

Abstract
Prorenin receptor (PRR) is a multi-functioning protein possessing at least four different roles: (1) working as a receptor for renin and prorenin producing angiotensin I from angiotensinogen thus enhancing the tissue renin-angiotensin system; (2) inducing intracellular signals when a ligand binds to PRR; (3) participating in the functions of vacuolar proton ATPase; and (4) constituting the Wnt signaling receptor complex. Here, the roles of PRR in kidney physiology and diabetic conditions as well as recent findings regarding a soluble form of PRR are discussed. We also propose the possible mechanism concerning diabetic nephropathy as “trade-off hypothesis” from a PRR point of view. In brief, under hyperglycemic conditions, injured podocytes degrade degenerated proteins and intracellular organelles which require V-ATPase and PRR for vesicle internal acidification. Sustained hyperglycemia overproduces PRR molecules, which are transported to the transmembrane and bind to increased serum prorenin in the diabetic condition. This enhances tissue renin-angiotensin system and PRR-mediated mitogen-activated protein kinase signals, resulting in increased injurious molecules such as transforming growth factor-β, cyclooxygenase2, interleukin-1β, and tumor necrosis factor-α ending in diabetic nephropathy progression. Although many findings led us to better PRR understanding, future works should elucidate which PRR functions, of the four discussed here, are dominant in each cell and kidney disease context.
Keywords: Prorenin receptor, Atp6ap2, Soluble prorenin receptor, Kidney, Diabetic nephropathy, Podocyte
Core tip: Prorenin receptor (PRR) has shown its multi-functionality in at least four different aspects. In this review, the roles of PRR in kidney physiology and diabetic conditions as well as recent findings regarding a soluble form of PRR are discussed. Additionally, we propose the possible mechanism concerning diabetic nephropathy as “trade-off hypothesis” from a PRR point of view.
INTRODUCTION
Prorenin receptor (PRR), also known as ATP 6-associated protein 2 (Atp6ap2), was cloned in 2002 as a single transmembrane protein whose ligand is renin and its precursor prorenin[1]. Initially, the roles of PRR were thought to enhance the tissue renin-angiotensin system (RAS) by binding PRR to its ligand, while also inducing intracellular signal transductions such as mitogen-activated protein kinase (MAPK) pathways independent of the RAS. However, recent findings have revealed additional aspects of PRR, including it functioning as an accessory protein of vacuolar proton ATPase (V-ATPase) and constituting the Wnt receptor complex. This review discusses PRR in kidney physiology and diabetic nephropathy, and in addition to recent findings regarding the kidney and a soluble form of PRR.
PRORENIN RECEPTOR IN KIDNEY PHYSIOLOGY
After its discovery by cloning a single transmembrane protein PRR a dozen years ago, PRR has shown its multi-functionality in at least four different aspects (Figure 1). One of these is to enhance angiotensin I production from angiotensinogen by non-proteolytically increasing catalyzing activity of renin or prorenin when bound to PRR, resulting in enhanced RAS (Figure 1A). Another is to induce MAPK signal transduction pathway when PRR is bound to its ligand renin or prorenin[1] (Figure 1B). PRR is located on the X chromosome and is distributed widely in the kidney, heart, brain, liver, placenta and pancreas, although its’ physiological role has not been elucidated until recently because of embryonic lethality of complete knockout of PRR in mice.
Figure 1.
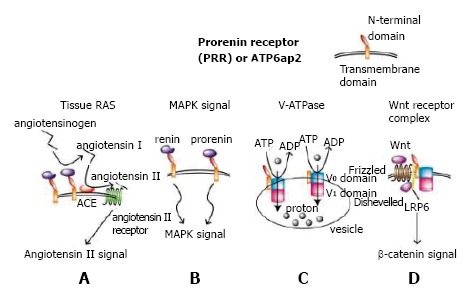
Four roles of prorenin receptor. A: When renin or prorenin binds to prorenin receptor (PRR), renin or prorenin enzymatic activity is enhanced through non-proteolytic conformational change, catalyzing angiotensinogen to angiotensin I. Produced angiotensin I is catalyzed by angiotensin-converting enzyme, yielding angiotensin II that induces angiotensin II receptor-mediated signal transduction, ending in enhanced tissue renin-angiotensin system (RAS)[45]; B: When PRR is bound to a ligand, renin, or prorenin, a mitogen-activated protein kinase (MAPK) signal is induced[1]; C: PRR, with or without the N-terminal domain, is required as a subunit of V-ATPase, which actively transports protons into vesicles such as endosomes, lysosomes, and autophagosomes using energy obtained by degrading ATP to ADP[9]. The V0 and V1 domains build up V-ATPase; D: PRR is required as an adaptor protein between V-ATPase and LRP6, which are members of the Wnt receptor complex[5,46].
