

Abstract
In Haemophilia B (HB) (factor IX (FIX) deficiency), F9 genotype largely determines clinical phenotype. Aimed to characterise Argentine families with HB, this study presents F9 genotype frequencies and their specific FIX inhibitor risk and 10 novel F9 mutations. Ninety-one DNA samples from HB patients and relatives were subjected to a new scheme: a primary screen for large deletions, a secondary screen for point mutations using conformation sensitive gel electrophoresis, DNA-sequencing and bioinformatic analysis.
Our unbiased HB population (N=52)(77% with severe, 11.5% moderate and 11.5% mild HB) showed 32 missense (61.5%) including three novel mutations predicting specific structural/functional defects in silico, 7 nonsense (13.5%)(one novel), 5 large deletions, 4 splice including three novel mutations affecting predicted splicing scores, 3 indels (two novel) and one Leiden mutation.
Our comprehensive HB population included five patients with long-lasting FIX inhibitors: three nonsense (p.E35* (novel), p.R75*, p.W240*) and two entire-F9 deletions. A further patient with an indel (p.A26Rfs*14) developed transient inhibitors.
A case-control analysis, based on our global prevalence of 3.05% for developing inhibitors in HB revealed that missense mutations were associated with a low risk odds ratio (OR) of 0.05 and a prevalence of 0.39%, whereas nonsense and entire-F9 deletions had significantly higher risks (OR 11.0 and 32.7) and prevalence (14.3% and 44.5%, respectively).
Our cost-effective practical approach enabled identification of the causative mutation in all 55 Argentine families with HB, analysis of the molecular pathology of novel F9 defects and determination of mutation-associated FIX inhibitor risks.
Keywords: F9, haemophilia B, mutation, FIX inhibitors, CSGE
INTRODUCTION
Haemophilia B (HB) (OMIM 306900) is an X-linked recessively-inherited coagulopathy caused by defects in the factor IX gene (F9). F9 is located at Xq27.1 and encodes a 2.8 kb mRNA comprising eight exons covering 34 kb (1). F9 encodes a primary polypeptide including a signal peptide, a pro-peptide and a mature polypeptide of 415 amino acids (aa) with four structural domains; Gla, epidermal growth factor (EGF)-1 and EGF-2, which together comprise the light chain (145 aa), and a catalytic heavy chain domain (235 aa, excluding the 35 aa activation peptide). Coagulation factor IX (FIX) is a serine protease, mainly synthesized in the liver via a vitamin K-dependent process. It is activated in plasma by either the factor VIIa/tissue factor complex or factor XIa in a calcium-dependent reaction (2, 3).
HB can be classified by the residual clotting activity of FIX into severe (FIX:C <1 IU/dl), moderate (1-5 IU/dl) and mild (5-40 IU/dl) disease (4). Biochemical and clinical HB severity are closely related to the particular causative mutations which are distributed throughout F9. HB has been associated with a high frequency of point mutations (83%), such as missense, nonsense and splice site mutations, and a smaller amount of small deletions or insertions (indel) (11.6 %) and gross deletions (6%) (5). The molecular and clinical features associated with HB mutations are compiled in a publicly accessible database (http://www.kcl.ac.uk/ip/petergreen/haemBdatabase.html) (6).
Data on the causative mutations in haemophilia is important to understand the patient-specific genotype-phenotype relationship, to predict specific risks associated with response to substitution therapy and to provide reliable information for genetic counseling to the affected family.
This paper presents the characterization of the F9 mutation in the first series of patients with HB in Argentina, which includes 10 novel F9 defects (i.e., three missense and three splicing mutations revealing association with structural/functional disturbances by analysis in silico, two nonsense and two indel associated with frameshifts) and six patients with HB who developed FIX inhibitors (representing virtually all patients with HB and inhibitors). The statistical analysis of the comprehensive series of 55 families with HB recruited countrywide allowed estimation of each F9 genotype inhibitor risk revealing significant increased risks for nonsense and entire F9 deletions and significant decreased risk for missense mutations.
MATERIAL AND METHODS
Studied populations and patient DNA samples
Fifty five apparently unrelated Argentine families affected by HB were studied: 43 families with severe HB (six of them with patients who developed inhibitors against therapeutic FIX), six with moderate HB and six with mild HB. The studied population included 91 individuals (53 probands, 16 proband's mothers and 22 female relatives). In addition, to investigate possible F9 polymorphic variants, 200 chromosomes from a DNA-sample collection from Argentine healthy blood donors were screened. This latter analysis was particularly significant to estimate the potential involvement of new missense, in-frame and intronic sequence variants in the HB phenotype. The study was approved by our Institutional Ethics Committee and written informed consent was obtained from all cases.
Genomic DNA was obtained from Tris/HCl:EDTA (10:1 mM) -washed leukocytes from 5-10 ml of peripheral blood using a standard salting out method (7). DNA sample quality and concentration was estimated by ethidium bromide stained agarose gel electrophoresis (1%).
Molecular protocol for F9 mutation characterisation
All relevant sequences of F9 (the promoter, 8 exons, exon-intron boundaries, splicing sequence consensus and polyadenylation sequence sites) were PCR amplified using a new protocol of 12 amplimers whose sizes, between 334 and 583 bp, and sensitive range were optimized for small mutation screening by conformation sensitive gel electrophoresis (CSGE) (primers listed in the Supplementary Table ST1). PCR reactions were performed using standard thermocycling conditions in a volume of 25 μl with 300 ng of genomic DNA, 200 μM of each dNTP, 0.5 μM of each primer, 0.5 U of Taq DNA polymerase (Promega, Argentina) and buffer provided by the manufacturer (1.5 mM MgCl2).
