

Abstract
Kit is a membrane-bound tyrosine kinase and receptor for stem cell factor (SCF) with a crucial role in hematopoiesis. Mutations of KIT occur in almost half of patients with core-binding factor leukemias where they have been associated with worse outcome. Development of new compounds targeting Kit may therefore hold promise for therapy. We investigated the activity and mechanism of action of APcK110, a novel Kit inhibitor, in the mastocytosis cell line HMC1.2 (KITV560G and KITD816V), AML lines OCIM2 and OCI/AML3 (both wild-type) and primary samples from patients with AML. We demonstrate that: (1) APcK110 inhibits proliferation of the mastocytosis cell line HMC1.2 and the SCF-responsive cell line OCI/AML3 in a dose dependent manner; (2) APcK110 is a more potent inhibitor of OCI/AML3 proliferation than the clinically used Kit inhibitors imatinib and dasatinib and at least as potent as cytarabine; (3) APcK110 inhibits the phosphorylation of Kit, Stat 3, Stat5, and Akt, in a dose-dependent fashion demonstrating activity of APcK110 on Kit and its downstream signaling pathways; (4) APcK110 induces apoptosis by cleavage of caspase 3 and PARP; (5) APcK110 inhibits proliferation of primary AML blasts in a clonogenic assay, but does not affect proliferation of normal colony-forming cells. Although APcK110 activity may partly depend on cytokine responsiveness (e.g. SCF) and not exclusively KIT mutation status, it remains a potent inhibitor of AML and mastocytosis cell lines and primary AML samples. APcK110 and similar compounds should be evaluated in clinical trials of patients with AML.
Keywords: Acute myeloid leukemia, c-Kit, targeted therapy, APcK110
Introduction
Acute myeloid leukemia (AML) is as a group of clonal diseases of hematopoietic progenitor cells. Non-random cytogenetic abnormalities occur in more than 50% of diagnostic samples of patients with adult AML and a number of molecular abnormalities have been identified that have shed further light on AML subtypes (1–3). Besides providing insights into the biology and pathophysiology of AML, cytogenetic and molecular genetic aberrations provide important independent prognostic information and in some cases determine treatment decisions (e.g. responsiveness of acute promyelocytic leukemia [APL] to all trans-retinoic acid [ATRA]; sensitivity of core binding factor [CBF] leukemias to cytarabine) (4). Despite progress in this area, possibilities at treating AML patients in a subtype-specific manner remain limited as molecular profiling is still inadequate and targeted drug therapies are often elusive.
Protein kinases play a crucial role in both normal and malignant cells and require tight control and regulation. Kit is a type III receptor protein tyrosine kinase and the transmembrane receptor for stem cell factor (SCF) (5,6). Binding of SCF to Kit leads to receptor dimerization, phosphorylation and activation of downstream intracellular signaling pathways involved in proliferation and survival (7). Gain-of-function mutations of KIT leading to ligand-independent activation have been described in some solid tumors as well as patients with acute myeloid leukemias (8–11). Although activating KIT mutations are rare in unselected patients with AML, they have been reported with frequencies of between 17% and 46.1% in core binding factor (CBF) leukemias such as those carrying translocations t(8;21), t(16;16)(p13;q22) and inv(16)(p13q22) (12–13).
Several recent studies of adults with inv(16) and t(8;21) AML have shown a negative prognostic impact of KIT mutations at codon 816 with respect to relapse rate and overall survival (14–16). Identification of KIT mutations however is not only important for prediction of outcome, but also for therapeutic decisions. Responses with receptor tyrosine kinase inhibitors in clinical studies have been reported and more specific Kit inhibitors may hold further promise for AML therapy. In the present study we have been evaluating the activity and possible mechanism of action of APcK110, a novel Kit inhibitor (17).
Materials and Methods
Drug
APcK110 [4-{7-(3,5-Dimethyloxyphenyl)-1H-pyrazolo[3,4-b]pyridin-6-yl}-4-fluoro]benzene (Fig. 1A) and imatinib mesylate were synthesized in Dr. Bornmann’s laboratory (Experimental Diagnostic Imaging, The University of Texas M.D. Anderson Cancer Center, Houston, TX) (17). Dasatinib was kindly provided by Dr. Francis Lee (Bristol-Myers Squibb, Princton, NJ). All drugs were dissolved in phosphate-buffered saline (PBS; GIBCO BRL, Grand Island, NY) to stock solutions at a final concentration of 50 mM.
Figure 1.
