called “CKD cardiomyopathy”, is believed to be the predominant pathophysiological mechanism. This may be compounded by salt and water overload caused by raised angiotensin II levels leading to elevated central venous pressure and organ congestion.
TRADITIONAL AND NON-TRADITIONAL RISK FACTORS FOR CARDIOVASCULAR DISEASE
Patients with cardiovascular disease and those with CKD share many ‘traditional’ risk factors for atheromatous plaque formation, such as hypertension, dyslipidaemia, diabetes mellitus, obesity and smoking, but it is increasingly recognised that these factors fail to account fully for the disproportionate increase in cardiovascular mortality risk in CKD compared to the general population[8,23,24,28]. In addition, evidence based treatment strategies such as the use of statins for treating hyperlipidaemia in type 2 diabetic patients which have reduced morbidity and mortality significantly[29], appear less effective in diabetic patients requiring dialysis[30]. Changes unique to CKD are the progressive accumulation of uraemic toxins, electrolyte abnormalities, metabolic acidosis, sympathetic nervous system and renin angiotensin aldosterone system activation, and volume overload that result in structural and functional abnormalities of the heart, termed uraemic cardiomyopathy[31].
Non-traditional risk factors include endothelial dysfunction, elevated plasma homocysteine, increased levels of inflammatory factors and oxidative stress, abnormal apolipoprotein levels, enhanced coagulability, albuminuria, increased arterial calcification and arterial stiffness, anaemia, and left ventricular hypertrophy[8,23,24,32]. Whether and how these and other, as yet unidentified, factors contribute to morbidity and mortality is not entirely clear, but is the subject of on-going research. In this review, we discuss current knowledge about emerging non-traditional risk factors and their complex interaction.
ENDOTHELIAL DYSFUNCTION
Endothelial dysfunction is considered to be one of the first mechanisms involved in the development of atherosclerosis[33]. In CKD, endothelial dysfunction has been defined by increased levels of von Willebrand factor, reduced nitric oxide levels (NO) and over expression of inflammatory cytokines and C-reactive protein (CRP)[34]. An excess of pro-inflammatory over anti-inflammatory cytokines has been reported in early atherosclerotic lesions as lipid particles accumulate[33]. Microalbuminuria, a marker of early kidney disease, in both diabetic and non-diabetic patients, correlates with altered endothelial function. NO levels decline with progressive kidney disease so that reduced vasodilatory activity contributes to the increased risk of developing hypertension[34]. Schiffrin and colleagues suggested that the combination of impaired endothelial function, low grade inflammation, and dyslipidaemia may interact to promote accelerated atherogenesis and progressive renal disease[32].
In health, endothelium-derived nitric oxide is an important anti-atherogenic molecule. Circulating NO levels fall as asymmetric dimethylarginine (ADMA) levels rise[34,35]. This competitive inhibitor of nitric oxide synthase (NOS) also blocks the entry of L-arginine into cells to reduce NO synthesis[36]. ADMA is excreted by the kidneys or metabolized by dimethylarginine dimethylaminohydrolases (DDAH) and this pathway is attractive as a potential modulator of the NOS system in dialysis patients. Plasma ADMA concentrations increase with deteriorating kidney function, and the highest levels are found in dialysis patients[34]. ADMA levels are raised in elderly hypertensive patients and are strongly predictive of acute coronary events and increased cardiovascular mortality risk in CKD[35,37-39].
The beneficial properties of NO are shown in Figure 3. Through NO inhibition, ADMA indirectly promotes vasoconstriction, raises blood pressure, impairs endothelium-dependent relaxation, and promotes platelet adhesion and aggregation and smooth muscle cell replication. It favours the release of superoxide radicals, formation of peroxynitrite and tyrosine nitration which cause vascular endothelial damage[40]. ADMA accumulation in CKD promotes atherogenesis and end-organ damage through sustained hypertension and other mechanisms[35]. In CKD, left ventricular hypertrophy, which is associated with premature death, may be a consequence of hypertension, partly triggered through prolonged inhibition of nitric oxide synthase. L-arginine supplementation should theoretically abrogate the effects of ADMA, but not all studies have found improvements in endothelial function[35]. One possible explanation is that chronically sustained high levels of ADMA induce irreversible vascular damage. There may be other mechanisms of action of ADMA other than NOS inhibition, yet to be identified, and not influenced by L-arginine supplementation. In an experimental model, DDAH activity of endothelial cells was decreased by almost half when incubated with oxidized LDL or TNF-α, and similar findings were observed in rabbits fed a high cholesterol diet[41]. These findings indicate that lipoproteins or cytokines may increase endothelial production of ADMA by reducing DDAH activity. This may be an important mechanism whereby local release and accumulation of intracellular ADMA inhibits NOS in hyperlipidaemia. The precise balance of pro and anti-inflammatory mediators present in the cellular milieu is likely to result in a variable overall effect which is directed towards or away from increased risk of atherosclerosis and atherothrombosis.
Figure 3.
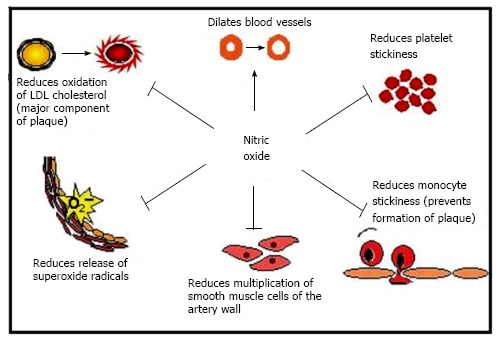
The physiological roles of nitric oxide on endothelial function.
HYPERHOMOCYSTEINAEMIA
Many studies have shown an association between elevated levels of homocysteine and increased risk of cardiovascular events, stroke and peripheral vascular disease in the general population[42]. High levels of homocysteine contribute to endothelial dysfunction, oxidative damage and thrombosis[42-45]. Patients with declining kidney function accumulate homocysteine due to decreased metabolism by the kidneys[43,46]. In one study, the finding of elevated homocysteine and fibrinogen levels in CKD was thought to account for 38% of the attributable mortality risk, whereas individuals with CKD and low homocysteine levels (< 10 μmol/L) had mortality rates similar to those with normal renal function[43]. Stuhlinger et al[45] have proposed that homocysteine exerts its atherogenic activity by indirectly suppressing NO production, through inhibition of dimethylarginine dimethylaminohydrolases (DDAH) that metabolise ADMA. Homocysteine is not obtained from the diet, but is synthesized from the amino acid, methionine. Homocysteine can be recycled back into methionine and deficiencies of the vitamins folic acid, pyridoxine (B6), or cyanocobalamin (B12) can lead to high homocysteine levels. Supplementation with these vitamins lowers homocysteine levels but has not been associated with a significant reduction in cardiovascular events[42]. However, fortification of grain products with folic acid in the United States since 1998 has seen a decrease in stroke incidence which, at least in part, is thought to be due to lower homocysteine levels in the population[44]. A recent meta-analysis failed to demonstrate a significant decrease in the risk for cardiovascular events, stroke and all-cause mortality among a CKD population[47]. Hyperhomocysteinaemia may be a marker rather than a direct cause of CVD[46].
EPIDEMIOLOGICAL LINKS BETWEEN ALBUMINURIA AND CARDIOVASCULAR EVENTS
Microalbuminuria refers to albumin excretion of between 30 and 300 mg in the urine over 24 h (20-200 μg/min) that derives from a glomerular or tubular abnormality in primary kidney disorders, or it may reflect a generalised increase in vascular permeability due to vascular endothelial dysfunction[8]. Microalbuminuria, most commonly seen in diabetes mellitus and hypertension, signifies risk of progressive renal impairment and predates any fall in eGFR[32]. In a prospective study over 7 years, the degree of microalbuminuria in diabetic patients was found to correlate strongly with cardiovascular events and mortality[48]. Microalbuminuria was found to be a stronger predictor of cardiovascular outcomes than smoking, hypertension and raised serum cholesterol, in men who had no pre-existing cardiovascular disease. This was one of many studies that confirmed the early findings of Schmitz et al[49] (Figure 4). Diabetic patients were divided into 3 groups, entering the study with microalbuminuria of less than 15 μg/min, 16-40 μg/min and 41-200 μg/min. At 10 years, the overall mortality was 58%, caused by CVD or stroke with a further 3% dying from end-stage renal failure. 10-year survival was 55% and 25% in the < 15 μg/min and 41-200 μg/min microalbuminuria patient groups.