Product size and identity of each F9 amplimer was analysed by agarose gel electrophoresis (2%) in a primary gross mutation screening. Large deletions (>100 bp) were defined by repeated and consistent absence of specific PCR amplification from contiguous F9 sequences. Each large deletion was confirmed by multiplex PCR experiments of the F9 amplimer along with a control amplimer from an uninvolved autosomal region within the β-globin gene (HBB) (240 bp) as reference (Supplementary Table ST1). Small mutation screening was performed by CSGE as described by Williams et al (1998) and modified by Rossetti et al (2007) (8, 9). Briefly, 5 μl of each PCR product from patients and controls were mixed, incubated for 5 min at 94° C and 30 min at 65° C to form heteroduplex and subjected to high resolution CSGE (41 cm) for 18 hr at 40 Watts. In some cases, pairs of F9 amplimers with different sizes and CSGE behaviours were loaded together on the gel to save time and costs (Supplementary Table ST1). To simplify F9 genotyping of proband's relatives from certain families where heteroduplex patterns showed exceptionally altered high resolution CSGE mobility profiles, F9 genotypes were resolved by medium (17 cm) or low (7 cm) resolution CSGE (Supplementary Fig. SF1). Polyacrylamide gels were silver stained, trans-illuminated by white light and their image digitalised. DNA samples displaying abnormal CSGE profiles relative to that obtained from normal control samples were PCR amplified, electrophoresed, purified by GFX™ Spin columns (Amersham, Argentina) and subjected to either radioactive/manual or fluorescent/automated DNA sequencing using fmol DNA Cycle Sequencing System (Promega, Argentina) and Dyenamic ET terminator Cycle sequencing kit (Amersham, Argentina), respectively.
Bioinformatic and genotype-phenotype analysis
DNA sequence analysis, exon-intron boundary and primer annotations were performed using SeqBuilder 7.1 (DNA Star Lasergene, USA). F9 genomic numbering (g.) follows the legacy notation from Yoshitake et al (1985) (1), while cDNA (c.) and protein (p.) nomenclature follows numbering and recommendations of the Human Genome Variation Society (HGVS) (10) using GenBank entries NM_000133.3 and NP_000124.1, respectively. Human FIX residues (Homo sapiens) (GenBank entry P00740) that involve novel missense defects were examined in silico using the UNIPROT (http://www.uniprot.org) alignment tool for their conservation in orthologous FIX: pig (Sus scorfa) P16293, mouse (Mus musculus) P16294, chicken (Gallus gallus) Q804X6, gorilla (Pan troglodytes) Q95ND7, rat (Rattus norvergicus) P16269 and rabbit (Oryctolagus cuniculus) P16292.
Theoretical 3D-structural analysis of new F9 missense mutations were performed assisted by the Swiss Model Workspace (http://swissmodel.expasy.org/workspace/), the software SWISS PDB Viewer version 4.0 (11), and the atom 3D coordinates deposited on files 1cfhA.pdb, 1edmC.pdb and 3kcgH.pdb to model the Gla domain (residues 47-93), the EGF1 (residues 92-130) and the catalytic domain of FIXa (residues 227-461), respectively.
In order to search and score potential splice site gain or loss in control and mutated F9 sequences, online software tools were applied: NNpredict (www.fruitfly.org/seq_tools/splice.html), NetGene2 (www.cbs.dtu.dk/services/NetGene2), SoftBerry SPL (www.softberry.com) and GeneSplicer (http://www.tigr.org/tdb/GeneSplicer/gene_spl.html) (12-14).
Data analysis and statistics
GraphPad Prism software (version 5.0, GraphPad Software, USA) was used to estimate inhibitor risks, such as mutation present/absent inhibitor odds ratio (IOR) and mutational absolute prevalence (IP). Fisher exact test was applied to study the association of FIX inhibitor development and F9 mutation type in HB. In order to improve the statistical estimation of the F9 mutation type-specific inhibitor risks in our population, only the best assessed parameters were used separately. First, the global inhibitor prevalence (3.05%) was extracted from the World Federation of Hemophilia (WFH) survey 2009 (eight out of 262, which represented all patients with HB in Argentina) (15). Second, the F9 mutation type frequencies (prevalence, P) in HB was estimated using an unbiased HB population (N=52), which was assembled by excluding three patients who were recruited to the Protocol for Mutation Characterisation of Haemophilia Patients with Inhibitors (period 2008-2010). This unbiased population showing the natural F9 mutation frequencies in Argentine patients with HB was recruited as consecutive families affected with HB that consulted for molecular diagnosis and genetic counselling. Third, the F9 mutation type-specific relative risk of inhibitor or inhibitor odds ratio (IOR) was estimated from a comprehensive population (N=55) including all five patients with permanent inhibitors (cases) and 50 without inhibitors (controls).
Absolute inhibitor prevalence of a specific HB mutation (IPMi) was calculated by the formula IPMi = IPHB IOR(Mi+/Mi-) / (1+ PMi/HB IOR(Mi+/Mi-) - PMi/HB), where IPHB is the estimated inhibitor prevalence in HB (i.e., 3.05%), IOR(Mi+/Mi-) is the calculated inhibitor odds ratio of a specific HB mutation type and PMi/HB is the estimated prevalence of a specific HB mutation type in patients with HB (estimated using frequency data from an unbiased population of HB).
RESULTS AND DISCUSSION
Haemophilia B mutation frequencies in the Argentine population
Between the years 2008 and 2010, our laboratory underwent a protocol aimed to characterise the disease causing defects in all Argentine patients with haemophilia A and B and their inhibitors against therapeutic FVIII and FIX, respectively. In order to estimate the natural frequencies of HB causative mutations in our population, the three patients from this protocol with HB and inhibitors registered within this time interval (2008-2010) were not considered for computing mutation frequencies (i.e., cases 33, 38 and 55, Table 1). Consequently, an unbiased population of 52 families affected with HB showed the following mutations; 32 missense (61.5%), seven nonsense (13.5%), four splice site (7.7%), one promoter region nucleotide substitution (Leiden mutation) (1.9%), three small indels associated with frameshifts (5.8%) and five large deletions (9.6%) (Table 1, FIX mutation location on Fig. 1). Forty cases had severe HB (77% of 52 cases), 47.5% were predicted to result in null alleles and 52.5% to result in missense mutations. All six cases with moderate HB phenotype had missense mutations, while five out of six cases with mild HB had missense mutations and the other one had a single nucleotide change in the F9 promoter (Table 1).
Table 1.