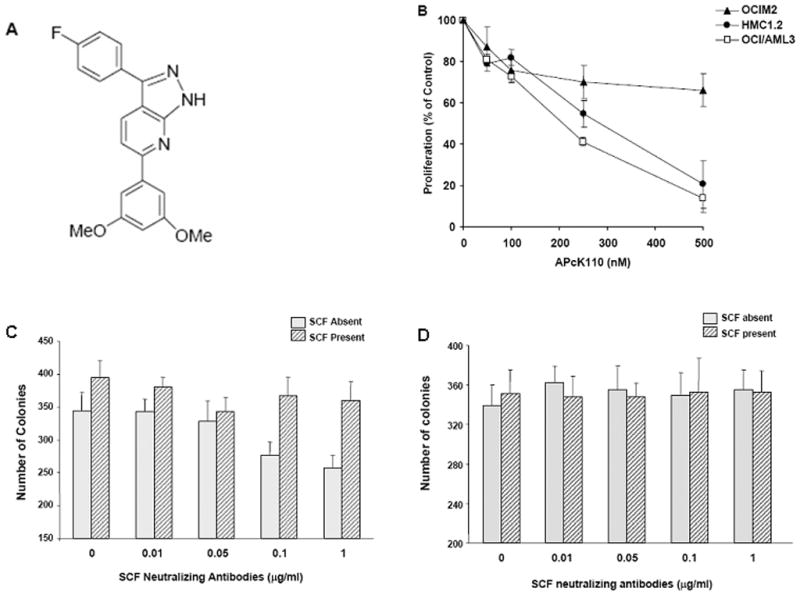
A) Structure of APcK110 [4-{7-(3,5-Dimethyloxyphenyl)-1H-pyrazolo[3,4-b]pyridin-6-yl}-4-fluoro]benzene; B) MTT assay demonstrates inhibition of proliferation of OCI/AML3, and HMC1.2 cells by increasing concentrations of APcK110. OCMM2 cells are marginally affected; C) Effect of SCF-neutralizing antibodies (at concentrations ranging from 0.01 to 1 μg/mL) on OCI/AML3 colony-forming capacity. The figure demonstrates significant stimulation by exogenous SCF (P = 0.043 by Mann-Whitney test) and inhibition of OCI/AML3 colony proliferation (P = 0.029 by Mann-Whitney test at a SCF neutralizing antibody concentrations of 0.1 and 1.0) as well as complete reversal of the inhibition by exogenous SCF; D) Effect of SCF-neutralizing antibodies on OCIM2 cells. SCF and SCF neutralizing antibodies do not affect OCIM2 cell proliferation.
APcK110 Kinase screening
A primary high-throughput screening of APcK110 at 10 μmol/L was conducted by Ambit Biosciences (San Diego, CA) against a T7-bacteriophage library displaying 240 human kinases, using imatinib screening as control. A rough estimation of the binding constant (Kd_1) for each assay was provided by the single-hit value in the primary screen at a single compound concentration. Kinase profiling was done using a bacteriophage library displaying fused human kinases that may attach at the ATP site to a fixed-ligand matrix, which, in turn, may be competitively displaced from binding by the tested compound. As expected, APcK110 demonstrated inhibition of Kit and KIT (D816V) mutant. In addition a small number of kinases were also inhibited which included TGFBR2, MEK2, p38 gamma, RET, MEK4, MAP4K5, MAP4K4, Aurora kinase B and PKC alpha.
Cell lines
The AML cell lines OCIM2 and OCI/AML3 were provided by M.D. Minden (Ontario Cancer Institute, Toronto, ON, Canada). OCI/AML3 was established from an AML patient and OCIM2 from a patient with erythroleukemia. Both cell lines proliferate in the presence of culture medium and fetal calf serum (FCS) without exogenous growth factors. Neither OCIM2 nor OCI/AML3 cells harbor KIT mutations or mutations of FLT3 (Albitar M., unpublished data). The mastocytosis cell line HMC1.2 and the murine, interleukin (IL)-3-dependent cell line, BaF3 were obtained from the American Type Culture Collection (ATCC; Rockville, MD). HMC1.2 cells are harboring V560G KIT as well as D816V KIT mutations (18). The BaF3 system included D816V KIT-expressing cells. Cells were maintained in RPMI1640 culture medium (Gibco, Grand Island, NY) supplemented with 10% FCS (Flow Laboratories, McLean, VA), grown in plastic tissue culture flasks (Falcon Plastics; Becton Dickinson, Oxnard, CA), and split twice weekly. BaF3 flasks also contained murine IL-3 (R&D Systems, Inc., Minneapolis, MN).
Patient samples
Fresh marrow samples were collected and analyzed from 12 patients with AML and 5 healthy volunteers (Table 1). Samples were obtained and studies were performed with the approval of the Institutional Review Board at The University of Texas M.D. Anderson Cancer Center. All patients signed the informed consent prior to sample collection.
Table 1.
Patient sample characteristics
| AML | BM Blasts (%) | CD117/CD34 (%)* | CG | FLT3 | KIT |
|---|---|---|---|---|---|
| 1 | 43 | NA | t(3;21)(q26q22), CBF+ | NA | NA |
| 2 | 72 | 98.6 | Diploid | 0 | NA |
| 3 | 92 | 56.7 | +8, t(3;17)(q21q25) | ITD | NA |
| 4 | 86 | 53.5 | t(9;21)(q31q22); del(4)(q12); add(21)(q22) | ITD | NA |
| 5 | 92 | 0.3 | t(9;11)(q22q23) | 0 | NA |
| 6 | 81 | 77.3 | t(3;3)(q21q26) | ITD | NA |
| 7 | 58 | 89 | t(4;12)(q12p13); der(13;15)(q10q10); del(20)(q11.2q13.3) | NA | NA |
| 8 | 85 | 51 | Diploid | ITD | 0 |
| 9 | 94 | 57 | +8, +10, +18, +22, +13 | ITD | 0 |
| 10 | 88 | 97.8 | +8, inv(16)(p13.l1q22) | 0 | D816V |
| 11 | 53 | 83.3 | t(8;21)(q22q22) | 0 | D816V |
| 12 | 65 | 95.2 | t(8;21)(q22q22); del(7)(q34) | 0 | D816V |
as measured by flow cytometry
BM, bone marrow; NA, not available; CG, cytogenetics; 0, no FLT3/KIT abnormalities detected; ITD, FLT3 internal tandem duplication; CBF, core binding factor
Cell surface marker analysis
Standard flow cytometric analysis was used to detect cell membrane-bound Kit (CD117) on OCIM2 and OCI/AML3 cells. Flow cytometry analysis was performed using FACSCaliber (Becton Dickinson Immunocytometry Systems [BDIS], San, Jose, CA). Data analysis was performed using CellQuest software (BDIS) and Modfit LT V2.0 (Verty Software House, Topsham, ME).