We previously generated floxed PRR mice and mated them with mice expressing Cre recombinase under the control of a podocyte-specific podocin promoter to create conditional PRR knockout mice in podocytes. Unexpectedly, these mice died of nephrotic syndrome and renal failure resulting from disturbed V-ATPase function. This provided evidence that, under physiological condition PRR is needed for maintaining vacuoles-such as endosomes, lysosomes, and autophagosomes- through normal V-ATPase function in mouse podocytes[2] (Figure 1C). Similar results in regard to podocytes[3] and cardiomyocytes[4] were obtained from another group and ours, respectively.
In Xenopus, PRR binds to V-ATPase and LRP6 to form a Wnt signaling receptor complex as an adaptor protein, showing that PRR is indispensable for normal Wnt signal transduction[5] (Figure 1D). The embryonic lethality of PRR full knockout in mice may be related to abrogated Wnt signals because Wnt signal is required for the formation of a primitive streak in early mouse embryogenesis[6]. PRR is also required for early embryogenesis in zebrafish[7]. PRR has been shown to be involved in nephrogenesis; mice with conditional PRR knockout in the ureteric bud developed renal hypodysplasia[8]. These mice were not embryonically lethal, presumably because the uretic bud is derived from the intermediate mesoderm, which develops long after the formation of the primitive streak.
Recently, the N-terminal of the PRR extracellular domain, which interacts with prorenin/renin, has been proven indispensable for V-ATPase biogenesis[9]. Although prorenin/renin does not influence overall V-ATPase activity[5,9], in vitro experiments using MCDK cells revealed prorenin and a handle region peptide that corresponds to the part of the prorenin responsible for binding to PRR and increases the initial linear phase of V-ATPase activity through binding to PRR[10]. Further investigations are expected to elucidate the roles of PRR in association with prorenin/renin, V-ATPase, and Wnt signals in physiological and developmental conditions in the kidney.
PRORENIN RECEPTOR IN DIABETIC NEPHROPATHY
In diabetes mellitus (DM), a sustained hyperglycemic state leads to diabetic nephropathy, causing end-stage renal disease. It has been reported that an elevated plasma prorenin concentration is associated with microalbuminuria[11] and diabetic nephropathy[12] in patients with DM. Tissue angiotensin II levels are also increased in the kidneys of DM rats[13-15], suggesting activated tissue RAS. This is consistent with the multiple clinical trials showing that angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARB) have reno-protective effects against diabetic nephropathy[16-19], although these effects were limited. Adversely, plasma renin activity is suppressed in patients with diabetic nephropathy[20,21], reflecting a suppressed systemic RAS possibly resulting from activated tissue RAS in the kidneys. Recently, podocytes have been thought to play an important role in diabetic nephropathy and albuminuria[22,23]. Identifying PRR has led to a better understanding of diabetic nephropathy.
Albuminuria is caused by the breakdown of the filtration barrier composed of endothelial cells, glomerular basement membranes, and podocytes. PRR has been heavily detected in mouse podocytes through immunoelectron microscopy[24]. Because transgenic rats overexpressing human PRR[25] developed slowly progressive proteinuria and enhanced MAPK signals mainly in the glomeruli podocytes[26], it is likely that podocytes are vulnerable to PRR-dependent signal transduction. The MAPK signals could be aggravated in the diabetic condition, in which both PRR and prorenin are upregulated, as discussed later in this article. This effect is believed to be independent of angiotensin II because rat prorenin bound to human PRR did not show renin activity.
In rats with streptozotocin-induced diabetic nephropathy, PRR protein up-regulation were observed in their kidneys[27]. High glucose increased mRNA and protein PRR levels in cultured podocytes[28]. This experimental evidence shows that PRR is up-regulated in the podocytes of DM; thus, in DM, both the ligand prorenin and its receptor PRR increase.