F9 mutations in Argentine patients with HB
| Case # |
Gene mutation notation *Legacy (g.); HGVS (c.) |
Protein mutation HGVS (p.) |
**No reports |
FIX Inh. | FIX Activity (%) |
Severity | F/S | Obs. |
|---|---|---|---|---|---|---|---|---|
| HB causative missense mutations | ||||||||
| 1. | g.117G>A; c.88G>A | p.V30I | 5 | N | <1 | S | F | |
| 2. | g.6364C>T; c.127C>T | p.R43W | >50 | N | <1 | S | S | CpG |
| 3. | g.6365G>A; c.128G>A | p.R43Q | >70 | N | <1 | S | F | CpG |
| 4. | g.6397G>C; c.160G>C | p.E54Q | 0 | N | <1 | S | S | g.6709A>G intron 3 |
| 5 | g.10419G>T; c.305G>T | p.C102F | 0 | N | <1 | S | F | |
| 6. | g.10430G>A; c.316G>A | p.G106S | 72 | N | 11 | Mi | S | CpG |
| 7. | g.10458A>G; c.344A>G | p.Y115C | 7 | N | <1 | S | F | |
| 8. | g.17699G>A; c.422G>A | p.C141Y | 5 | N | <1 | S | S | |
| 9. | g.20375G>A; c.533G>A | p.C178Y | 5 | N | <1 | S | S | |
| 10. | g.20413C>T; c.571C>T | p.R191C | 54 | N | 2.5 | Md | S | CpG |
| 11. | g.20519G>A; c.677G>A | p.R226E | 44 | N | <1 | S | S | OC, CpG |
| 12. | g.30076G>A; c.761G>A | p.G254D | 2 | N | <1 | S | S | |
| 13. | g.30112C>T; c.797C>T | p.A266V | 3 | N | 6.6 | Mi | S | |
| 14. | g.30150G>A; c.835G>A | p.A279T | 69 | N | 10 | Mi | F | CpG |
| 15. | g.30864G>A; c.881G>A | p.R294Q | 85 | N | 3.5 | Md | F | CpG |
| 16. | g.30864G>A; c.881G>A | p.R294Q | 85 | N | 12 | Mi | F | CpG |
| 17. | g.31008C>T; c.1025C>T | p.T342M | 125 | N | 3 | Md | F | CpG |
| 18. | g.31051G>T; c.1068G>T | p.W356C | 1 | N | <1 | S | F | OC |
| 19. | g.31112G>T c.1129G>T | p.V377F | 1 | N | <1 | S | S | |
| 20. | g.31119G>A; c.1136G>A | p.R379Q | 63 | N | 6 | Mi | S | CpG |
| 21. | g.31119G>A; c.1136G>A | p.R379Q | 63 | N | 2 | Md | S | CpG |
| 22 | g.31128G>A c.1145G>A | p.C382Y | 5 | N | <1 | S | S | |
| 23. | g.31176G>A; c.1193G>A | p.G398D | 6 | N | <1 | S | S | |
| 24. | g.31176G>A; c.1193G>A | p.G398N | 6 | N | <1 | S | S | |
| 25. | g.31187G>A; c.1204G>A | p.G402R | 6 | N | 2 | Md | S | |
| 26. | g.31217G>T; c.1234G>T | p.G412W | 1 | N | <1 | S | F | |
| 27. | g.31227A>G; c.1244A>G | p.H415R | 2 | N | <1 | S | F | |
| 28. | g.31257T>C c.1274T>C | p.L425S | 0 | N | <1 | S | S | |
| 29. | g.31262G>A; c.1279G>A | p.G427R | 2 | N | <1 | S | S | |
| 30. | g.31274T>C; c.1291T>C | p.W341R | 6 | N | 3 | Md | F | |
| 31. | g.31274T>A; c.1291T>A | p.W341R | 1 | N | <1 | S | F | |
| 32. | g.31282A>G; c.1306G>A | p.A436T | 7 | N | <1 | S | S | |
| HB causative nonsense mutations | ||||||||
| 33. | g.6340G>T; c.103G>T | p.E35* | 0 | + HR | <1 | S | F | |
| 34. | g.6460C>T; c.223C>T | p.R75* | 50 | N | <1 | S | S | CpG |
| 35. | g.6460C>T; c.223C>T | p.R75* | 50 | + HR | <1 | S | S | CpG |
| 36. | g.20377G>T; c.535G>T | p.G179* | 0 | N | <1 | S | S | |
| 37. | g.20497C>T; c.655C>T | p.Q219* | 1 | N | <1 | S | S | |
| 38. | g.20562G>A; c.719G>A | p.W240* | 6 | + HR | <1 | S | S | |
| 39. | g.30875C>T c.892C>T | p.R298* | 48 | N | <1 | S | S | CpG |
| 40. | g.30875C>T c.892C>T | p.R298* | 48 | N | <1 | S | S | CpG |
| 41. | g.31118C>T; c.1135C>T | p.R379* | 46 | N | <1 | S | F | CpG |
| HB associated splicing site mutations | ||||||||
| 42. | g.6491T>G; c.252+2T>G | 0 | N | <1 | S | S | ||
| 43. | g.6491T>C; c.252+2T>C | 0 | N | <1 | S | S | ||
| 44. | g.6676A>G; c.253-2A>G | 1 | N | <1 | S | F | ||
| 45. | g.10506insT; c.391+1insT | 0 | N | <1 | S | F | ||
| HB causative Promoter region mutation | ||||||||
| 46. | g.-6G>A; c.-35G>A | 29 | N | 6 | Mi | S | CpG | |
| HB causative small Ins/Del | ||||||||
| 47. | g.118_119delGTTT; c.88+1_4delGTTT | 5 | N | <1 | S | F | ||
| 48. | g.105_106delGinsAGATATCCTAAA; c.76_77delGinsAGATATCCTAAA | p.A26Rfs*14 | 0 | Transient LR | <1 | S | S | |
| 49. | g.30845_30846delGA; c.862_863delGA | p.E288Tfs*22 | 0 | N | <1 | S | F | |
| HB causative gross deletions | ||||||||
| 50. | Deletion exon 7_8 | S | N | <1 | S | S | ||
| 51. | Deletion exon 6 | p.Ala173_Gly242delfs*5 | S | N | <1 | S | S | |
| 52. | Deletion exon 4 | p.Val92_Asp131del | S | N | <1 | S | F | |
| 53. | Deletion exon 1_8 | S | N | <1 | S | F | ||
| 54. | Deletion exon 1_8 | S | + HR | <1 | S | S | ||
| 55. | Deletion exon 1_8 | S | + HR | <1 | S | S | ||
Fifty five patients (Case #) with haemophilia B whose F9 causative mutation was characterized are shown. Notation (c.) and (p.) corresponds to the recommendations of the HGVS and Goodeve et al (2011)
Number of reports in the HB international database (URL, http://www.kcl.ac.uk/ip/petergreen/Dec-2011).