Cell proliferation and survival assay
The MTT [3,(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] assay was performed using an MTT-based cell proliferation/cytotoxicity assay system from Promega (Madison, WI). OCIM2 and OCI/AML3 cells were washed twice in RPMI containing 10% FCS by centrifugation at 300 x g for 5 min. Cell concentration and viability were determined by hemocytometer counts of cells with 0.1% trypan blue dye. Cells were suspended in medium and plated immediately at 5 × 104 cells per well in a total volume of 100 μl in 96-well flat bottomed plates (Linbro; Flow Laboratories, McLean, VA), and incubated for 48–72 h at 37°C in a humidified 5% CO2 atmosphere. Following incubation, 20 μl of CellTiter96 One Solution Reagent (Promega, Madison, WI) was added per well. The plates were then incubated for an additional 60 minutes at 37°C in a humidified 5% CO2 atmosphere. Immediately after incubation, absorbance was read using a 96-well plate reader at a wavelength of 490 nm. Each data point was determined six times prior to analysis.
Cell line clonogenic assay
The cell line clonogenic assay was performed as described previously (19). Colonies were counted after 7 days by using an inverted microscope. A colony was defined as a cluster of more than 40 cells.
KIT mutation analysis
The total nucleic acid was extracted from cells using NucliSens (BioMérieux, Boxtel, The Netherlands) extraction kit. For KIT mutation analysis, two primer pairs were designed to amplify the juxtamembrane and tyrosine kinase domains of kit (Kit R1-F: ATT GTA GAG CAA ATC CAT CCC C; c-Kit R1-R: GCC CCT GTT TCA TAC TGA CCA; Kit R2-F: CCT CCA ACC TAA TAG TGT ATT CAC AG; c-Kit R2-R: TCA CAT GCC CCA AAA TTA CA). A 351 bp PCR product (Kit-R1) was amplified from DNA sample spans exon 10, intron 10 and exon 11 containing codon 560 and a 380 bp PCR product (Kit-R2) was amplified spans exon 17, intron 17 containing codon 816 mutation. The PCR products were then filtration purified and sequenced in both forward and reverse directions using ABI PRISM 3100 genetic Analyzer. Sequencing data were base-called by Sequencing Analysis software and assembled and analyzed by ABI Prism® SeqScape software using GenBank accession number U63834 as reference. For NPM1 testing, the NPM gene PCR amplification was performed using NPM intron 11 forward primer (5′-TTA ACT CTC TGG TGG TAG AAT GAA-3′) and 6-fam labeled NPM exon 12 reverse primer (5′-FAM-TGT TAC AGA AAT GAA ATA AGA CGG-3′). The NPM mutated or wild type products were verified by determining the size of PCR products using ABI3100 Genetic Analyzer. The wild type displayed a 212bp peak while the NPM mutant display an extra 216bp peak in addition to the NPM wt peak. The percent of mutant to total (peak height) was calculated.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed with the SCF ELISA kit (Amersham Life Science, Arlington Heights, IL) in accordance with the manufacturer’s instructions. In brief, cell lysates and standard dilutions of SCF were added to test wells in duplicate and incubated for 2 hours at 37ºC. The test wells were then washed 3 times with PBS, incubated with mouse SCF antiserum for 2 hours, washed, and incubated for 30 minutes with goat antimouse IgG conjugated to horseradish peroxidase. The test wells were washed, and o-phenylenediamine dissolved in 3% hydrogen peroxide solution and 4 N sulfuric acid were added. The color intensity was read within 15 minutes at a wavelength of 490 nm with a microplate autoreader (model EL-309; Biotec, Winooski, VT). The average net optical density (OD) of the standard SCF concentration was then plotted, and the amount of the SCF in each sample was determined by interpolation from the standard curve.
Western immunoblotting
Cell lysates (from 5 × 105 cells) were used as described. The following antibodies were used to detect the relevant proteins: mouse antihuman Stat3 and 5, phosphorylated (p)-Stat3 and pSTAT5 antibodies (Upstate Cell Signaling Solution, Charlottesville, VA), monoclonal mouse antihuman CPP32 (Transduction Laboratories, Lexington, KY) for detection of procaspase 3, rabbit antihuman cleaved caspase 3 (New England Bio Labs, Beverly, MA), mouse antihuman poly(adenosine diphosphate [ADP]-ribose)polymerase (PARP; Pharmingen, San Diego, CA), mouse antihuman Kit and pKit (Pharmingen). Normal mouse immunoglobulin G (IgG) and rabbit IgG (Sigma) were used as controls.