Electron microscopic analysis in DM mice revealed that podocytes go through a hypertrophic state before the atrophic state. This hypertrophy reflects the occurrence of many vesicles (presumably lysosomes and autophagosomes) resulting from injured intracellular organelles[29]. Also, Golgi apparatus, rough endoplasmic reticulum, and free ribosome develop because of increased protein production demand resulting from cell injury[29] (Figure 2B). It is possible that these increased intracellular organelles might call for PRR up-regulation because PRR is required for V-ATPase activity, which acidifies intracellular vesicles (Figure 2C). V-ATPase is required for both maturation of synthesized proteins and degradation by lysosomes and autophagosomes that affects proteins and organelles.
Figure 2.
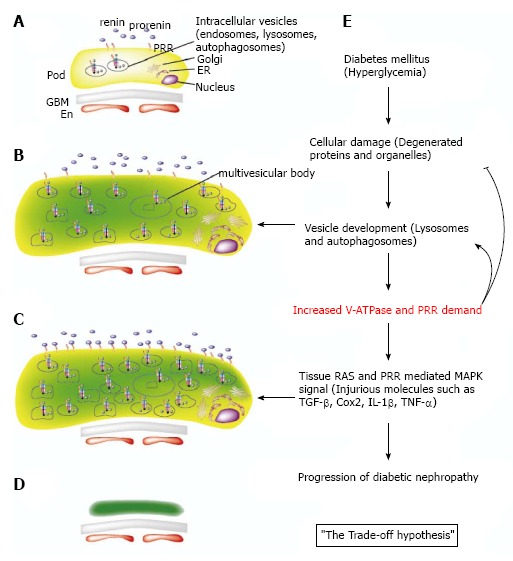
The trade-off hypothesis in diabetic nephropathy from the prorenin receptor perspective. A: Normal podocyte intracellular structure; B: Under hyperglycemic conditions, injured podocytes degrade degenerated proteins and intracellular organelles, forming vesicles such as lysosomes and autophagosomes, which require V-ATPase and prorenin receptor (PRR) for internal acidification; C: Sustained hyperglycemia overproduces PRR molecules, which are transported to the transmembrane and bind to increased serum prorenin in the diabetic condition. This enhances tissue renin-angiotensin system (RAS) and PRR-mediated mitogen-activated protein kinase (MAPK) signals; D: Atrophic or apoptotic podocytes after long-term hyperglycemic conditions. The changes in the glomerular basement membrane and endothelial cells are not shown; E: The schematic view of the trade-off hypothesis. In diabetes mellitus, podocyte injury occurs by producing degenerated proteins and organelles, which in turn are degraded in lysosomes and autophagosomes. This process requires V-ATPase and PRR production for internal acidification of the vesicles. PRR overproduction enhances tissue RAS and PRR-mediated MAPK signals, resulting in increased injurious molecules such as transforming growth factor-β, cyclooxygenase2, interleukin-1β, and tumor necrosis factor-α. Progression and deterioration of diabetic nephropathy occurs at the end. En: Endothelial cell; ER: Endoplasmic reticulum; GBM: Glomerular basement membrane; Pod: Podocyte.
As discussed previously, the serum prorenin level is increased in DM patients. Inhibition of prorenin binding to PRR by subcutaneous administration of a handle region peptide prevents diabetic nephropathy development, characterized by the inhibition of albuminuria and glomerulosclerosis in DM rats[15]. Moreover, nephropathy regression was observed in the peptide-treated handle region, but not inhibitor-treated ACE, in DM rats[30]. Handle region peptide treatment inhibited diabetic nephropathy in angiotensin II type 1a receptor-deficient mice, suggesting the effectiveness of PRR signal transduction blockade in inhibiting diabetic nephropathy apart from angiotensin II effects. This is consistent with the results of clinical trials showing a partial effect of ACE inhibitors or ARB in diabetic nephropathy in humans[16-19]. Other in vivo data showed that PRR increases transforming growth factor-β1[31], cyclooxygenase2[32], and cytokines such as interleukin-Iβ and tumor necrosis factor-α[33] in diabetic nephropathy. Our hypothesis here is that overproduced PRR counteracting cell injury could exhibit adverse effects through PRR-distinctive signal transductions, angiotensin II-mediated effects, and other injurious molecules contributing to the progression of diabetic nephropathy. We term this the “trade-off hypothesis” (Figure 2E). According to our preliminary data using human PRR over-expression in transgenic rats, albuminuria was not seen in the early stages of diabetic nephropathy, whereas albuminuria was seen in wild-type rats. This experimentally supports our hypothesis.