Inhibitor development is classified in categories (N, without inhibitors; +, long-lasting inhibitors; HR, high response; LR, low response, Transient, inhibitors that disappeared before the sixth month). FIX activity (%). HB phenotype severity (Severity) is classified in three categories: severe (S), moderate (Md) and mild (Mi). F/S indicates the familial (F) or sporadic (S) origin of haemophilia. Obs., observations, CpG, indicates a point mutation occurring at the hypermutable CpG dinucleotide, and OC, obligate carrier.
Figure 1. Spectrum and location of HB causative mutations.
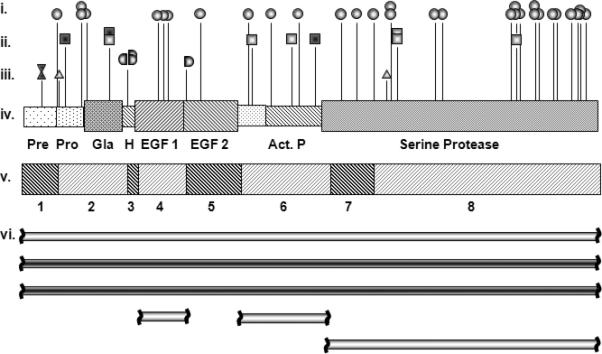
(i) Missense mutations (circles); (ii) nonsense mutations (squares); (iii) splice mutations (donor or acceptor right or left handled semicircles, respectively), small deletions (triangle), small insertions (inverted triangle). A dark center indicates association with inhibitors and light center, absence of inhibitors; (iv) scheme of FIX protein in which each domain is indicated: Pre: pre-peptide, Pro: pro-peptide, Gla: γ-carboxy glutamic domain, H: hydrophobic domain, EGF-1 and -2: epidermal growth factor 1 and 2, respectively, Act. P: activation peptide and Serine Protease: catalytic domain. (v) F9 scheme including exons 1-8; (vi) large deletions are represented as horizontal bars: three entire F9 deletion (exons 1-8), two deletions of a single exon (exon 4 and exon 6) and one two-exon deletion (exons 7-8).
FIX inhibitor risks in haemophilia B
A global survey from the World Federation of Hemophilia in 2009, indicated that Argentina recorded 262 patients with HB including eight (3.05%) who developed FIX inhibitors (15). The literature has shown that the incidence of inhibitor development in HB ranges from 1.4-11.7% in full series, and 3.7-24% when only severe HB cases were considered, depending on the study group (Table 2). In agreement with these international figures, our unbiased series of patients with HB, which are suitable for comparison with other series, showed FIX inhibitor prevalence of 3.8% (2/52) for all patients and 5% (2/40) for severe cases, respectively. The comprehensive series includes six patients with severe HB who developed FIX inhibitors. Three of them showed nonsense mutations, p.E35* and p.R75* affecting exon 2; and p.W240* affecting exon 6. The latter two mutations (p.R75*; p.W240*) were reported in the HB database and both were occasionally associated with inhibitors, whereas p.E35* in the FIX pro-peptide has not been previously reported (Table 1 cases: 33, 35 and 38, respectively, Fig. 1). A recent paper addressed the expression of FIX in HB patients with nonsense mutations and found evidence of trace levels of full length FIX even in patients with premature stop codons by ribosome readthrough (24). Additionally, they also demonstrated the involvement of nonsense mediated decay (NMD) for mutations located within upstream exons (e.g., p.L103* in exon 3) and absence of NMD for nonsense in the last F9 exon (p.R294* and p.R298* in exon 8) (24).
Table 2.
Inhibitor development in international patients with HB
| Series | Cases with inhibitors/ Total | Cases with inhibitors/ severe cases | HB mutations associated with inhibitor (N) | Origin | Reference |
|---|---|---|---|---|---|
| 1 | 6/217 (2.8%) | 6/68 (8.8%) | Large F9 deletion (4) Small indel (1) Nonsense (1) |
UK | Saad et al, 1994 (16) |
| 2 | 7/62 (11.3%) | 7/30 (23.3%) | Total F9 deletion (2) Partial F9 deletion (2) Nonsense (3) |
Sweden | Lung et al, 1995 (17) |
| 3 | 1/15 (6.6%) | 1/7 (14.3%) | Total F9 deletion (1) | France | Parquet et al, 1999 (18) |
| 4 | 9/77 (11.7%) | 9/38 (24%) | Total F9 deletion (2) Partial F9 deletion (1) Small F9 deletion (2) Nonsense (4) |
Sweden | Ljung et al, 2001 (19) |
| 5 | 8/236 (3.4%) | 8/152 (5.3%) | Large F9 deletion (3) Nonsense (5) |
Italy | Belvini et al, 2005 (20) |
| 6 | 8/302 (2.6%) | 8/211 (4%) | Large F9 deletion (3) Nonsense (5) |
Italy | Tagariello et al, 2007 (21) |
| 7 | 1/33 (3%) | 1/27 (3.7%) | Large F9 deletion (1) | Korea | Kwon et al, 2008 (22) |
| 8 | 2/144 (1.4%) | 2/47 (4.2%) | Large F9 deletion (2) | USA | Miller et al, 2012 (23) |
It is known that specific protein complexes are deposited on mRNAs 20-25 nucleotides upstream of exon junctions (EJC), which should be physiologically removed by the readthrough of the translational machinery. In mRNAs bearing premature stop codons (i.e., those located upstream of EJC) all EJC are not removed and therefore might trigger NMD, whereas termination codons located in the 3’ terminal exon are generally interpreted as proper stop codons and do not trigger NMD (reviewed in 25). These findings allowed us to suggest that due to their location our three nonsense mutations with FIX inhibitors (two in exon 2 and one in exon 6) may undergo NMD not mitigated by ribosome read-through thus representing genuine null mutations. Moreover, none of our three patients with nonsense mutations in F9 exon 8 developed inhibitors (two patients with p.298* and one patient with p.R379*, cases 39, 40 and 41, Table 1). This reasoning relies on the notion that traces of residual FIX reduce the risks of inhibitor development in patients with HB.