Figure 4.
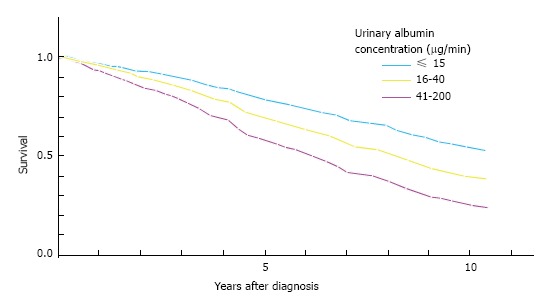
Microalbuminuria as a risk factor for death in type 2 diabetes. Reproduced from Schmitz et al[49], 1988.
In diabetic patients, the development of microalbuminuria indicates microvascular disease which is associated with extracellular matrix expansion in the glomeruli of the kidney[50]. The natural history results in progressive glomerulosclerosis, increasing proteinuria, and chronic renal failure. But how does microalbuminuria, a marker of diabetic nephropathy, or a slightly reduced eGFR predict cardiovascular events and mortality before there is any evidence of overt coronary artery disease? Endothelial cell dysfunction seems to be the common pathophysiological pathway linking renal disease and CVD. Interplay between low-grade inflammation, elevated ADMA, increased circulating pro-inflammatory cytokines, dyslipidaemia, oxidative stress, sympathetic system over-activity and inappropriate activation of the renin angiotensin aldosterone system, is implicated for generalised endothelial cell dysfunction[51].
Large epidemiological studies have demonstrated that microalbuminuria in non-diabetic individuals is also associated with coronary, peripheral and cerebral vascular events[52]. Here again, the premise is that microalbuminuria is a marker of generalised endothelial cell dysfunction. Microalbuminuria is associated with increased trans-capillary leakage of albumin and increased von Willebrand factor and other markers of endothelial dysfunction[50,51]. Inhibition of the renin-angiotensin-aldosterone system with either angiotensin converting enzyme inhibitors (ACE-I) or angiotensin receptor antagonists (AT1 receptor antagonists) can reduce albuminuria in both diabetic and non-diabetic individuals, to slow down the progression of renal disease and provide cardioprotection[53]. The underlying pathophysiology relates to the ability of ACE-I and AT1 receptor blockers to lower intra-glomerular capillary pressure by decreasing efferent arteriolar pressure and the reduction in transglomerular pressure decreases albuminuria. These drug classes have reduced progression of diabetic renal disease by reversing microalbuminuria to normoalbuminuria. Further long-term epidemiological studies are necessary to define its extent and enduring impact. The heart outcomes and prevention evaluation study (HOPE) is one of many studies showing that the ACE inhibitors reduce the progression of albuminuria and, at the same time, are effective in decreasing cardiovascular mortality in both diabetic and non-diabetic patients with normal blood pressure[54]. The Irbesartan Diabetic Nephropathy Trial demonstrated concomitant reductions in urinary protein excretion and cardiovascular endpoints in hypertensive type 2 diabetic participants[55].
Proteinuria is widely accepted as an independent risk factor for cardiovascular morbidity and mortality. Microalbuminuria progressing to overt proteinuria confers increasing cardiovascular mortality risk and concomitant CKD is associated with worsening cardiovascular and all-cause mortality, acting as risk multipliers across the CKD continuum[1,56,57]. Only a minority of the stage 3-5 CKD population progress to end-stage dialysis-requiring disease, most succumbing to premature cardiovascular death. Proteinuria surpasses blood pressure and cholesterol as a predictor of adverse clinical outcome[58]. Furthermore, the same group reported that the risk of acute myocardial infarction in subjects with CKD and proteinuria was similar to or exceeded the cardiovascular risk associated with diabetes mellitus[59]. The PREVEND study found a significant independent association between microalbuminuria and myocardial ischaemia found on ECG[60]. Microalbuminuria is accompanied by raised inflammatory markers such as CRP and a fall in adiponectin levels[61]. Endothelial dysfunction has been implicated in the aetiopathology linking proteinuria with ADMA[62]. Since the detection of subclinical atherosclerosis is difficult and approximately 50% of cardiac events arise from the rupture of a vulnerable plaque in non-occlusive coronary disease, it may be that microalbuminuria associated with endothelial dysfunction is also acting as a marker of subclinical atherosclerosis. In this respect, studies have demonstrated that microalbuminuria independently predicts an increased carotid artery intima-media thickness which is a marker of subclinical atherosclerosis[63].
INFLAMMATION, DYSLIPIDAEMIA, ADVANCED GLYCATION END-PRODUCTS AND OXIDATIVE STRESS
Systemic inflammation is fundamental to the development of atherosclerosis[33,64]. Inflammatory markers including CRP, fibrinogen, soluble adhesion molecules and pro-inflammatory cytokines correlate with the future development of CVD and risk of sudden death in the general population[65].
Progressive renal insufficiency is characterised by a chronic inflammatory state represented by higher concentrations of CRP, interleukin-6 (IL-6), tumour necrosis factor α (TNF-α) and fibrinogen, and with lower levels of albumin[66]. But disorders commonly associated with CKD such as diabetes and hypertension exhibit low grade inflammation long before there is evidence of renal damage, and in the absence of any discernable reduction in serum albumin concentration. Irrespective of the mechanisms that trigger inflammation, there is no doubt that chronic inflammation induces vascular injury, initiating accelerated atherosclerosis[64], which is more pronounced in the younger patient with CKD. The causes of inflammation in renal disease can be partly attributed to reduced clearance of pro-inflammatory cytokines such as IL-6 and TNF-α, to the production of reactive oxygen species and the contribution of co-morbid conditions such as diabetes mellitus and heart failure[23,32,67]. Accumulation of advanced glycation end-products (AGE), normally cleared by the kidneys, may be important in the generalised chronic inflammatory process[68,69]. It has been established that AGEs are naturally formed in all tissues as part of ageing but at an increased rate in certain pathological processes such as diabetes and CKD[69]. Studies have demonstrated that up to 50% of patients with CKD have raised serum levels of CRP, fibrinogen, IL-1, IL-6, TNF-α, D-dimer and the soluble adhesion molecules E-selectin, VCAM-1 and ICAM-1[32,33], and research is focussed on the mechanisms of interaction that promote oxidative stress and atherosclerosis[64].
Correlation between raised serum CRP levels and future cardiovascular events in the general population is strong[65], but the association is more robust in CKD, in which it also closely predicts progression of kidney disease[67,70]. In a prospective study involving over 1000 dialysis patients with a median follow up of 2.5 years, 22% of patients died due to sudden cardiac death[71]. The highest third of high sensitivity CRP and IL-6 levels had twice the risk of sudden cardiac death. Moreover, those with a reduced serum albumin level had a 35% increased risk of suffering sudden cardiac death. IL-6 acts as a major stimulus for release of CRP by the liver. Ramkumar et al[72] identified increased production of IL-6 by intra-abdominal adipocytes, which provides a possible link between obesity and increased inflammation in CKD. In haemodialysis patients, other factors stimulating inflammation include repeated exposure to bio-incompatible dialysis membranes, exposure to foreign materials such as dialysis catheters and infection[23,32]. Infection in this group of patients is often subclinical being related to the presence of polytetrafluoroethylene (PTFE) central venous haemodialysis and peritoneal dialysis catheters.