Cell cycle analysis
Cell cycle analysis was performed according to standard protocols. Briefly, 5 × 106 cells were incubated with APck110 and pelleted. The cell pellets were washed and resuspended in 2 mL of 1% paraformaldehyde in phosphate-buffered saline (PBS; Gibco BRL, Grand Island, NY). Cells were incubated for 15 minutes at 4°C and then washed again in PBS, resuspended in 2 mL absolute ethanol, and stored at − 20°C until staining. The stored cells were washed twice in PBS, resuspended in 0.5 mL propidium iodide (PI) staining buffer (50 mg/mL PI, 10 μg/mL RNase in PBS), and then incubated for 1 hour at room temperature in the dark. Flow cytometric analysis was performed as described above.
Annexin V assay
The annexin V-FITC assay (Pharmingen, San Diego, CA) was used as described previously (20). Cells were analyzed using Annexin V FITC (BD Pharmingen) and flow cytometry with a FACSCalibur flow cytometer and the CellQuest software program. Data analysis was performed using the CellQuest and ModFit LT (version 2.0) software programs. To determine whether OCI/AML3-induced apoptosis was caspase-dependent, OCI/AML3 cells were preincubated with 20 μM of the pan-caspase inhibitor Z-VAD-FMK (BD Pharmingen).
AML blast colony assay
The AML blast colony assay was performed as previously described (21,22). APcK110 was added at the initiation of the cultures at concentrations ranging from 50 to 500 nM. The cultures were incubated in 35-mm Petri dishes in duplicate or triplicate for 7 days at 37°C in a humidified atmosphere of 5% CO2 in air. AML blast colonies were microscopically evaluated on day 7 of culture. A blast colony was defined as a cluster of 20 or more cells. Individual colonies were plucked, smeared on glass slides, and stained to confirm their leukemic cell composition.
Colony forming unit granulocyte-macrophage (CFU-GM) culture assay
The CFU-GM clonogenic assay was performed as follows: 2 × 105 low density bone marrow cells were cultured in 0.8% methylcellulose in Iscove’s modified Dulbecco’s medium (Gibco) supplemented with 10% FCS, 50 ng/mL rhGM-CSF (Bentax) with or without 50 nM rhSCF (Amgen), in the presence or absence of APcK110. All cultures were evaluated after 14 days for the number of CFU-GMs, defined as clusters of more than 40 granulocytes and/or monocyte/macrophage cells.
Results
APcK110 inhibits AML cell viability and proliferation – differential response of SCF-responsive and non-responsive AML cells
We first performed a cell surface marker analysis to establish whether OCIM2 and OCI/AML3 lines express Kit. Using flow cytometry we found expression of CD117 (Kit) in 21% of OCIM2 and 15% of OCI/AML3 cells, respectively. We then examined the antiproliferative effect of APcK110 on OCIM2, OCI/AML3 cells and on HMC1.2 cells that harbor an activating KIT mutation. Cells were incubated for 72 hours without or in the presence of APcK110 at concentrations of 100, 200, 300, 400, and 500 nM and their viability and rate of multiplication were determined by the MTT assay. As shown in Fig. 1B, growth inhibition was most significant in OCI/AML3 and HMC1.2 cells. In both cell lines, 80% percent inhibition of proliferation was achieved at APcK110 concentrations of 500 nM. In contrast, proliferation of OCIM2 cells was far less affected with inhibition of proliferation not exceeding 25% at APcK110 concentrations of 500 nM.
Because OCIM2 responded poorly to APcK110 and OCI/AML3 cells were as sensitive to APcK110 as HMC1.2 cells, we asked whether the sensitivity of OCI/AML3 to APcK110 results form an activating mutation(s) similar to that of HMC1.2 cells (in whom KITV560G and KITD816V mutations were found) (18). To look for mutations in OCIM2 and OCI/AML3 lines, we performed a mutation analysis using a 351 bp PCR product amplified from patient DNA samples spanning exons 10, 11, and 17 as well as introns 10 and 17. We did not detect KIT mutations in either of the two AML cell lines including at codons V560D, N822K, and D816V. Although neither OCIM2 nor OCI/AML3 cells have detectable KIT mutations, their proliferation response to APcK110 was significantly different. Furthermore, inhibition of proliferation of OCI/AML3, a cell line without KIT mutations, was similar to inhibition of
HMC1.2, a cell line with mutated KIT. To delineate these differences, we measured SCF levels in both cell line lysates using ELISA. We found that both OCIM2 and OCI/AML3 produce 10 pg of SCF per 107 cells. Then we investigated whether these AML cell lines proliferate in response to the endogenously produced SCF. We incubated OCIM2 and OCI/AML3 cells with SCF neutralizing antibodies (R&D Systems, Minneapolis, MN) at concentrations of 0.01, 0.05, 0.1 and 1 μg/mL in the presence or absence of 50 ng/mL SCF and evaluated colony-forming cell growth using a clonogenic assay. Whereas SCF neutralizing antibodies significantly inhibited OCI/AML3 colony proliferation, the inhibitory effect was completely reversed by exogenous SCF. Furthermore, SCF stimulated OCI/AML3 colony-forming cell proliferation (Fig. 1C). In contrast, no change in colony numbers of OCIM2 cells occurs in the presence of SCF neutralizing antibodies, and there was no further effect to addition of exogenous SCF (Fig. 1D). The results therefore suggest that OCI/AML3 is an SCF-responsive cell line whose growth is stimulated by SCF in an autocrine manner whereas OCIM2 cells produce SCF and express Kit but do not proliferate in response to SCF.