V-ATPase is indispensable for the normal function of endosomes, lysosomes, and autophagosomes, whereas diabetic nephropathy is associated with endoplasmic reticulum stress[34] and autophagy[35]. Future experiments are needed to investigate the pathophysiological functions of PRR both in V-ATPase and signal aspects in diabetic nephropathy.
SOLUBLE PRORENIN RECEPTOR AND KIDNEY DISEASE
PRR is a single transmembrane protein with a proteinase cleavage site at the N-terminal domain near the transmembrane domain[36,37]. Cleavage of PRR by either furin[36] or ADAM19[38] results in the dissociation of the N-terminal fragment as soluble PRR (sPRR).
In healthy subjects, plasma sPRR did not exhibit circadian or posture variation or correlate with renin, prorenin or aldosterone levels[39], although serum sPRR negatively correlated with an estimated glomerular filtration rate independent of age, blood pressure, and glucose metabolism in essential hypertension and normotensive subjects[40]. sPRR correlated positively with urinary angiotensinogen[40], which is a biomarker of intrarenal RAS[41]. Moreover, in patients with chronic kidney disease, serum sPRR negatively correlated with estimated glomerular filtration rate and chronic kidney disease stage[42]. In human kidneys with end-stage renal disease, intrarenal PRR was immunostained mainly in tubules, suggesting a possible contribution of increased renal PRR expression to elevated sPRR in end-stage renal disease[43]. These findings suggest that sPRR might reflect the intrarenal RAS status.
However, in addition to the changes in sPRR levels being modest and not offering a set cut-off line for clinical use, these levels are affected by many other factors including, RAS inhibitor administration[39], lipid metabolites such as high-density lipoprotein cholesterol and triglycerides[40], age[40], and obstructive sleep apnea syndrome[44]. Moreover, the correlation between sPRR level and the activity of furin or ADAM19 has not yet been investigated under these conditions. Further investigations are expected to determine the relationships, if any, between kidney disease and sPRR, PRR, tissue RAS, V-ATPase and Wnt.
SUMMARY
In physiological and developmental conditions, it has been suggested from the experiments discussed previously that PRR primarily exhibits as V-ATPase or Wnt signaling instead of RAS enhancement or PRR-distinct MAPK signal transduction. These former functions are related to fundamental cellular survival and embryo development. On the other hand, in DM, inhibition of prorenin binding to PRR has the beneficial effect of inhibiting diabetic nephropathy, which is not achieved in Ang II blockade. In DM, PRR primarily works as an RAS enhancement or MAPK signal on top of V-ATPase. Involvement of Wnt signals in diabetic nephropathy has not been investigated throughly.
The shift of PRR’s function from a physiological condition to a DM condition could be attributed to altered PRR demand. As in the trade-off hypothesis, a hyperglycemic state leads to increased PRR demand caused by increased V-ATPase activity to recycle or degrade intracellular organelles and proteins. The overproduced PRR is transported to the cell membrane, which triggers RAS enhancement and PRR-dependent MAPK signals via prorenin binding. As a result, diabetic nephropathy progression occurs.
The significance of sPRR in kidney disease is not clearly defined because sPRR changes in kidney disease are too modest to set a cut-off line for clinical use. Yet, it is possible that sPRR reflects intrarenal RAS status.
CONCLUSION
Rigorous work has uncovered PRR physiology and pathophysiology in the kidneys. The multi-functioning protein PRR can shift its role from the physiological condition to the DM condition depending on underlying cellular conditions. V-ATPase is believed to help cells maintain a clean environment; however, in DM, it may have adverse effects in terms of overproduced PRR. This functional shift may occur because, unlike other molecules, PRR not only is important in fundamental cellular survival but also in disease progression. According to the trade-off hypothesis, over-expression of PRR may have beneficial effects in the very early stages of diabetic nephropathy, although PRR may have harmful effects in the late stages. Future works should elucidate which PRR functions, of the four discussed here, are dominant in each cell and kidney disease context.