Hyperfibrinogenaemia is observed with declining renal function and raised levels of this acute phase protein parallel those of other inflammatory markers. Hyperfibrinogenaemia predisposes to coronary thrombosis[72,73]. However, there is some uncertainty in defining the extent to which this factor contributes to overall mortality because fibrinogen is closely linked with other factors, such as dyslipidaemia[8].
Dyslipidaemia in CKD is a major factor in the inflammatory response and to atherosclerosis[74]. In health, high-density lipoprotein (HDL) cholesterol acts as an anti-atherogenic molecule in a number of ways. It reverses cholesterol transport, and has anti-thrombotic, anti-inflammatory and anti-oxidant properties, reducing oxidised LDL cholesterol[32,74]. It also promotes endothelial repair by decreasing the expression of adhesion molecules by vascular endothelial cells induced by cytokines[74]. HDL levels fall progressively as renal function declines, and the HDL cholesterol that is produced is dysfunctional. Figure 5 depicts the maturation of HDL and the protective effect of the apolipoprotein-A (apoA-I). In renal failure, apoA-I, synthesized by the liver, decreases so HDL cholesterol levels fall[32,74]. ApoA-I acts via ABCG1 taking up cholesterol from macrophages. The enzyme, lecithin-cholesterol acyltransferase (LCAT) esterifies cholesterol in the maturation of HDL cholesterol. The resulting HDL3 isoform and, to a lesser extent HDL2, are rich in anti-oxidative enzymes including paraoxonase 1 (PON 1). With progressive renal dysfunction, there is decreased LCAT activity and consequently less HDL. ApoA-I that normally comprises half of the proteins in HDL is replaced by serum amyloid A. This form of HDL cholesterol has a reduced ability to counteract the effects of oxidised LDL[75] and decreased capacity to protect against cytokine action on vascular endothelium. Vaziri et al[76] showed that an apolipoprotein A-I mimetic peptide could reduce the effect of oxidised LDL on cultured aortic endothelial cells to produce the cytokine monocyte chemoattractant protein 1 (MCP-1), which may ultimately provide a therapeutic pathway to overcome dysfunctional HDL present in CKD (Figure 5).
Figure 5.
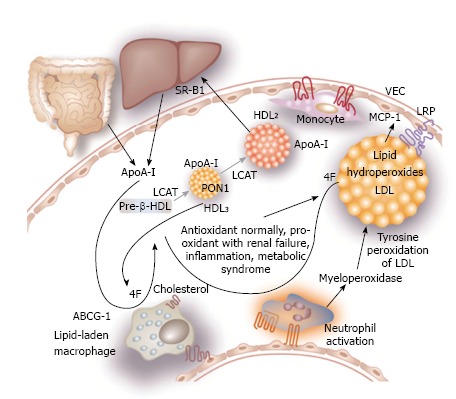
Maturation of high-density lipoprotein and the protective effect of apoA-I mimetic peptide 4F. (Apo)A-I: Apolipoprotein A-I; ABCG1: Adenosine triphosphate–binding cassette transporter G-1 protein; LCAT: Lecithin–cholesterol acyltransferase; SR-B1: Scavenger receptor B1; PON1: Paraoxonase-1; LRP: Lipoprotein-like receptor; VEC: Vascular endothelial cells; MCP-1: Monocyte chemoattractant protein-1; HDL: High-density lipoprotein. After Kaysen[74], 2009.
Higher levels of LDL cholesterol and triglycerides are found with declining kidney function[8]. The impact on coronary artery disease is significantly greater than in the general population without renal impairment[8,74]. Accumulation of small dense atherogenic LDL cholesterol activates the renin-angiotensin-aldosterone system and also up-regulates the angiotensin type 1 receptor (AT1). This increases the burden of oxidative stress and inflammation leading to endothelial dysfunction and atherosclerosis[32,33,75]. Not only does angiotensin II cause hypertension (directly linked to atherosclerosis) but also activates vascular NADPH oxidase which induces superoxide anion generation (O2-). The superoxide anion inactivates nitric oxide which causes increased smooth muscle hypertrophy and proliferation, leading to hypertension and atherosclerosis[75].
Myeloperoxidase (MPO) is an enzyme present in leucocytes, particularly neutrophils, monocytes and tissue macrophages, which may play an important role in vascular injury and atherosclerosis in patients with advanced kidney disease[32]. Figure 5 shows how MPO promotes tyrosine peroxidation of LDL cholesterol. Leucocytes, which are activated in acute and chronic inflammation, secrete MPO into the blood which binds to vascular endothelium where it interferes with nitric oxide[74].
Lipoprotein a [Lp(a)] is a potent risk factor for CVD and serum levels increase progressively with declining renal function[8]. Lp(a) concentrations are highest in proteinuric states and fall after kidney transplantation.
Inflammation is a key component in the malnutrition-inflammation-atherosclerosis syndrome, observed across the spectrum of CKD, especially in young adults and is associated with substantial mortality[77]. Systemic inflammation, low serum HDL cholesterol and activation of angiotensin II provide the rationale for use of anti-inflammatory agents such as aspirin, statins, angiotensin-converting enzyme inhibitors, angiotensin II receptor blocking agents and antioxidants in combating endothelial dysfunction and atheroma[33,67,75].
SYMPATHETIC NERVOUS SYSTEM OVERACTIVITY, ANAEMIA AND LEFT VENTRICULAR HYPERTROPHY
Sympathetic nervous system over-activity in CKD is deleterious and results from renal ischaemia, raised angiotensin II levels, and suppression of nitric oxide, causing hypertension, left ventricular hypertrophy and eventually left ventricular dilatation[24]. The prevalence of LVH was found to be 31% in those with GFR 25-49 mL/min and 45% in those with GFR < 25 mL/min[78]. There is evidence from epidemiological studies such as Framingham that LVH is independently associated with increased risk of fatal and non-fatal cardiovascular events[79]. ACE inhibitors and angiotensin II receptor antagonists have been widely used to overcome sympathetic overdrive and to inhibit the renin angiotensin aldosterone system. However, a recent meta-analysis has demonstrated that although pharmacological intervention reduces LV mass it has no significant impact on reducing the risk of fatal and non-fatal cardiovascular events[80].
In the general population, LV dilatation and heart failure are generally the end result of end-organ damage sustained as a consequence of chronic hypertension and coronary atheroma. Although there is a higher prevalence of these conditions in young adult patients with CKD, cardiac myocyte death is accelerated by increased oxidative stress and anaemia[7,67,81]. Cardiac failure leads to premature death (Figure 6)[81].
Figure 6.
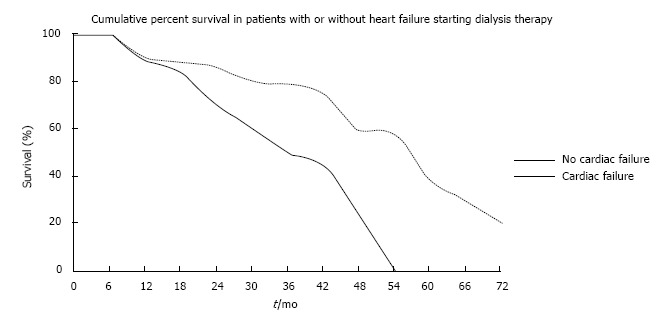
Premature death related to heart failure in haemodialysis. After Harnett et al[81], 1995.
Anaemia appears in CKD stage 3 or 4 when erythropoietin production by the kidney diminishes[23]. If left untreated, the pathophysiological response is increased cardiac output, cardiomegaly, left ventricular structural abnormalities and, ultimately, congestive heart failure[24]. Correction of anaemia in CKD with erythropoietin results in regression of LVH and improved survival[23]. Timely intervention with iron and erythropoietin supplementation is important.