APcK110 is a more potent inhibitor of the proliferation of OCI/AML3 cells than imatinib and dasatinib and is at least an equally potent inhibitor as is cytarabine
Imatinib and dasatinib are potent tyrosine kinase inhibitors with broad activity against BCR-ABL, PDGFR, and Kit kinases, and in the case of dasatinib also against SRC kinases. We incubated OCI/AML3 cells for 72 hours in the presence of increasing concentrations of imatinib, dasatinib and APcK110 and, using the MTT assay, compared the antiproliferative effect of these kinase inhibitors. As shown in Fig. 2A we demonstrate stronger inhibition with APcK110. At 72 hours and at concentrations of 250 nM for imatinib, dasatinib, and APcK110, respectively, cell viability was 52%, 48%, and 35% of the control sample. Average IC50 values at 72 hours were 175 nM for APcK110. As cytarabine is the backbone nucleoside analog for treatment of AML, we also compared the antiproliferative effects of cytarabine with APcK110 in OCI/AML3 cells. As shown in Fig. 2B we demonstrate at least equal if not stronger inhibition with rising concentrations of APcK110 when compared to cytarabine. Maximum inhibition of OCI/AML3 proliferation was reached at concentration of 500 nM of APcK110 (17% of control), 2 mM of cytarabine (31% of control), and 250 nM APcK110 with 2 mM cytarabine (12% of control).
Figure 2.

Inhibition of cell line proliferation by APcK110. A) Dose-dependent antiproliferative effect of APcK110, imatinib, and dasatinib on OCI/AML3 cells. Using the MTT assay we demonstrate that APcK110 is a more potent inhibitor than imatinib and dasatinib. B) Dose-dependent antiproliferative effect of APcK110, cytarabine, and APcK110 plus cytarabine on OCI/AML3 cells. APcK110 is an at least equally potent inhibitor of AML cell proliferation as is cytarabine. C) Comparison of antiproliferative effect in wild-type versus mutant KIT –expressing BaF3 cells. We demonstrate preferential inhibition of KIT supported by the fact that incubation of mutant KIT-expressing BaF3 cells with IL-3 was not able to reverse the inhibition.
APcK110 is preferentially inhibiting KIT mutated BaF3 cell lines
To further explore the sensitivity of APcK110 towards its putative target Kit we incubated growth factor (IL-3)-dependent murine BaF3 cells with increasing concentrations of APcK110 and compared the proliferative activity by MTT assay with that in mutant KIT expressing BaF3 cells (Fig. 2C). We show that although APcK110 interferes with IL-3 mediated signaling (wild-type BaF3 cells), inhibition of cell proliferation is stronger in the mutated KIT expressing cells especially at APcK110 concentrations of 250 nM to 500 nM. Addition of IL-3 to the mutated BaF3 cell line did not reverse inhibition supporting selective targeting of KIT-mutated cells as compared to BaF3 wild-type cells. It should however be noted that the dose-response curves are close and therefore suggest a possibly narrow therapeutic window regarding APcK110 and its affinity to mutated KIT.
APcK110 inhibits the phosphorylation of Kit, Stat3, Stat5, and Akt
Kit signaling is transmitted through several pathways including signal transducer and activator of transcription (STAT), phosphatidyl-inositol-3-kinase (PI3K), SRC, and RAS (7). Due to its presumed relevance, we chose the Stat and PI3K pathways to determine the effect of APcK110 on Kit downstream phosphorylation and activation events (Fig. 3A). Using Western immunoblotting, we found a dose-dependent inhibition of the phosphorylation of Kit, Stat3, Stat5, and Akt, a downstream effector of PI3K. The decrease in levels of the respective phosphoproteins in both OCI/AML3 and HMC1.2 cell lines started at doses of 50 to 100 nM. We demonstrate the same inhibition of phosphorylation of Kit, Stat3, Stat5, and Akt in the primary AML sample from patient UPN 8 (see Table 1) suggesting that the findings of APcK110 are not isolated on cell lines (Fig. 3B).
Figure 3.