Footnotes
P- Reviewer: Gharaee-Kermani M, Isaka Y, Tsilibary PEC S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
References
- 1.Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–1427. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oshima Y, Kinouchi K, Ichihara A, Sakoda M, Kurauchi-Mito A, Bokuda K, Narita T, Kurosawa H, Sun-Wada GH, Wada Y, et al. Prorenin receptor is essential for normal podocyte structure and function. J Am Soc Nephrol. 2011;22:2203–2212. doi: 10.1681/ASN.2011020202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riediger F, Quack I, Qadri F, Hartleben B, Park JK, Potthoff SA, Sohn D, Sihn G, Rousselle A, Fokuhl V, et al. Prorenin receptor is essential for podocyte autophagy and survival. J Am Soc Nephrol. 2011;22:2193–2202. doi: 10.1681/ASN.2011020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kinouchi K, Ichihara A, Sano M, Sun-Wada GH, Wada Y, Kurauchi-Mito A, Bokuda K, Narita T, Oshima Y, Sakoda M, et al. The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ Res. 2010;107:30–34. doi: 10.1161/CIRCRESAHA.110.224667. [DOI] [PubMed] [Google Scholar]
- 5.Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, Boutros M, Niehrs C. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science. 2010;327:459–463. doi: 10.1126/science.1179802. [DOI] [PubMed] [Google Scholar]
- 6.Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A. Requirement for Wnt3 in vertebrate axis formation. Nat Genet. 1999;22:361–365. doi: 10.1038/11932. [DOI] [PubMed] [Google Scholar]
- 7.Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci U S A. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song R, Preston G, Ichihara A, Yosypiv IV. Deletion of the prorenin receptor from the ureteric bud causes renal hypodysplasia. PLoS One. 2013;8:e63835. doi: 10.1371/journal.pone.0063835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinouchi K, Ichihara A, Sano M, Sun-Wada GH, Wada Y, Ochi H, Fukuda T, Bokuda K, Kurosawa H, Yoshida N, et al. The role of individual domains and the significance of shedding of ATP6AP2/(pro)renin receptor in vacuolar H(+)-ATPase biogenesis. PLoS One. 2013;8:e78603. doi: 10.1371/journal.pone.0078603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu X, Garrelds IM, Wagner CA, Danser AH, Meima ME. (Pro)renin receptor is required for prorenin-dependent and -independent regulation of vacuolar H⁺-ATPase activity in MDCK.C11 collecting duct cells. Am J Physiol Renal Physiol. 2013;305:F417–F425. doi: 10.1152/ajprenal.00037.2013. [DOI] [PubMed] [Google Scholar]
- 11.Deinum J, Rønn B, Mathiesen E, Derkx FH, Hop WC, Schalekamp MA. Increase in serum prorenin precedes onset of microalbuminuria in patients with insulin-dependent diabetes mellitus. Diabetologia. 1999;42:1006–1010. doi: 10.1007/s001250051260. [DOI] [PubMed] [Google Scholar]
- 12.Luetscher JA, Kraemer FB, Wilson DM, Schwartz HC, Bryer-Ash M. Increased plasma inactive renin in diabetes mellitus. A marker of microvascular complications. N Engl J Med. 1985;312:1412–1417. doi: 10.1056/NEJM198505303122202. [DOI] [PubMed] [Google Scholar]
- 13.Nishiyama A, Nakagawa T, Kobori H, Nagai Y, Okada N, Konishi Y, Morikawa T, Okumura M, Meda I, Kiyomoto H, et al. Strict angiotensin blockade prevents the augmentation of intrarenal angiotensin II and podocyte abnormalities in type 2 diabetic rats with microalbuminuria. J Hypertens. 2008;26:1849–1859. doi: 10.1097/HJH.0b013e3283060efa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Awad AS, Webb RL, Carey RM, Siragy HM. Increased renal production of angiotensin II and thromboxane B2 in conscious diabetic rats. Am J Hypertens. 2005;18:544–548. doi: 10.1016/j.amjhyper.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 15.Ichihara A, Hayashi M, Kaneshiro Y, Suzuki F, Nakagawa T, Tada Y, Koura Y, Nishiyama A, Okada H, Uddin MN, et al. Inhibition of diabetic nephropathy by a decoy peptide corresponding to the “handle” region for nonproteolytic activation of prorenin. J Clin Invest. 2004;114:1128–1135. doi: 10.1172/JCI21398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 17.Parving HH, Lehnert H, Bröchner-Mortensen J, Gomis R, Andersen S, Arner P. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med. 2001;345:870–878. doi: 10.1056/NEJMoa011489. [DOI] [PubMed] [Google Scholar]