VASCULAR CALCIFICATION AND ARTERIAL STIFFNESS
Declining kidney function is associated with alterations in calcium and phosphorus metabolism that result in mineral bone disease (renal osteodystrophy), characterised by soft tissue and vascular calcification[82,83]. Secondary hyperparathyroidism, the physiological response aimed at correcting hyperphosphatemia by promoting phosphaturia, and vitamin D deficiency are both associated inflammation, vascular risk and cardiovascular mortality[83]. The situation is more complex with the discovery of fibroblast growth factor 23 (FGF-23) which is secreted by osteocytes. It acts as a phosphaturic hormone, inhibits production and secretion of parathyroid hormone, and reduces 1,25 vitamin D synthesis by interfering with vitamin D metabolism (Figure 7)[82]. α-Klotho is a hormone synthesised by the kidneys which seems to be essential for normal physiological functioning of FGF-23[84]. It is phosphaturic and may also have antioxidant and vasoprotective activity. As kidney function deteriorates, FGF-23 concentrations rise and early evidence indicates a strong association with left ventricular hypertrophy, independent of α-Klotho[85,86].
Figure 7.
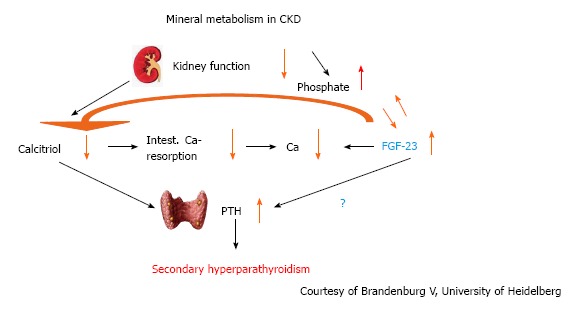
Interplay between calcium, phosphate, calcitriol and Fibroblast growth factor-23 in chronic kidney disease. FGF: Fibroblast growth factor; CKD: Chronic kidney disease.
Coronary artery calcification is known to be important in the formation of atherosclerotic plaque[87]. Vascular calcification can take two forms, involving the tunica intima and media[82]. While it is not clear whether they follow a common pathogenesis, both are stimulated by CKD. Intimal calcifications develop in more than 80% of atherosclerotic plaques that occlude the vessel lumen causing ischaemia and myocardial necrosis[88]. Calcification of the tunica media involves smooth muscle cells and the elastic lamina[82,88,89], and is prevalent in more than 40% of patients with CKD stage 3B, who have accelerated coronary artery calcification[89]. Diabetic patients are particularly prone to calcific atheroma[32,67,89]. Calcification of the tunica media increases vascular rigidity and decreases compliance. Systolic hypertension occurs contributing to LVH. Increasing vascular calcification and stiffness, particularly of the aorta, measured by pulse-wave velocity has been shown to predict cardiovascular mortality in CKD patients[90,91].
Current theory suggests that tunica media calcification is an inflammatory process transforming vascular smooth muscle cells into osteoblast-like cells that express phosphorus transporter Pit-1 which concentrates calcium and phosphorus in an extracellular matrix that ultimately mineralises[87,92]. Vascular calcification is a complex process dependent upon the balance between promoters and inhibitors of calcification summarized in Table 2[87]. For example, a high calcium-phosphate product, raised parathyroid hormone levels and bone morphogenetic protein 2 (BMP-2) are pro calcific, whereas inhibitory factors include Fetuin-A, BMP-7, osteopontin, pyrophosphate and osteoprotegerin (OPG)[87,92].
Table 2.
Promoters and inhibitors of vascular calcification
| Promoters of vascular calcification | Inhibitors of vascular calcification |
| Traditional factors | Circulating inhibitors |
| Older age, male gender, hypertension, diabetes, smoking, high LDL cholesterol, low HDL cholesterol, genetic predisposition | Fetuin-A, bone morphogenetic protein-7, parathyroid hormone-related peptide, HDL cholesterol, magnesium, |
| Uraemia-related factors | |
| Uraemia, hyperphosphatemia, | Locally acting inhibitors |
| increased Ca x P product, exogenous vitamin D therapy, elevated parathyroid hormone level, duration of dialysis, calcium load and hypercalcaemia, chronic inflammation, warfarin, elevated leptin levels | Matrix Gla protein, α-klotho, osteopontin, pyrophosphate, osteoprotegerin, genetic predisposition |
Adapted from Qunibi[87], 2005. HDL: High-density lipoprotein.
Fetuin-A, produced by the liver, is a negative acute phase protein, its levels falling with rising CRP. Conversely, as CRP levels fall, fetuin-A levels may rise. This provides a link between inflammation and vascular calcification[93]. Matrix-Gla protein inhibits calcification of vascular smooth muscle and a negative correlation has been found with coronary artery calcification[32]. Osteoprotegerin regulates osteoclast activity and its deficiency has been associated with vascular calcification[82,87]. Osteoprotegerin deficiency is associated with increased mortality in dialysis patients[94]. Leptin is normally associated with nutritional state and satiety, but in the context of renal failure, in which its levels are increased, it promotes calcification[87].
SUDDEN CARDIAC DEATH AND DIALYSIS
Dialysis patients have a 10-20 times increased risk of cardiovascular death than do age and sex-matched controls in the general population[5,7-9], with the largest relative excess mortality seen in younger patients on dialysis. Sudden, unexpected death accounts for 25% of mortality on haemodialysis[95,96]. In most cases, acute coronary occlusion (the end-product of “accelerated atherosclerosis”), which was previously implicated[7], is not the underlying pathology. Instead, sudden cardiac death is explained by the composite effects of left ventricular hypertrophy caused by long-standing hypertension, left ventricular dilatation attributed to volume overload, chronic inflammation, over activation of the renin angiotensin aldosterone pathway, and increased propensity to ventricular dysrhythmias caused by electrolyte abnormalities[95-97].
The structural and functional abnormalities that occur through the interaction of numerous factors presented in this review, gives rise to stiff and non-compliant vasculature which has to negotiate myocardial ischaemia imposed by repeated haemodynamic stresses of haemodialysis therapy. Echocardiographic evaluation and positron emission tomography have shown that repeated haemodialysis adversely affects cardiac perfusion caused by transient myocardial stunning, but over time this leads to fixed regional myocardial wall abnormalities and fibrosis, culminating in heart failure and death[98,99]. In a recent pilot study, our group have demonstrated a possible relationship between endothelial dysfunction and the development of LVH in non-dialysis CKD patients[100]. The CRASH-ILR study is currently underway which is prospectively using implantable loop recorders in patients on haemodialysis to ascertain the frequency of cardiac arrhythmia. In an interim analysis of 18 patients monitored for 124 to 512 h there were 3 significant cardiac events including 1 bradyarrhythmia requiring pacing, 1 sudden cardiac death due to ventricular fibrillation and one onset of atrial tachycardia requiring anti-arrhythmic drug therapy. The final results of this study are awaited[101].
CONCLUSION
Cardiovascular disease is the most important cause for the high morbidity and mortality seen in patients with chronic kidney disease, especially younger people. Traditional risk factors play a major role, but coronary heart disease is somewhat different in CKD patients with increased contribution made by oxidative stress, low grade chronic inflammation and vascular calcification. The interaction of many of these pathophysiological processes is, if not unique to, most marked in CKD. Haemodialysis imparts the greatest mortality risk because of the challenges imposed on the heart by the repeated haemodynamic stresses that accelerate development of heart failure and dilated cardiomyopathy. Limited benefit of standard pharmacological interventions in the dialysis population emphasises the different pathophysiology which has evolved. The complexities of the association between chronic kidney disease and cardiovascular events are summarised in Figure 8. A better appreciation of the interplay and relative contribution of the various mechanisms discussed, along with new pathogenetic mechanisms yet to be discovered over the next few years, will enable new therapies to be developed. This will, hopefully, curb the excessive morbidity and mortality which seems to be particularly pronounced in young dialysis patients. Data from implantable loop recorders may give further insights to the precise events which result in sudden cardiac death and allow development of strategies to predict risk and reduce events.
Figure 8.