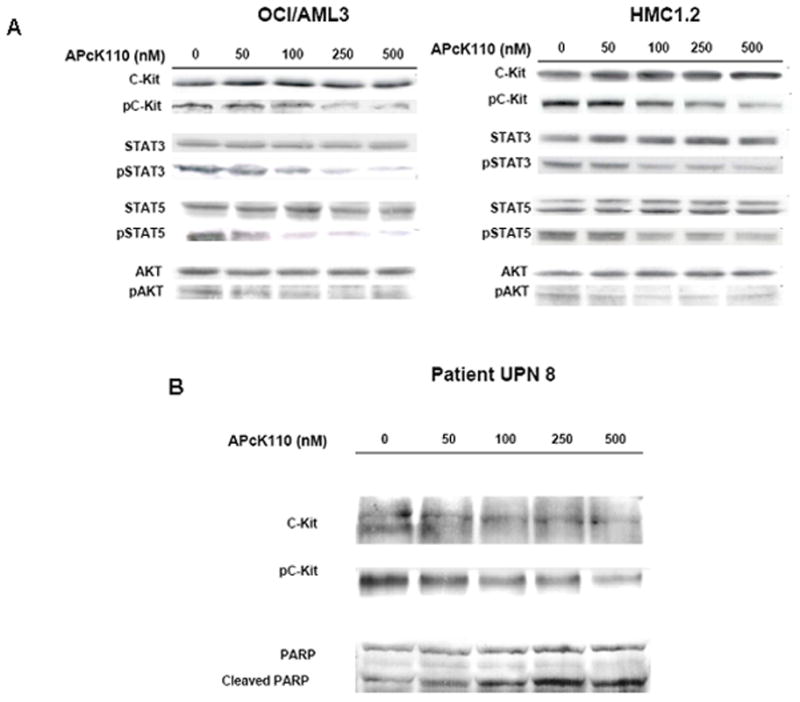
A) Effect of APcK110 on phosphorylation levels of Kit, Stat3, Stat5, and Akt in OCI/AML3 and HMC1.2 cells. Concentration-dependent inhibition of phosphorylation of Kit, Stat3, Stat5, and Akt is depicted. Cells were incubated with APcK110 for 30 minutes and then analyzed by Western immunoblotting. B) The same experiment has been repeated in primary samples from patient UPN8 suggesting that inhibition of phosphorylation of Kit, Stat3, Stat5, and Akt is not restricted to cell lines.
APcK110 induces caspase-dependent apoptosis in OCI/AML3 cells
We further analyzed whether inhibition of phosphorylation of the Stat and PI3K pathways would lead to induction of apoptosis in OCI/AML3 cells. Cell cycle analysis using propidium iodide staining of OCI/AML3 cells shows a shift of cells into sub-G0 following a 2 hour incubation with APck110 at a concentration of 500 nM consistent with an increase in the apoptotic cell fraction as well (Fig. 4A). OCI/AML3 cells were then incubated with 20 mM of the caspase inhibitor Z-VAD-FMK for one hour in their normal growth media. APcK110 was added at a concentration of 500 nM and the cells were incubated overnight (Fig. 4B). Incubation of OCI/AML3 cells with Z-VAD-FMK alone did not affect their apoptotic rate whereas preincubation of these cells with Z-VAD-FMK prior to treatment with APcK110 lowered their apoptotic rate from 73% to 26% (background rate of 21% in this experiment), suggesting that APcK110 induces apoptosis by activation the caspase pathway. Indeed in a Western immunblot analysis we found a dose-dependent increase of levels of the active form of caspase 3 with a concurrent increase in the levels of cleaved PARP thus confirming induction of apoptosis by a caspase 3-dependent pathway (Fig. 4C).
Figure 4.

Effect of APcK110 on the progression through the cell cycle and survival of OCI/AML3 cells. A) Cell cycle analysis using propdium iodide staining. OCI/AML3 cells were incubated for 2 hours with APcK110 at 500 nM. An increase in the sub-G0 fraction of cells at 2 hours to 27% from 10% (control) is demonstrated. B) Induction of caspase-dependent apoptosis. The Fig. shows apoptosis in the absence of presence of the caspase inhibitor Z-VAD-FMK (Annexin V assay). Concurrent incubation of OCI/AML3 cells with Z-VAD-FMK and APcK110 decreases apoptotic cell fraction from 73% to 26% (baseline activity 21%). C) Induction of apoptosis by activation of caspase 3 and cleavage of PARP. Western immunoblot analysis demonstrates concentration-dependent increase in caspase 3 with concurrent increase in levels of cleaved PARP.
APcK110 inhibits AML blast colony-forming cell proliferation
To investigate the effect of APcK110 on AML primary samples, we incubated diagnostic marrow cells from 12 patients with AML and 5 normal controls (see Table 1 for details of AML patients) with increasing doses of the Kit inhibitor APcK110. We studied the activity of APcK110 with or without SCF (50 nM) using the AML and the CFU-GM (for the control normal marrow) colony culture assays. As shown in Fig. 5A we found that APcK110 did not reduce GM-CSF-induced colony proliferation unless SCF was absent from the medium. Sample “Normal 1” did not show any response to either SCF or APcK110 (including at the highest concentration of 500 nM). Interestingly, this was very similar to the sample of AML patient 5 in Figure 5B whose blasts were characterized by absence of SCF receptor (CD117/CD34 negative by flow cytometry, Table 1). In both cases, absence of SCF from the medium did not reduce the GM-CSF-induced colony proliferation. In all remaining AML samples, a concentration-dependent inhibition of AML colony-forming cell proliferation occurred. In all of these cases, lack of exogenous SCF in the medium also resulted in inhibition of colony growth supporting the notion that the proliferation of these cells is SCF-responsive similar to what we found in our previous experiments with OCI/AML3 cells.
Figure 5.

A) Effect of APcK110 and SCF on normal marrow CFU-GM. B) and C) Effect of APcK110 and SCF on AML marrow leukemia blast colony-forming cells. Figure B shows experiments without SCF in the presence and absence of APcK110. In figure C, AML cells are incubated with 50 nM of SCF and increasing concentrations of APcK110. CFU-GM and AML-CFU are presented as percent of control (50 nM SCF and no APcK110).