- 18.Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851–860. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 19.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329:1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 20.Antonipillai I, Tan SY, Suzuki S, Franco-Saenz R, Mulrow PJ. Active and inactive renin in low renin states: studies in human plasma. J Clin Endocrinol Metab. 1981;53:694–697. doi: 10.1210/jcem-53-4-694. [DOI] [PubMed] [Google Scholar]
- 21.Price DA, Porter LE, Gordon M, Fisher ND, De’Oliveira JM, Laffel LM, Passan DR, Williams GH, Hollenberg NK. The paradox of the low-renin state in diabetic nephropathy. J Am Soc Nephrol. 1999;10:2382–2391. doi: 10.1681/ASN.V10112382. [DOI] [PubMed] [Google Scholar]
- 22.Lai KN, Leung JC, Tang SC. The renin-angiotensin system. Contrib Nephrol. 2011;170:135–144. doi: 10.1159/000325649. [DOI] [PubMed] [Google Scholar]
- 23.Diez-Sampedro A, Lenz O, Fornoni A. Podocytopathy in diabetes: a metabolic and endocrine disorder. Am J Kidney Dis. 2011;58:637–646. doi: 10.1053/j.ajkd.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ichihara A, Kaneshiro Y, Takemitsu T, Sakoda M, Itoh H. The (pro)renin receptor and the kidney. Semin Nephrol. 2007;27:524–528. doi: 10.1016/j.semnephrol.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 25.Kaneshiro Y, Ichihara A, Takemitsu T, Sakoda M, Suzuki F, Nakagawa T, Hayashi M, Inagami T. Increased expression of cyclooxygenase-2 in the renal cortex of human prorenin receptor gene-transgenic rats. Kidney Int. 2006;70:641–646. doi: 10.1038/sj.ki.5001627. [DOI] [PubMed] [Google Scholar]
- 26.Kaneshiro Y, Ichihara A, Sakoda M, Takemitsu T, Nabi AH, Uddin MN, Nakagawa T, Nishiyama A, Suzuki F, Inagami T, et al. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J Am Soc Nephrol. 2007;18:1789–1795. doi: 10.1681/ASN.2006091062. [DOI] [PubMed] [Google Scholar]
- 27.Durvasula RV, Shankland SJ. Activation of a local renin angiotensin system in podocytes by glucose. Am J Physiol Renal Physiol. 2008;294:F830–F839. doi: 10.1152/ajprenal.00266.2007. [DOI] [PubMed] [Google Scholar]
- 28.Li C, Siragy HM. High glucose induces podocyte injury via enhanced (pro)renin receptor-Wnt-β-catenin-snail signaling pathway. PLoS One. 2014;9:e89233. doi: 10.1371/journal.pone.0089233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandache E, Penescu M. Nanostructural features of diabetic podocytopathy. Rom J Morphol Embryol. 2012;53:23–27. [PubMed] [Google Scholar]
- 30.Takahashi H, Ichihara A, Kaneshiro Y, Inomata K, Sakoda M, Takemitsu T, Nishiyama A, Itoh H. Regression of nephropathy developed in diabetes by (Pro)renin receptor blockade. J Am Soc Nephrol. 2007;18:2054–2061. doi: 10.1681/ASN.2006080820. [DOI] [PubMed] [Google Scholar]