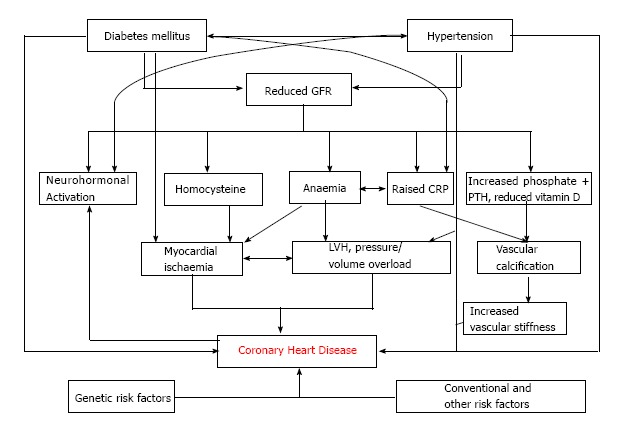
The association between chronic kidney disease and coronary heart disease. Adapted from Hage FG 2009[67].
Footnotes
P- Reviewer: Friedman EA, Hatamizadeh P, Lopez-Hernandez FJ, Olowu WA S- Editor: Ji FF L- Editor: A
E- Editor: Lu YJ
References
- 1.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 2.Ross L, Banerjee D. Cardiovascular complications of chronic kidney disease. Int J Clin Pract. 2013;67:4–5. doi: 10.1111/ijcp.12069. [DOI] [PubMed] [Google Scholar]
- 3.Soucie JM, McClellan WM. Early death in dialysis patients: risk factors and impact on incidence and mortality rates. J Am Soc Nephrol. 1996;7:2169–2175. doi: 10.1681/ASN.V7102169. [DOI] [PubMed] [Google Scholar]
- 4.Bradbury BD, Fissell RB, Albert JM, Anthony MS, Critchlow CW, Pisoni RL, Port FK, Gillespie BW. Predictors of early mortality among incident US hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study (DOPPS) Clin J Am Soc Nephrol. 2007;2:89–99. doi: 10.2215/CJN.01170905. [DOI] [PubMed] [Google Scholar]
- 5.de Jager DJ, Grootendorst DC, Jager KJ, van Dijk PC, Tomas LM, Ansell D, Collart F, Finne P, Heaf JG, De Meester J, et al. Cardiovascular and noncardiovascular mortality among patients starting dialysis. JAMA. 2009;302:1782–1789. doi: 10.1001/jama.2009.1488. [DOI] [PubMed] [Google Scholar]
- 6.Steenkamp R, Shaw C, Feest T. UK Renal Registry 15th annual report: Chapter 5 survival and causes of death of UK adult patients on renal replacement therapy in 2011: national and centre-specific analyses. Nephron Clin Pract. 2013;123 Suppl 1:93–123. doi: 10.1159/000353324. [DOI] [PubMed] [Google Scholar]
- 7.Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32:S112–S119. doi: 10.1053/ajkd.1998.v32.pm9820470. [DOI] [PubMed] [Google Scholar]
- 8.Baigent C, Burbury K, Wheeler D. Premature cardiovascular disease in chronic renal failure. Lancet. 2000;356:147–152. doi: 10.1016/S0140-6736(00)02456-9. [DOI] [PubMed] [Google Scholar]
- 9.Cheung AK, Sarnak MJ, Yan G, Berkoben M, Heyka R, Kaufman A, Lewis J, Rocco M, Toto R, Windus D, et al. Cardiac diseases in maintenance hemodialysis patients: results of the HEMO Study. Kidney Int. 2004;65:2380–2389. doi: 10.1111/j.1523-1755.2004.00657.x. [DOI] [PubMed] [Google Scholar]
- 10.Jardine AG, Gaston RS, Fellstrom BC, Holdaas H. Prevention of cardiovascular disease in adult recipients of kidney transplants. Lancet. 2011;378:1419–1427. doi: 10.1016/S0140-6736(11)61334-2. [DOI] [PubMed] [Google Scholar]
- 11.Foley RN, Collins AJ. The USRDS: what you need to know about what it can and can’t tell us about ESRD. Clin J Am Soc Nephrol. 2013;8:845–851. doi: 10.2215/CJN.06840712. [DOI] [PubMed] [Google Scholar]
- 12.Jaber BL. Bacterial infections in hemodialysis patients: pathogenesis and prevention. Kidney Int. 2005;67:2508–2519. doi: 10.1111/j.1523-1755.2005.00364.x. [DOI] [PubMed] [Google Scholar]
- 13.Liu JW, Su YK, Liu CF, Chen JB. Nosocomial blood-stream infection in patients with end-stage renal disease: excess length of hospital stay, extra cost and attributable mortality. J Hosp Infect. 2002;50:224–227. doi: 10.1053/jhin.2001.1162. [DOI] [PubMed] [Google Scholar]
- 14.Slinin Y, Foley RN, Collins AJ. Clinical epidemiology of pneumonia in hemodialysis patients: the USRDS waves 1, 3, and 4 study. Kidney Int. 2006;70:1135–1141. doi: 10.1038/sj.ki.5001714. [DOI] [PubMed] [Google Scholar]
- 15.Martín-Peña A, Luque Márquez R, Guerrero MJ, Espinosa N, Blanco Y, Ibeas J, Ríos-Villegas MJ, Cisneros JM. Tunneled hemodialysis catheter-related bloodstream infections: a prospective multicenter cohort study from Spain. J Vasc Access. 2012;13:239–245. doi: 10.5301/jva.5000034. [DOI] [PubMed] [Google Scholar]
- 16.Sarnak MJ, Jaber BL. Pulmonary infectious mortality among patients with end-stage renal disease. Chest. 2001;120:1883–1887. doi: 10.1378/chest.120.6.1883. [DOI] [PubMed] [Google Scholar]
- 17.Kato S, Chmielewski M, Honda H, Pecoits-Filho R, Matsuo S, Yuzawa Y, Tranaeus A, Stenvinkel P, Lindholm B. Aspects of immune dysfunction in end-stage renal disease. Clin J Am Soc Nephrol. 2008;3:1526–1533. doi: 10.2215/CJN.00950208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dantas LG, Cruz C, Rocha M, Moura JA, Paschoalin E, Paschoalin S, Marcilio de Souza C. Prevalence and predictors of nonadherence to hemodialysis. Nephron Clin Pract. 2013;124:67–71. doi: 10.1159/000355866. [DOI] [PubMed] [Google Scholar]
- 19.Heaf JG, Wehberg S. Relative survival of peritoneal dialysis and haemodialysis patients: effect of cohort and mode of dialysis initiation. PLoS One. 2014;9:e90119. doi: 10.1371/journal.pone.0090119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neovius M, Jacobson SH, Eriksson JK, Elinder CG, Hylander B. Mortality in chronic kidney disease and renal replacement therapy: a population-based cohort study. BMJ Open. 2014;4:e004251. doi: 10.1136/bmjopen-2013-004251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krediet RT, Balafa O. Cardiovascular risk in the peritoneal dialysis patient. Nat Rev Nephrol. 2010;6:451–460. doi: 10.1038/nrneph.2010.68. [DOI] [PubMed] [Google Scholar]
- 22.de Moraes TP, Figueiredo AE, Campos LG, Olandoski M, Barretti P, Pecoits-Filho R. Characterization of the brazpd ii cohort and description of trends in peritoneal dialysis outcome across time periods. Perit Dial Int. 2014 doi: 10.3747/pdi.2013.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fort J. Chronic renal failure: a cardiovascular risk factor. Kidney Int Suppl. 2005;(99):S25–S29. doi: 10.1111/j.1523-1755.2005.09906.x. [DOI] [PubMed] [Google Scholar]
- 24.Parfrey PS, Foley RN. The clinical epidemiology of cardiac disease in chronic renal failure. J Am Soc Nephrol. 1999;10:1606–1615. doi: 10.1681/ASN.V1071606. [DOI] [PubMed] [Google Scholar]
- 25.Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52:1527–1539. doi: 10.1016/j.jacc.2008.07.051. [DOI] [PubMed] [Google Scholar]
- 26.McCullough PA, Kellum JA, Haase M, Müller C, Damman K, Murray PT, Cruz D, House AA, Schmidt-Ott KM, Vescovo G, et al. Pathophysiology of the Cardiorenal Syndromes: Executive Summary from the Eleventh Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Blood Purif. 2014;37 Suppl 2:2–13. [Google Scholar]