Discussion
Molecular profiling of AML blasts has identified a number of genes, which have been associated with prognosis (23). These observations will ultimately lead to clues about their function and biologic significance in leukemogenesis and thus open the possibility of new therapeutic avenues. Mutations of KIT are among those molecular markers that are increasingly recognized in AML, and hence are being pursued for their relevance as therapeutic targets (14–16). In addition, the receptor is expressed on more than 10% blasts in 64% of de novo AMLs and 95% of patients with relapsed AML (24). In an in-vitro study using KG1, a chemotherapy-resistant AML cell line, the combination of anti-SCF with low doses of cytarabine and daunorubicin resulted in a synergistic increase of chemotherapy-induced apoptosis (25). A combination of low-dose cytarabine with imatinib was investigated in a study including 34 patients with Kit-positive AML (26). In 6 of these 38 patients, a “blast” response was observed and 8 more patients had stable disease. Among mutations of a number of exons within KIT, those at exon 17 affecting codon D816 were crucial for expression of an adverse prognostic phenotype of patients with AML harboring inv(16) and translocations t(8;21). Anecdotal responses with the tyrosine kinase inhibitor imatinib have been reported in patients with CBF AML as well (27,28). However, imatinib remained ineffective for those patients with D816 mutations, whereas a number of newer compounds (e.g. PKC412, dasatinib) have been identified as potent inhibitors of wild-type Kit and KITD816V with reported clinical activity in patients with mast cell leukemias harboring KITD816V mutations (29–31).
APcK110 is the result of a structure-based drug design of Kit inhibitors based on the available crystal structure of Kit. The design resulted from in-silico screening of several targeted libraries via docking to the crystal structure of Kit, followed by aggressive post-filtering by several criteria to determine the most prominent candidates for synthesis (17). Five compounds were selected for further in vitro testing, of which we chose APcK110 for its favorable IC50 value in Kit kinase assays and in AML cell proliferation assays.
We investigated the activity of APcK110 in two AML cell lines lacking KIT mutations and one mastocytosis line harboring KITV560G and KITD816V mutations. We found growth inhibition in all cell lines, suggesting activity against both wild-type (wt) and mutated KIT. However, inhibition of proliferation was most marked in OCI/AML3 cells with wild-type KIT when compared with both OCIM2 (wt) and HMC1.2 (mutated) cells. When compared with growth inhibition of HMC1.2, activity of APcK110 may indeed be slightly higher in cells with wt KIT than those with KITV560G or KITD816V mutations. The difference of activity between OCIM2 and OCI/AML3 still raises the question as to why activity differs in these two wt KIT lines. What may explain this difference is the responsiveness to SCF (Fig. 1). Whereas both lines express surface CD117 (Kit) and produce SCF, only OCI/AML3 cells proliferate in response to SCF (hence SCF-responsive), likely in an autocrine manner. On the other hand, OCIM2 cells could not be stimulated by increasing concentrations of exogenous SCF and the growth of these cells was not inhibited by SCF neutralizing antibodies. Thus, either endogenously produced SCF by OCIM2 cells is surface bound and could already stimulate cellular proliferation to its maximum without being affected by exogenous SCF or SCF neutralizing antibodies (in that case OCIM2 cells should have responded to the intracellular Kit inhibitor APcK110), or OCIM2 cells respond poorly to SCF rendering it SCF non responsive.
In both OCM/AML3 (wt) and HMC1.2 (mutated) cell lines we demonstrate downstream decrease of the levels of phosphorylated Kit, Stat3, Stat5, and Akt. The decrease in the levels of phosphorylated Kit in particular confirms activity of APcK110 on its target with subsequent downregulation of downstream components of at the Stat and PI3K-Akt pathways. Downregulation of phosphorylated Kit requires a higher dose of APcK110 in HMC1.2 cells than OCI/AML3 cells underlining the results from the MTT assay that APcK110 may be more active against SCF responsive cells with wt Kit. We observed the same results in the marrow of one patient with AML, findings which require extended evaluation in a large number of patient samples.
We extended our observations from cell lines to primary samples from patients with AML (see Table 1 for characteristics). An interesting pattern emerged. Normal marrow cells from healthy volunteers remained unaffected including by the highest concentration of APcK110 used unless SCF was deprived from the serum. The one exception to this observation were the leukemia blasts from patient AML5 where there was no expression of SCF receptor (Kit) and which was also the only sample that was not SCF responsive. The proliferation rate was unaffected by absence of SCF in the growth medium nor was there any decrease in AML blast colony numbers irrespective of the concentration of APcK110. In all other samples, absence of SCF caused a decrease in growth, whereas addition of SCF in the absence of APcK110 stimulated growth, which in turn was again inhibited in a dose-dependent manner by APcK110 although with a great amount of heterogeneity between samples. The observation in patient samples thus confirms the observation in the cell line models in that those blasts whose proliferation responds to SCF are also the most amenable to inhibition by APcK110.
Given the narrowness of the dose-response curve in Figure 2C, questions remain as to the target specificity of APcK110. Although our data suggest preferential inhibition of KIT, it is likely that other kinases are affected as well. Whether this is clinically relevant or not remains to be seen. In general, inclusion within the spectrum of activity of small molecule inhibitors of additional kinases can also provide a therapeutic advantage, especially in an as complex and heterogeneous disease as AML.
In conclusion, Kit inhibitors such as ApcK110 should be incorporated into future trials of patients with AML.