- 31.Huang J, Matavelli LC, Siragy HM. Renal (pro)renin receptor contributes to development of diabetic kidney disease through transforming growth factor-β1-connective tissue growth factor signalling cascade. Clin Exp Pharmacol Physiol. 2011;38:215–221. doi: 10.1111/j.1440-1681.2011.05486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng H, Fan X, Moeckel GW, Harris RC. Podocyte COX-2 exacerbates diabetic nephropathy by increasing podocyte (pro)renin receptor expression. J Am Soc Nephrol. 2011;22:1240–1251. doi: 10.1681/ASN.2010111149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matavelli LC, Huang J, Siragy HM. (Pro)renin receptor contributes to diabetic nephropathy by enhancing renal inflammation. Clin Exp Pharmacol Physiol. 2010;37:277–282. doi: 10.1111/j.1440-1681.2009.05292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cunard R, Sharma K. The endoplasmic reticulum stress response and diabetic kidney disease. Am J Physiol Renal Physiol. 2011;300:F1054–F1061. doi: 10.1152/ajprenal.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka Y, Kume S, Kitada M, Kanasaki K, Uzu T, Maegawa H, Koya D. Autophagy as a therapeutic target in diabetic nephropathy. Exp Diabetes Res. 2012;2012:628978. doi: 10.1155/2012/628978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L, Nguyen G. Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension. 2009;53:1077–1082. doi: 10.1161/HYPERTENSIONAHA.108.127258. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen G. Renin, (pro)renin and receptor: an update. Clin Sci (Lond) 2011;120:169–178. doi: 10.1042/CS20100432. [DOI] [PubMed] [Google Scholar]
- 38.Yoshikawa A, Aizaki Y, Kusano K, Kishi F, Susumu T, Iida S, Ishiura S, Nishimura S, Shichiri M, Senbonmatsu T. The (pro)renin receptor is cleaved by ADAM19 in the Golgi leading to its secretion into extracellular space. Hypertens Res. 2011;34:599–605. doi: 10.1038/hr.2010.284. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen G, Blanchard A, Curis E, Bergerot D, Chambon Y, Hirose T, Caumont-Prim A, Tabard SB, Baron S, Frank M, et al. Plasma soluble (pro)renin receptor is independent of plasma renin, prorenin, and aldosterone concentrations but is affected by ethnicity. Hypertension. 2014;63:297–302. doi: 10.1161/HYPERTENSIONAHA.113.02217. [DOI] [PubMed] [Google Scholar]
- 40.Morimoto S, Ando T, Niiyama M, Seki Y, Yoshida N, Watanabe D, Kawakami-Mori F, Kobori H, Nishiyama A, Ichihara A. Serum soluble (pro)renin receptor levels in patients with essential hypertension. Hypertens Res. 2014;37:642–648. doi: 10.1038/hr.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobori H, Alper AB, Shenava R, Katsurada A, Saito T, Ohashi N, Urushihara M, Miyata K, Satou R, Hamm LL, et al. Urinary angiotensinogen as a novel biomarker of the intrarenal renin-angiotensin system status in hypertensive patients. Hypertension. 2009;53:344–350. doi: 10.1161/HYPERTENSIONAHA.108.123802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamada K, Taniguchi Y, Shimamura Y, Inoue K, Ogata K, Ishihara M, Horino T, Fujimoto S, Ohguro T, Yoshimoto Y, et al. Serum level of soluble (pro)renin receptor is modulated in chronic kidney disease. Clin Exp Nephrol. 2013;17:848–856. doi: 10.1007/s10157-013-0803-y. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi K, Yamamoto H, Hirose T, Hiraishi K, Shoji I, Shibasaki A, Kato I, Kaneko K, Sasano H, Satoh F, et al. Expression of (pro)renin receptor in human kidneys with end-stage kidney disease due to diabetic nephropathy. Peptides. 2010;31:1405–1408. doi: 10.1016/j.peptides.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 44.Nishijima T, Tajima K, Takahashi K, Sakurai S. Elevated plasma levels of soluble (pro)renin receptor in patients with obstructive sleep apnea syndrome: association with polysomnographic parameters. Peptides. 2014;56:14–21. doi: 10.1016/j.peptides.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 45.Ichihara A, Sakoda M, Kurauchi-Mito A, Kaneshiro Y, Itoh H. Renin, prorenin and the kidney: a new chapter in an old saga. J Nephrol. 2009;22:306–311. [PubMed] [Google Scholar]
- 46.Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13:767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]