- 27.Ljungman S, Laragh JH, Cody RJ. Role of the kidney in congestive heart failure. Relationship of cardiac index to kidney function. Drugs. 1990;39 Suppl 4:10–21; discussion 22-24. doi: 10.2165/00003495-199000394-00004. [DOI] [PubMed] [Google Scholar]
- 28.Ortiz A, Covic A, Fliser D, Fouque D, Goldsmith D, Kanbay M, Mallamaci F, Massy ZA, Rossignol P, Vanholder R, et al. Epidemiology, contributors to, and clinical trials of mortality risk in chronic kidney failure. Lancet. 2014;383:1831–1843. doi: 10.1016/S0140-6736(14)60384-6. [DOI] [PubMed] [Google Scholar]
- 29.Nilsson PM. ACCORD and Risk-Factor Control in Type 2 Diabetes. N Engl J Med. 2010;362:1628–1630. doi: 10.1056/NEJMe1002498. [DOI] [PubMed] [Google Scholar]
- 30.Wanner C, Krane V, März W, Olschewski M, Mann JF, Ruf G, Ritz E. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med. 2005;353:238–248. doi: 10.1056/NEJMoa043545. [DOI] [PubMed] [Google Scholar]
- 31.Mark PB, Johnston N, Groenning BA, Foster JE, Blyth KG, Martin TN, Steedman T, Dargie HJ, Jardine AG. Redefinition of uremic cardiomyopathy by contrast-enhanced cardiac magnetic resonance imaging. Kidney Int. 2006;69:1839–1845. doi: 10.1038/sj.ki.5000249. [DOI] [PubMed] [Google Scholar]
- 32.Schiffrin EL, Lipman ML, Mann JF. Chronic kidney disease: effects on the cardiovascular system. Circulation. 2007;116:85–97. doi: 10.1161/CIRCULATIONAHA.106.678342. [DOI] [PubMed] [Google Scholar]
- 33.Methe H, Weis M. Atherogenesis and inflammation--was Virchow right? Nephrol Dial Transplant. 2007;22:1823–1827. doi: 10.1093/ndt/gfm112. [DOI] [PubMed] [Google Scholar]
- 34.Thambyrajah J, Landray MJ, McGlynn FJ, Jones HJ, Wheeler DC, Townend JN. Abnormalities of endothelial function in patients with predialysis renal failure. Heart. 2000;83:205–209. doi: 10.1136/heart.83.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vallance P, Leiper J. Cardiovascular biology of the asymmetric dimethylarginine: dimethylarginine dimethylaminohydrolase pathway. Arterioscler Thromb Vasc Biol. 2004;24:1023–1030. doi: 10.1161/01.ATV.0000128897.54893.26. [DOI] [PubMed] [Google Scholar]
- 36.Bogle RG, MacAllister RJ, Whitley GS, Vallance P. Induction of NG-monomethyl-L-arginine uptake: a mechanism for differential inhibition of NO synthases? Am J Physiol. 1995;269:C750–C756. doi: 10.1152/ajpcell.1995.269.3.C750. [DOI] [PubMed] [Google Scholar]
- 37.Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. 2004;15:1983–1992. doi: 10.1097/01.ASN.0000132474.50966.DA. [DOI] [PubMed] [Google Scholar]
- 38.Zoccali C, Bode-Böger S, Mallamaci F, Benedetto F, Tripepi G, Malatino L, Cataliotti A, Bellanuova I, Fermo I, Frölich J, et al. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet. 2001;358:2113–2117. doi: 10.1016/s0140-6736(01)07217-8. [DOI] [PubMed] [Google Scholar]
- 39.Tripepi G, Mattace Raso F, Sijbrands E, Seck MS, Maas R, Boger R, Witteman J, Rapisarda F, Malatino L, Mallamaci F, et al. Inflammation and asymmetric dimethylarginine for predicting death and cardiovascular events in ESRD patients. Clin J Am Soc Nephrol. 2011;6:1714–1721. doi: 10.2215/CJN.11291210. [DOI] [PubMed] [Google Scholar]
- 40.Amirmansour C, Vallance P, Bogle RG. Tyrosine nitration in blood vessels occurs with increasing nitric oxide concentration. Br J Pharmacol. 1999;127:788–794. doi: 10.1038/sj.bjp.0702590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ito A, Tsao PS, Adimoolam S, Kimoto M, Ogawa T, Cooke JP. Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. Circulation. 1999;99:3092–3095. doi: 10.1161/01.cir.99.24.3092. [DOI] [PubMed] [Google Scholar]
- 42.Clarke R, Halsey J, Lewington S, Lonn E, Armitage J, Manson JE, Bønaa KH, Spence JD, Nygård O, Jamison R, et al. Effects of lowering homocysteine levels with B vitamins on cardiovascular disease, cancer, and cause-specific mortality: Meta-analysis of 8 randomized trials involving 37 485 individuals. Arch Intern Med. 2010;170:1622–1631. doi: 10.1001/archinternmed.2010.348. [DOI] [PubMed] [Google Scholar]
- 43.Shishehbor MH, Oliveira LP, Lauer MS, Sprecher DL, Wolski K, Cho L, Hoogwerf BJ, Hazen SL. Emerging cardiovascular risk factors that account for a significant portion of attributable mortality risk in chronic kidney disease. Am J Cardiol. 2008;101:1741–1746. doi: 10.1016/j.amjcard.2008.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Q, Botto LD, Erickson JD, Berry RJ, Sambell C, Johansen H, Friedman JM. Improvement in stroke mortality in Canada and the United States, 1990 to 2002. Circulation. 2006;113:1335–1343. doi: 10.1161/CIRCULATIONAHA.105.570846. [DOI] [PubMed] [Google Scholar]
- 45.Stühlinger MC, Oka RK, Graf EE, Schmölzer I, Upson BM, Kapoor O, Szuba A, Malinow MR, Wascher TC, Pachinger O, et al. Endothelial dysfunction induced by hyperhomocyst(e)inemia: role of asymmetric dimethylarginine. Circulation. 2003;108:933–938. doi: 10.1161/01.CIR.0000085067.55901.89. [DOI] [PubMed] [Google Scholar]
- 46.Chao MC, Hu SL, Hsu HS, Davidson LE, Lin CH, Li CI, Liu CS, Li TC, Lin CC, Lin WY. Serum homocysteine level is positively associated with chronic kidney disease in a Taiwan Chinese population. J Nephrol. 2014 doi: 10.1007/s40620-013-0037-9. [DOI] [PubMed] [Google Scholar]
- 47.Pan Y, Guo LL, Cai LL, Zhu XJ, Shu JL, Liu XL, Jin HM. Homocysteine-lowering therapy does not lead to reduction in cardiovascular outcomes in chronic kidney disease patients: a meta-analysis of randomised, controlled trials. Br J Nutr. 2012;108:400–407. doi: 10.1017/S0007114511007033. [DOI] [PubMed] [Google Scholar]
- 48.Mattock MB, Barnes DJ, Viberti G, Keen H, Burt D, Hughes JM, Fitzgerald AP, Sandhu B, Jackson PG. Microalbuminuria and coronary heart disease in NIDDM: an incidence study. Diabetes. 1998;47:1786–1792. doi: 10.2337/diabetes.47.11.1786. [DOI] [PubMed] [Google Scholar]
- 49.Schmitz A, Vaeth M. Microalbuminuria: a major risk factor in non-insulin-dependent diabetes. A 10-year follow-up study of 503 patients. Diabet Med. 1988;5:126–134. doi: 10.1111/j.1464-5491.1988.tb00958.x. [DOI] [PubMed] [Google Scholar]
- 50.Lane JT. Microalbuminuria as a marker of cardiovascular and renal risk in type 2 diabetes mellitus: a temporal perspective. Am J Physiol Renal Physiol. 2004;286:F442–F450. doi: 10.1152/ajprenal.00247.2003. [DOI] [PubMed] [Google Scholar]
- 51.Amann K, Wanner C, Ritz E. Cross-talk between the kidney and the cardiovascular system. J Am Soc Nephrol. 2006;17:2112–2119. doi: 10.1681/ASN.2006030204. [DOI] [PubMed] [Google Scholar]