Two additional cases with FIX inhibitors had large deletions spanning the entire F9 gene (Table 1 cases 54 and 55, Supplementary Fig. SF1) confirming the highest inhibitor risk for this type of severe FIX null mutations as was originally reported (26). All five cases developed persistent high responding FIX inhibitors (present >6 months, historical peak >5 Bethesda units (BU)). In contrast, a novel indel frameshift (p.A26Rfs*14) in exon 1 (Fig. 1, case 48 in Table 1) had transient low-responding inhibitors. In agreement with predictions from the literature on HB mutations, we did not find HB cases with inhibitors associated with splicing defects or missense mutations (27).
The identification of the causative mutation in our series of patients with HB including all registered cases with FIX inhibitors allowed us to make estimations about relative and absolute risks of inhibitor development by HB causative F9 mutation type by the combined use of the unbiased HB population frequencies of F9 mutation types, the absolute inhibitor prevalence in HB (3.05%) and the relative risks (IOR) of each mutation type (present/absent) versus inhibitor status (yes/no) applying a case-control analysis on a comprehensive series of 55 Argentine patients with HB. Table 3 shows estimations of the F9 mutational odds ratio IOR and absolute prevalence IP for developing FIX inhibitors. Noteworthy, missense mutations presented a very significantly low IOR of 0.05 (CI 0.0027-0.9902) and an IP of 0.4% (CI 0.02-3.04%), whereas nonsense mutations presented a significantly high risk IOR of 11.0 (CI 1.5-79.9) and an IP of 14.3% (CI 4.3-20.3%)(Table 3). Splice site mutations, frameshifts and the Leiden F9 promoter mutation revealed the same non-significant IOR of 0.94 accompanied by extremely wide confidence intervals encompassing unity (null hypothesis).
Table 3.
FIX inhibitor risks in Argentine patients with HB.
| Mutation Type | HB1 [%] | Cases2 N=5 | Controls3 N=50 | IOR4(CI95) | IP5(CI95) [%] | P value6 |
|---|---|---|---|---|---|---|
| Missense | 61.5 | 0 | 32 | 0.05(0.003-0.99) | 0.4(0.02-3.04) | 0.0097** |
| Splicing defect | 7.7 | 0 | 4 | 0.94(0.04-19.9) | 2.9(0.15-24.8) | 1.0000 |
| Frameshift indel & Leiden | 7.7 | 0 | 4 | 0.94(0.04-19.9) | 2.9(0.15-24.8) | 1.0000 |
| Nonsense | 13.5 | 3 | 6 | 11(1.51-79.88) | 14.3(4.3-21) | 0.0267* |
| All-large deletions | 9.6 | 2 | 4 | 7.67(0.97-60.2) | 14.3(3.0-27.5) | 0.0864 |
| Entire F9 deletions | 3.8 | 2 | 1 | 32.7(2.26-471) | 45.0(6.6-75.4) | 0.0194* |
HB [%]: Artificially made unbiased HB population excluding three patients of the Inhibitor Molecular Protocol to estimate the natural mutation type frequencies in Argentine patients with HB.
Cases: Long-lasting inhibitor positive cases (high and low responders).
Controls: Inhibitor negative or transient.
IOR: Inhibitor Likelihood Odds Radio (Mutation positive/Mutation negative), (CI95): Confidence Interval 95%.
IP [%]: Absolute inhibitor prevalence, (CI95): Confidence interval 95%. Null hypothesis is 3.05%, which represents the global HB inhibitor prevalence in Argentina.
P values: Fisher exact test P value
P<0.05 significant
P<0.001 highly significant.
The group of large deletions showed non-significant differences for inhibitor development (P = 0.086, Table 3) although it included three deletions of the entire gene (two of them associated with inhibitors) and three large partial deletions without inhibitors, which showed similar inhibitor risks to those obtained for F9 nonsense mutations. However, if only deletions spanning the entire F9 were computed as a single group, significantly increased risks were obtained (P = 0.019), i.e., an IOR of 32.7 (CI 2.3-471.3) and an IP of 45.0% (CI 6.6-75.4%) (Table 3).
The observation of HB cases with equal clinical severity caused by the same causative mutation and different inhibitor status highlighted the fact that there are still additional risk factors for inhibitor development to reveal (genetic and/or environmental factors). Our series showed two patients with the same nonsense mutation (i.e., p.R75*) (cases 34 and 35) and three patients with deletions of the entire F9 (cases: 53, 54 and 55, Table 1), who showed discordant inhibitor status.
It has been observed that a subgroup of patients with severe HB caused by deletion of the entire F9 gene and particular nonsense mutations developed inhibitors and anaphylaxis, suggesting the association of these two clinical signs in response to therapeutic exogenous FIX (28, 29). Although one of our patients with severe HB due to an entire F9 deletion mutation developed high responding (HR) FIX inhibitory antibodies along with a strong allergic response during his early treatment, it was not associated with anaphylaxis (case 54, Table 1). Our other patient with severe HB and HR inhibitors caused by a nonsense mutation (p.R75*) suffered strong repeated infections in his knee arthropathies (case 35, Table 1). However, these immunological and clinical features were not specifically associated with FIX inhibitor development in HB as these features also affected other patients with severe HB without inhibitors (data not shown).
Molecular analysis shortly after laboratory diagnosis of severe HB may identify children at risk of developing inhibitors. The close monitoring of these patients at risk of anaphylaxis and allergic reactions, associated with entire F9 deletions or nonsense mutations, during early replacement therapy with FIX products enables clinicians to apply appropriate treatment when a first indication of life-threatening emergency happens.
Spectrum of Haemophilia B-causative F9 defects and polymorphic variants
Missense mutations
Our group of 32 unrelated cases with missense mutations included three novel single nucleotide changes associated with severe HB: c.160G>C in exon 2 predicting p.E54Q in the Gla domain, c.305G>T in exon 4 predicting p.C102F in the EGF-1 domain and c.1274T>C in exon 8 predicting p.L425S in the serine-protease catalytic domain of mature FIX (cases 4, 5 and 28, respectively, Table 1). Sixty five % (21/32) of the missense mutations were associated with clinical and biochemical levels of severe HB, 19% (6/32) moderate and 16% (5/32) mild HB. The fact that all three novel mutations associated with severe disease agrees with the high turnover (gain and loss) rates observed among severe disease for X-linked recessive disorders (each severe mutation presents a low persistence through the generations), in contrast to moderate or mild disease (Haldane's rule) (30). Consequently, severe HB mutations tend not to be restricted to particular ethnic or geographic groups. Therefore, Argentine novel severe HB mutations may also appear in other parts of the world as a consequence of a general pathological mechanism.