- 52.Donnelly R, Yeung JM, Manning G. Microalbuminuria: a common, independent cardiovascular risk factor, especially but not exclusively in type 2 diabetes. J Hypertens Suppl. 2003;21:S7–12. [PubMed] [Google Scholar]
- 53.Jerums G, MacIsaac RJ. Treatment of microalbuminuria in patients with type 2 diabetes mellitus. Treat Endocrinol. 2002;1:163–173. doi: 10.2165/00024677-200201030-00004. [DOI] [PubMed] [Google Scholar]
- 54.Mann JF, Gerstein HC, Yi QL, Lonn EM, Hoogwerf BJ, Rashkow A, Yusuf S. Development of renal disease in people at high cardiovascular risk: results of the HOPE randomized study. J Am Soc Nephrol. 2003;14:641–647. doi: 10.1097/01.asn.0000051594.21922.99. [DOI] [PubMed] [Google Scholar]
- 55.Parving HH, Lehnert H, Bröchner-Mortensen J, Gomis R, Andersen S, Arner P. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med. 2001;345:870–878. doi: 10.1056/NEJMoa011489. [DOI] [PubMed] [Google Scholar]
- 56.Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med. 2004;164:659–663. doi: 10.1001/archinte.164.6.659. [DOI] [PubMed] [Google Scholar]
- 57.Di Angelantonio E, Chowdhury R, Sarwar N, Aspelund T, Danesh J, Gudnason V. Chronic kidney disease and risk of major cardiovascular disease and non-vascular mortality: prospective population based cohort study. BMJ. 2010;341:c4986. doi: 10.1136/bmj.c4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bello AK, Hemmelgarn B, Lloyd A, James MT, Manns BJ, Klarenbach S, Tonelli M. Associations among estimated glomerular filtration rate, proteinuria, and adverse cardiovascular outcomes. Clin J Am Soc Nephrol. 2011;6:1418–1426. doi: 10.2215/CJN.09741110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tonelli M, Muntner P, Lloyd A, Manns BJ, Klarenbach S, Pannu N, James MT, Hemmelgarn BR. Risk of coronary events in people with chronic kidney disease compared with those with diabetes: a population-level cohort study. Lancet. 2012;380:807–814. doi: 10.1016/S0140-6736(12)60572-8. [DOI] [PubMed] [Google Scholar]
- 60.Diercks GF, van Boven AJ, Hillege HL, Janssen WM, Kors JA, de Jong PE, Grobbee DE, Crijns HJ, van Gilst WH. Microalbuminuria is independently associated with ischaemic electrocardiographic abnormalities in a large non-diabetic population. The PREVEND (Prevention of REnal and Vascular ENdstage Disease) study. Eur Heart J. 2000;21:1922–1927. doi: 10.1053/euhj.2000.2248. [DOI] [PubMed] [Google Scholar]
- 61.Tsioufis C, Dimitriadis K, Chatzis D, Vasiliadou C, Tousoulis D, Papademetriou V, Toutouzas P, Stefanadis C, Kallikazaros I. Relation of microalbuminuria to adiponectin and augmented C-reactive protein levels in men with essential hypertension. Am J Cardiol. 2005;96:946–951. doi: 10.1016/j.amjcard.2005.05.052. [DOI] [PubMed] [Google Scholar]
- 62.Yilmaz MI, Sonmez A, Saglam M, Qureshi AR, Carrero JJ, Caglar K, Eyileten T, Cakir E, Oguz Y, Vural A, et al. ADMA levels correlate with proteinuria, secondary amyloidosis, and endothelial dysfunction. J Am Soc Nephrol. 2008;19:388–395. doi: 10.1681/ASN.2007040461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kong X, Jia X, Wei Y, Cui M, Wang Z, Tang L, Li W, Zhu Z, Chen P, Xu D. Association between microalbuminuria and subclinical atherosclerosis evaluated by carotid artery intima-media in elderly patients with normal renal function. BMC Nephrol. 2012;13:37. doi: 10.1186/1471-2369-13-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 65.Albert CM, Ma J, Rifai N, Stampfer MJ, Ridker PM. Prospective study of C-reactive protein, homocysteine, and plasma lipid levels as predictors of sudden cardiac death. Circulation. 2002;105:2595–2599. doi: 10.1161/01.cir.0000017493.03108.1c. [DOI] [PubMed] [Google Scholar]
- 66.Stenvinkel P, Heimbürger O, Lindholm B, Kaysen GA, Bergström J. Are there two types of malnutrition in chronic renal failure? Evidence for relationships between malnutrition, inflammation and atherosclerosis (MIA syndrome) Nephrol Dial Transplant. 2000;15:953–960. doi: 10.1093/ndt/15.7.953. [DOI] [PubMed] [Google Scholar]
- 67.Hage FG, Venkataraman R, Zoghbi GJ, Perry GJ, DeMattos AM, Iskandrian AE. The scope of coronary heart disease in patients with chronic kidney disease. J Am Coll Cardiol. 2009;53:2129–2140. doi: 10.1016/j.jacc.2009.02.047. [DOI] [PubMed] [Google Scholar]
- 68.Miyata T, Wada Y, Cai Z, Iida Y, Horie K, Yasuda Y, Maeda K, Kurokawa K, van Ypersele de Strihou C. Implication of an increased oxidative stress in the formation of advanced glycation end products in patients with end-stage renal failure. Kidney Int. 1997;51:1170–1181. doi: 10.1038/ki.1997.160. [DOI] [PubMed] [Google Scholar]
- 69.Leurs P, Lindholm B. The AGE-RAGE pathway and its relation to cardiovascular disease in patients with chronic kidney disease. Arch Med Res. 2013;44:601–610. doi: 10.1016/j.arcmed.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 70.Arici M, Walls J. End-stage renal disease, atherosclerosis, and cardiovascular mortality: is C-reactive protein the missing link? Kidney Int. 2001;59:407–414. doi: 10.1046/j.1523-1755.2001.059002407.x. [DOI] [PubMed] [Google Scholar]
- 71.Parekh RS, Plantinga LC, Kao WH, Meoni LA, Jaar BG, Fink NE, Powe NR, Coresh J, Klag MJ. The association of sudden cardiac death with inflammation and other traditional risk factors. Kidney Int. 2008;74:1335–1342. doi: 10.1038/ki.2008.449. [DOI] [PubMed] [Google Scholar]
- 72.Ramkumar N, Cheung AK, Pappas LM, Roberts WL, Beddhu S. Association of obesity with inflammation in chronic kidney disease: a cross-sectional study. J Ren Nutr. 2004;14:201–207. [PubMed] [Google Scholar]
- 73.Zoccali C, Mallamaci F, Tripepi G, Cutrupi S, Parlongo S, Malatino LS, Bonanno G, Rapisarda F, Fatuzzo P, Seminara G, et al. Fibrinogen, mortality and incident cardiovascular complications in end-stage renal failure. J Intern Med. 2003;254:132–139. doi: 10.1046/j.1365-2796.2003.01180.x. [DOI] [PubMed] [Google Scholar]
- 74.Kaysen GA. Potential restoration of HDL function with apolipoprotein A-I mimetic peptide in end-stage renal disease. Kidney Int. 2009;76:359–361. doi: 10.1038/ki.2009.205. [DOI] [PubMed] [Google Scholar]
- 75.Kaysen GA, Eiserich JP. The role of oxidative stress-altered lipoprotein structure and function and microinflammation on cardiovascular risk in patients with minor renal dysfunction. J Am Soc Nephrol. 2004;15:538–548. doi: 10.1097/01.asn.0000111744.00916.e6. [DOI] [PubMed] [Google Scholar]