Missense mutations were distributed in almost all of the FIX domains (Table 1 and Fig. 1). None of the 32 patients had FIX inhibitors.
In order to estimate the potential consequences of the residue changes resulting from novel missense mutations, the human FIX sequence was aligned with its orthologues. The analysis showed that all three novel missense changes involved a highly conserved amino acid (Supplementary Fig. SF2). Furthermore, these missense changes were not observed in 200 X-chromosomes from the Argentine healthy population reducing the possibility of prevalent HB-neutral single nucleotide polymorphisms in our population.
The potential structural and functional changes associated with our novel FIX missense mutations were analysed using 3D structural analysis software. However, it is advisable to exercise caution about the possible functional consequences predicted by this theoretical approach based on the data of FIX activity and clinical severity and without FIX antigen data to rule out, for example, intracellular degradation of mutated FIX, reduced protein secretion, instability in the circulation of a correctly secreted product, or possible pleiotropic effects of the mutation on protein biology affecting both secretion and FIX function.
Missense mutation p.E54Q is a non-conservative change (negative polar to neutral polar residue) associated with severe HB and was predicted to introduce modifications in the hydrogen bond network of the Gla domain and more importantly, it may impede gamma carboxylation of both Glu54 and Glu53 (as it is predicted to be involved in a putative hydrogen bond with Gln54) (Supplementary Fig. SF3A). These are the first and second Glu residues of a series of 12 glutamic acid residues (Supplementary Fig. SF2A) that should be modified for proper interaction between the FIX Gla-domain and the platelet surface, which is essential for tenase complex activity.
Missense change p.C102F was predicted to disrupt the disulfide bridge between Cys102 and Cys117, which is part of a triad of disulfide bonds (i.e., Cys97-Cys108, Cys102-Cys117 and Cys119-Cys128) that maintain the structural scaffold of the FIXa EGF-1 domain, thus potentially compromising its function (Supplementary Fig. SF2B). The HB database reports other missense defects affecting the same FIX residues Glu54 and Cys102 (p.E54G and p.C102R), both associated with a severe phenotype, indicating the important roles of these residues for FIX activity.
Other unreported missense mutation affecting a previously uninvolved residue, p.L425S, was hypothesised to distort a key region of the catalytic domain that holds in place Ser411, one of the three amino acids of the catalytic triad (Ser411, His267 and Asp315) essential for the function of serine proteases by means of a subtle mechanism. This non-conservative missense change at a highly conserved residue, Leu425 was predicted by in silico 3D modelling (validated in Supplementary Fig. SF4A) to affect neither the hydrogen bond network nor the molecular surface at the FIX catalytic domain (Supplementary Fig. SF3C). Moreover, Leu425 is deeply buried in the catalytic domain and is predicted to interact with four residues Ile334, Pro414, Val263 and Val447 forming a hydrophobic core that allows the normal β-sheet structure of the center of the catalytic domain, to hold in place the catalytic triad (Supplementary Fig. SF4B). In missense p.L425S, the putative replacement of Leu (a large branched aliphatic residue) by Ser (a small polar residue) may disturb this hydrophobic core by introducing an alcohol group thus preventing these hydrophobic interactions and particularly disturbing the β-sheet loop delimited by Cys407 and Cys435 that holds in place Ser411 (Supplementary Fig. SF4) (1). Our hypothesis is that p.L425S may introduce a structural instability in loop Cys407-Cys435 in the catalytic domain impacting on the protease activity of FIXa. In addition to the presented theoretical structural analysis of these novel missense mutations, alternative explanations to support the severe HB phenotype, such as defective secretion of FIX from the hepatocyte or correct secretion but reduced stability in plasma, cannot be excluded without FIX antigen data.
Although most of our spectrum of patients affected by F9 missense mutations presented similar clinical and laboratory severities to those reported on the HB database, two out of 18 severe defects (20%), two out of six moderate and four out of six mild (67%) escaped this rule. This increasing inconsistency observed in milder phenotypes might be associated with the wide diversity of missense changes and their dependence on subtle differences in their molecular interactions, which become particularly prevalent in non-severe structurally-conserved missense mutations and also upon inter-laboratory differences in determining FIX level.
Nonsense mutations
Two of the nine patients with HB and nonsense mutations had novel single nucleotide changes: c.103G>T within exon 2 (p.E35*), and c.535G>T exon 6 (p.G179*) (cases 33 and 36, respectively, Table 1).
In agreement with unreported point mutations leading to missense and nonsense mutations, none of our new single nucleotide changes involved a CpG dinucleotide and all of them corresponded to nucleotide transversions (6).
Intronic point mutations
Four patients showed intronic point mutations affecting splicing consensus sequences, all of them with clinical features of severe disease, but without FIX inhibitors (Table 1). Three were novel mutations: two cases had nucleotide substitutions both affecting the intron 2 donor splice site; c.252+2T>G and c.252+2T>C (cases 42, 43, respectively, Table 1), and one case had a single nucleotide insertion at the intron 4 donor splicing site, c.391+3insT (case 45, Table 1). The impact of the mutations were analysed by bioinformatics tools that score and predict splicing sites. Most of these tools recognised the F9 wild type splicing sites with high scores, indicating that the analysis was informative (4/4 for c.252+2T>G and c.252+2T>C, and 3/4 for c.391+3intT, Supplementary Fig. SF5), whereas all mutated sequences presented score values below the software detection thresholds (Supplementary Fig. SF5). Moreover, mutations c.252+2T>G and c.252+2T>C were unequivocally associated with splicing defects as any mutation at the conserved dinucleotide GT (+1/+2) of donor splice sites have been experimentally proven to be deleterious to splicing function (14).
Although the direct experimental determination of splicing defects is the gold standard to link genotype and phenotype, the computational analysis has proven to be valuable to achieve accurate predictions when patient's RNA is not available (31).
Nucleotide mutations in 5’ untranslated regions
Our series included a young patient (seven years) with mild HB, who had a well-known nucleotide transition in the F9 promoter region, c.-35G>A (g.-6G>A). This specific mutation has been reported in HB Leiden and predicts a hormonal-dependent increase in factor IX levels after puberty associated with an important improvement in patient's clinical features, normalization that is predicted to be more significant than for other mutations in this region of the F9 promoter (20, 32, 33).
Large F9 deletions
Our series comprised six cases with large F9 deletions, all of them associated with severe disease. Three cases showed a consistent absence of all F9 amplimers (cases 53, 54 and 55), case 50 showed an absence of four adjacent F9 amplimers (exons 7-8 plus the poly-A consensus site) (Table 1) and two cases showed an absence of one exon (exon 6 and 4, in cases 51 and 52, respectively, Table 1, Fig. 1). In all cases, large F9 deletions were analysed using multiplex-PCR amplification of a F9 specific amplimer along with a HBB specific amplimer, as an internal control.
The deletion of exon 4 predicted an in-frame shortened protein (c.276_392del; p.Val92_Asp131del) with loss of all EGF-1 domain residues. These are essential for the interaction between FIX-FVIIIa and FIX-FX, and for FIX activation by FVIIIa and tissue factor (34). The exon 6 deletion predicted a frameshift introducing a premature stop codon (c.520_724del; p.Ala173_Gly242delfs*5).
Small indel mutations
Two unreported small indel were found. The first was identified in a severe HB patient with a low responding transient inhibitor, whose indel predicted a frameshift in exon 1, c.76delins12; p.A26Rfs*14 (case 48). A 2 bp deletion mutation in exon 8, c.862delGA; p.E288Tfs*22 was found in a severe HB patient without inhibitors (case 49, Table 1).
This work presents the first molecular series of HB causative mutations in Argentina. It applied a cost effective laboratory algorithm for F9 genotyping adapted to the characteristics of a developing country that started with detection of gross deletions by multiplex PCR amplification, followed by rapid small mutation screening based on high resolution CSGE, and concluded by characterization of the specific HB-causative mutation by DNA sequencing. These procedures enabled provision of reliable information for genetic counselling in 55 HB families.
Our series of 55 families showed 34 cases (62%) of sporadic HB (27 severe and 7 moderate/mild) and 21 cases (38%) showed familial HB (16 severe and 5 moderate/mild) (Table 1). Among 13 studied proband's mothers (38%) from families with sporadic HB, five were negative for the causative mutation in peripheral blood leukocytes. All these five mutations (i.e., two missense: c.127C>T, c.1306G>A, two nonsense: c.535G>T, c.655C>T, one splice defect: c.252+2T>C) were associated with severe disease and originated from either a de novo mutation affecting a single maternal germ cell or from germinal mosaicism involving partially or totally the female gonad. A former study from Ketterling et al (1993) revealed the parental origin of HB mutations: 25 out of 39 cases occurred in the female germ line and 18 in the male germ line indicating a male/female ratio of 3.8-fold (p<0.002) taking into account the number of X-chromosomes involved by Bayesian analysis (35). Our high proportion (38%) of peripheral blood mutation-negative mothers revealed that, at least in our series of HB mutations, de novo mutations were not associated with the same extent of male bias as F8 inversions in HA, where virtually all proband's mothers were carriers (>98% in our series of F8 inversions and previous reports) (36).
The data on the molecular pathology of HB presented here constitute a clear example of how the use of freely available bioinformatic tools to provide rational explanations of clinical characteristics and, perhaps more importantly to help in making adjusted predictions about the potential outcomes associated with specific F9 genotypes.
Supplementary Material
Acknowledgements
We wish to thank patients, relatives and the haemophilia care staff; and the scientists Derrick Bowen, Amy Curto and Eduardo Tizzano for their kind help in different phases of the work.
Funding: This study was supported by grants from the René Barón Foundation, the Alberto J. Roemmers Foundation, the Florencio Fiorini Foundation, Novo Nordisk Argentina, the Academia Nacional de Medicina de Buenos Aires, the National Research Council (CONICET), the National Agency for Science and Technology Promotion (ANPCyT) and the Word Federation of Hemophilia.
Footnotes
Disclosures
The authors reported no potential conflicts of interest.
REFERENCES
- 1.Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry. 1985;24:3736–50. doi: 10.1021/bi00335a049. [DOI] [PubMed] [Google Scholar]
- 2.Di Scipio RG, Kurachi K, Davie EW. Activation of human factor IX (Christmas factor). J Clin Invest. 1978;61:1528–38. doi: 10.1172/JCI109073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anson DS, Choo KH, Rees DJ, et al. The gene structure of human anti-haemophilic factor IX. EMBO J. 1984;3:1053–60. doi: 10.1002/j.1460-2075.1984.tb01926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker I, Pai M, Akabutu J, et al. The Canadian hemophilia registry as the basis for a national system for monitoring the use of the factor concentrates. Transfusion. 1995;35:548–51. doi: 10.1046/j.1537-2995.1995.35795357875.x. [DOI] [PubMed] [Google Scholar]
- 5.Oldenburg J, Schroder J, Brackmann HH, et al. Environmental and genetic factors influencing inhibitor development. Semin Hematol. 2004;41:82–8. doi: 10.1053/j.seminhematol.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 6.Giannelli F, Green PM, Sommer SS, et al. Haemophilia B: database of point mutations and short additions and deletions--eighth edition. Nucleic Acids Res. 1998;26:265–8. doi: 10.1093/nar/26.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lahiri DK, Nuremberg JI. A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 1991;19:5444. doi: 10.1093/nar/19.19.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams IJ, Abuzenadah A, Winship PR, et al. Precise carrier diagnosis in families with haemophilia A: use of conformation sensitive gel electrophoresis for mutation screening and polymorphism analysis. Thromb Haemost. 1998;79:723–6. [PubMed] [Google Scholar]