- 76.Vaziri ND, Oveisi F, Ding Y. Role of increased oxygen free radical activity in the pathogenesis of uremic hypertension. Kidney Int. 1998;53:1748–1754. doi: 10.1046/j.1523-1755.1998.00947.x. [DOI] [PubMed] [Google Scholar]
- 77.Zoccali C, Mallamaci F, Tripepi G. Inflammatory proteins as predictors of cardiovascular disease in patients with end-stage renal disease. Nephrol Dial Transplant. 2004;19 Suppl 5:V67–V72. doi: 10.1093/ndt/gfh1059. [DOI] [PubMed] [Google Scholar]
- 78.Levin A, Singer J, Thompson CR, Ross H, Lewis M. Prevalent left ventricular hypertrophy in the predialysis population: identifying opportunities for intervention. Am J Kidney Dis. 1996;27:347–354. doi: 10.1016/s0272-6386(96)90357-1. [DOI] [PubMed] [Google Scholar]
- 79.Levy D. Clinical significance of left ventricular hypertrophy: insights from the Framingham Study. J Cardiovasc Pharmacol. 1991;17 Suppl 2:S1–S6. doi: 10.1097/00005344-199117002-00002. [DOI] [PubMed] [Google Scholar]
- 80.Tai DJ, Lim TW, James MT, Manns BJ, Tonelli M, Hemmelgarn BR. Cardiovascular effects of angiotensin converting enzyme inhibition or angiotensin receptor blockade in hemodialysis: a meta-analysis. Clin J Am Soc Nephrol. 2010;5:623–630. doi: 10.2215/CJN.07831109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harnett JD, Foley RN, Kent GM, Barre PE, Murray D, Parfrey PS. Congestive heart failure in dialysis patients: prevalence, incidence, prognosis and risk factors. Kidney Int. 1995;47:884–890. doi: 10.1038/ki.1995.132. [DOI] [PubMed] [Google Scholar]
- 82.Hruska KA, Mathew S, Lund RJ, Memon I, Saab G. The pathogenesis of vascular calcification in the chronic kidney disease mineral bone disorder: the links between bone and the vasculature. Semin Nephrol. 2009;29:156–165. doi: 10.1016/j.semnephrol.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vervloet MG, Massy ZA, Brandenburg VM, Mazzaferro S, Cozzolino M, Ureña-Torres P, Bover J, Goldsmith D. Bone: a new endocrine organ at the heart of chronic kidney disease and mineral and bone disorders. Lancet Diabetes Endocrinol. 2014;2:427–436. doi: 10.1016/S2213-8587(14)70059-2. [DOI] [PubMed] [Google Scholar]
- 84.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 85.Jovanovich A, Ix JH, Gottdiener J, McFann K, Katz R, Kestenbaum B, de Boer IH, Sarnak M, Shlipak MG, Mukamal KJ, et al. Fibroblast growth factor 23, left ventricular mass, and left ventricular hypertrophy in community-dwelling older adults. Atherosclerosis. 2013;231:114–119. doi: 10.1016/j.atherosclerosis.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutiérrez OM, Aguillon-Prada R, Lincoln J, Hare JM, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qunibi WY. Reducing the burden of cardiovascular calcification in patients with chronic kidney disease. J Am Soc Nephrol. 2005;16 Suppl 2:S95–102. doi: 10.1681/ASN.2005060666. [DOI] [PubMed] [Google Scholar]
- 88.Goodman WG, London G, Amann K, Block GA, Giachelli C, Hruska KA, Ketteler M, Levin A, Massy Z, McCarron DA, et al. Vascular calcification in chronic kidney disease. Am J Kidney Dis. 2004;43:572–579. doi: 10.1053/j.ajkd.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 89.Russo D, Palmiero G, De Blasio AP, Balletta MM, Andreucci VE. Coronary artery calcification in patients with CRF not undergoing dialysis. Am J Kidney Dis. 2004;44:1024–1030. doi: 10.1053/j.ajkd.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 90.Pannier B, Guérin AP, Marchais SJ, Safar ME, London GM. Stiffness of capacitive and conduit arteries: prognostic significance for end-stage renal disease patients. Hypertension. 2005;45:592–596. doi: 10.1161/01.HYP.0000159190.71253.c3. [DOI] [PubMed] [Google Scholar]
- 91.Sigrist MK, Taal MW, Bungay P, McIntyre CW. Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin J Am Soc Nephrol. 2007;2:1241–1248. doi: 10.2215/CJN.02190507. [DOI] [PubMed] [Google Scholar]
- 92.Moe SM, Chen NX. Mechanisms of vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2008;19:213–216. doi: 10.1681/ASN.2007080854. [DOI] [PubMed] [Google Scholar]
- 93.Shroff RC, Shanahan CM. The vascular biology of calcification. Semin Dial. 2007;20:103–109. doi: 10.1111/j.1525-139X.2007.00255.x. [DOI] [PubMed] [Google Scholar]
- 94.Morena M, Terrier N, Jaussent I, Leray-Moragues H, Chalabi L, Rivory JP, Maurice F, Delcourt C, Cristol JP, Canaud B, et al. Plasma osteoprotegerin is associated with mortality in hemodialysis patients. J Am Soc Nephrol. 2006;17:262–270. doi: 10.1681/ASN.2005030260. [DOI] [PubMed] [Google Scholar]
- 95.Green D, Roberts PR, New DI, Kalra PA. Sudden cardiac death in hemodialysis patients: an in-depth review. Am J Kidney Dis. 2011;57:921–929. doi: 10.1053/j.ajkd.2011.02.376. [DOI] [PubMed] [Google Scholar]
- 96.Poulikakos D, Banerjee D, Malik M. Risk of Sudden Cardiac Death in Chronic Kidney Disease. J Cardiovasc Electrophysiol. 2013:Nov 20; Epub ahead of print. doi: 10.1111/jce.12328. [DOI] [PubMed] [Google Scholar]
- 97.Poulikakos D, Banerjee D, Malik M. Major arrhythmic events and T wave morphology descriptors in hemodialyzed patients. J Electrocardiol. 2014;47:240–243. doi: 10.1016/j.jelectrocard.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 98.Burton JO, Jefferies HJ, Selby NM, McIntyre CW. Hemodialysis-induced cardiac injury: determinants and associated outcomes. Clin J Am Soc Nephrol. 2009;4:914–920. doi: 10.2215/CJN.03900808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Assa S, Dasselaar JJ, Slart RH, de Jong PE, Voors AA, Tio RA, Franssen CF. Comparison of cardiac positron emission tomography perfusion defects during stress induced by hemodialysis versus adenosine. Am J Kidney Dis. 2012;59:862–864. doi: 10.1053/j.ajkd.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 100.Poulikakos D, Ross L, Recio-Mayoral A, Cole D, Andoh J, Chitalia N, Sharma R, Carlos Kaski J, Banerjee D. Left ventricular hypertrophy and endothelial dysfunction in chronic kidney disease. Eur Heart J Cardiovasc Imaging. 2014;15:56–61. doi: 10.1093/ehjci/jet120. [DOI] [PubMed] [Google Scholar]
- 101.Roberts PR, Zachariah D, Borman N, Morgan JM, Yue AM, Greenwood EF, Philips PC, Green D, Philip KA, Lewis RJ, et al. Cardio Renal Arrhythmia Study in Hemodialysis Patients Using Implantable Loop Recorders (CRASH-ILR) Circulation. 2013;128:A10581. [Google Scholar]