- 9.Rossetti LC, Radic CP, Candela M, et al. Sixteen novel hemophilia A causative mutations in the first Argentinian series of severe molecular defects. Haematologica. 2007;92:842–5. doi: 10.3324/haematol.11112. [DOI] [PubMed] [Google Scholar]
- 10.Goodeve AC, Reitsma PH, McVey JH. Working Group on Nomenclature of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Nomenclature of genetic variants in hemostasis. J Thromb Haemost. 2011;9:852–5. doi: 10.1111/j.1538-7836.2011.04191.x. [DOI] [PubMed] [Google Scholar]
- 11.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 12.Pertea M, Lin X, Salzberg S. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001;29:1185–90. doi: 10.1093/nar/29.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vorechovský I. Aberrant 3' splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2006;34:4630–41. doi: 10.1093/nar/gkl535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buratti E, Chivers M, Královic̆ová J, et al. Aberrant 50 splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007;35:4250–63. doi: 10.1093/nar/gkm402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.World Federation of Hemophilia . Report on the annual global survey 2009. René Lévesque Boulevard West; Montreal, Quebec H3G1T7: 2009. pp. 15–19. [Google Scholar]
- 16.Saad S, Rowley G, Tagliavacca L, et al. First report on UK database of haemophilia B mutations and pedigrees. Thromb Haemost. 1994;71:563–70. [PubMed] [Google Scholar]
- 17.Ljung RCR. Gene mutations and inhibitor formation in patients with hemophilia B. Acta Haematologica. 1995;94:49–52. doi: 10.1159/000204029. [DOI] [PubMed] [Google Scholar]
- 18.Parquet A, Laurian Y, Rothschild C, et al. Incidence of factor IX inhibitor development in severe haemophilia B patients treated with only one brand of high purity plasma derived factor IX concentrate. Thromb Haemost. 1999;82:1247–9. [PubMed] [Google Scholar]
- 19.Ljung R, Petrini P, Tengborn L, Sjörin E. Haemophilia B mutations in Sweden: a population-based study of mutational heterogeneity. Br J Haematol. 2001;113:81–6. doi: 10.1046/j.1365-2141.2001.02759.x. [DOI] [PubMed] [Google Scholar]
- 20.Belvini D, Salviato R, Radossi P, et al. Molecular genotyping of the Italian cohort of patients with haemophilia B. Haematologica. 2005;90:635–42. [PubMed] [Google Scholar]
- 21.Tagariello G, Belvini D, Salviato R, et al. The Italian haemophilia B mutation database: a tool for genetic counselling, carrier and prenatal diagnosis. Blood Transfus. 2007;5:158–63. doi: 10.2450/2007.0024-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwon MJ, Yoo KY, Kim HJ, Kim SH. Identification of mutations in the F9 gene including exon deletion by multiplex ligation-dependent probe amplification in 33 unrelated Korean patients with haemophilia B. Haemophilia. 2008;14:1069–75. doi: 10.1111/j.1365-2516.2008.01796.x. [DOI] [PubMed] [Google Scholar]
- 23.Miller CH, Benson J, Ellingsen D, et al. F8 and F9 mutations in US haemophilia patients: correlation with history of inhibitor and race/ethnicity. Haemophilia. 2012;18:375–82. doi: 10.1111/j.1365-2516.2011.02700.x. [DOI] [PubMed] [Google Scholar]
- 24.Pinotti M, Caruso P, Canella A, et al. Ribosome Readthrough Accounts for Secreted Full-Length Factor IX in Hemophilia B Patients with NonsenseMutations. Hum Mutat. 2012 May 22; doi: 10.1002/humu.22120. doi: 10.1002/humu.22120. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 25.Bhuvanagiri M, Schlitter AM, Hentze MW, et al. NMD: RNA biology meets human genetic medicine. Biochem. J. 2010;430:365–77. doi: 10.1042/BJ20100699. [DOI] [PubMed] [Google Scholar]
- 26.Giannelli F, Choo KH, Rees DJ, et al. Gene deletions in patients with haemophilia B and anti-factor IX antibodies. Nature. 1983;303:181–2. doi: 10.1038/303181a0. [DOI] [PubMed] [Google Scholar]
- 27.Lillicrap D. The molecular basis of haemophilia B. Haemophilia. 1998;4:350–7. doi: 10.1046/j.1365-2516.1998.440350.x. [DOI] [PubMed] [Google Scholar]
- 28.Warrier I, Ewenstein BM, Koerper MA, et al. Factor IX inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol. 1997;19:23–7. doi: 10.1097/00043426-199701000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Tagariello G, Belvini D, Salviato R, et al. The Italian haemophilia B mutation database: a tool for genetic counselling, carrier detection and prenatal diagnosis. Blood Transfus. 2007;5:158–63. doi: 10.2450/2007.0024-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogel F, Motulsky AG. Human Genetics. Problems and Approaches. 3rd ed. Springer-Verlag; Berlin: 1997. [Google Scholar]
- 31.Hartmann l, Theiss S, Niederacher D, Schaal H. Diagnostics of pathogenic splicing mutations: does bioinformatics cover all bases? Front Biosci. 2008;13:3252–72. doi: 10.2741/2924. [DOI] [PubMed] [Google Scholar]
- 32.Morgan GE, Figueiredo MS, Winship PR, et al. The high frecuency of the -6G>A factor IX promoter mutation is the result both of a founder effect and recurrent mutation at a CpG dinucleotide. Br J Haematol. 1995;89:672–4. doi: 10.1111/j.1365-2141.1995.tb08388.x. [DOI] [PubMed] [Google Scholar]
- 33.Vidaud D, Tartary M, Costa JM, et al. Nucleotide substitutions at the -6 position in the promoter region of the factor IX gene result in different severity of hemophilia B Leyden: Consequences for genetic counseling. Hum Genet. 1993;91:241–4. doi: 10.1007/BF00218264. [DOI] [PubMed] [Google Scholar]
- 34.Lenting PJ, Christophe OD, Maat H, et al. Ca2+ binding to the first epidermal growth factor-like domain of human blood coagulation factor IX promotes enzyme activity and factor VIII light chain binding. J Biol Chem. 1996;271:25332–7. doi: 10.1074/jbc.271.41.25332. [DOI] [PubMed] [Google Scholar]
- 35.Ketterling RP, Vielhaber E, Bottema CDK, et al. Germ-Line Origins of Mutation in Families with Hemophilia B: The Sex Ratio Varies with the Type of Mutation. Am J Hum Genet. 1993;52:152–66. [PMC free article] [PubMed] [Google Scholar]
- 36.Tizzano EF, Domènech M, Baiget M. Inversion of intron 22 in isolated cases of severe hemophilia A. Thromb Haemost. 1995;73:6–9. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.