References
- 1.Byrd JC, Mrózek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100:4325–36. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 2.Marcucci G, Mrózek K, Ruppert AS, et al. Abnormal cytogenetics at date of morphologic complete remission predicts shorter overall and disease-free survival, and higher relapse rate in adult acute myeloid leukemia: results from Cancer and Leukemia Group B Study 8461. J Clin Oncol. 2004;22:2410–8. doi: 10.1200/JCO.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 3.Fröhling S, Schlenk RF, Kayser S, et al. Cytogenetics and age are major determinants of outcome in intensively treated acute myeloid leukemia patients older than 60 years: results from AMLSG trial AML HD98-B. Blood. 2006;108:3280–8. doi: 10.1182/blood-2006-04-014324. [DOI] [PubMed] [Google Scholar]
- 4.Fröhling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23:6285–94. doi: 10.1200/JCO.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 5.Kitamura Y, Hirota S. Kit as a human oncogenic tyrosine kinase. Cell Mol Life Sci. 2004;61:2924–31. doi: 10.1007/s00018-004-4273-y. [DOI] [PubMed] [Google Scholar]
- 6.Roskoski R., Jr Structure and regulation of Kit protein-tyrosine kinase – the stem cell factor receptor. Biochem Biophys Res Comm. 2005;338:1307–15. doi: 10.1016/j.bbrc.2005.09.150. [DOI] [PubMed] [Google Scholar]
- 7.Rönnstrand L. Signal transduction via the stem cell factor receptor/c-Kit. Cell Mol Life Sci. 2004;61:2535–48. doi: 10.1007/s00018-004-4189-6. [DOI] [PubMed] [Google Scholar]
- 8.Nagata H, Worobec AS, Oh CK, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci. 1995;92:10560–4. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubin BP, Singer S, Tsao C, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–21. [PubMed] [Google Scholar]
- 10.Kemmer K, Corless CL, Fletcher JA, et al. KIT mutations are common in testicular seminomas. Am J Pathol. 2004;164:305–13. doi: 10.1016/S0002-9440(10)63120-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nanri T, Matsuno N, Kawakita T, et al. Mutations in the receptor tyrosine kinase pathway are associated with clinical outcome in patients with acute myeloblastic leukemia harboring t(8;21)(q22;q22) Leukemia. 2005;19:1361–6. doi: 10.1038/sj.leu.2403803. [DOI] [PubMed] [Google Scholar]
- 12.Du J, Schlenk RF, Corbacioglu A, et al. Detection of RAS, KIT and FLT3 gene mutations in t(8;21)-positive acute myeloid leukemia (AML) : evaluation of the clinical relevance. Blood. 2006;108:652a. [Google Scholar]
- 13.Du J, Schlenk RF, Corbacioglu A, et al. RAS, KIT and FLT3 gene mutations in inv(16)/t(16;16)-positive acute myeloid leukemia (AML) : incidence and relevance on clinical outcome. Blood. 2006;108:653a. [Google Scholar]
- 14.Cairoli R, Beghini A, Grillo G, et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood. 2006;107:3463–8. doi: 10.1182/blood-2005-09-3640. [DOI] [PubMed] [Google Scholar]
- 15.Schnittger S, Kohl TM, Haferlach T, et al. KIT-D816 mutations in AML-ETO-positive AML are associated with impaired event-free and overall survival. Blood. 2006;107:1791–9. doi: 10.1182/blood-2005-04-1466. [DOI] [PubMed] [Google Scholar]
- 16.Paschka P, Marcucci G, Ruppert AS, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21) : a Cancer and Leukemia Group B Study. J Clin Oncol. 2006;107:3904–11. doi: 10.1200/JCO.2006.06.9500. [DOI] [PubMed] [Google Scholar]
- 17.Maxwell DS, Pal A, Peng Z, et al. Structure-based drug design of c-Kit inhibitors for use in the treatment of acute myeloid leukemia. Blood. 2006;106:540a. [Google Scholar]
- 18.Butterfield JH, Weiler D, Dewald G, Gleich GJ. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res. 1988;12:345–55. doi: 10.1016/0145-2126(88)90050-1. [DOI] [PubMed] [Google Scholar]
- 19.Estrov Z, Manna SK, Harris D, et al. Phenylarsine oxide blocks interleukin-1-induced activation of the nuclear transcription factor NF-κB, inhibits proliferation, and induces apoptosis of acute myelogenous leukemia cells. Blood. 1999;94:2844–53. [PubMed] [Google Scholar]
- 20.Tuyt LML, Bregman K, Lummen C, et al. Differential binding activity of the transcription factor LIL-STAT in immature and differentiated normal and leukemia myeloid cells. Blood. 1998;92:1364–73. [PubMed] [Google Scholar]
- 21.Minden MD, Buick RN, McCulloch EA. Separation of blast cells and T lymphocyte progenitors in the blood of patients with acute myeloblastic leukemia. Blood. 1979;54:186–95. [PubMed] [Google Scholar]
- 22.Estrov Z, Grunberger T, Dube ID, et al. Detection of residual acute lymphoblastic leukemia cells in cultures of bone marrow obtained during remission. N Engl J Med. 1986;315:538–42. doi: 10.1056/NEJM198608283150902. [DOI] [PubMed] [Google Scholar]
- 23.Radmacher MD, Marcucci G, Ruppert AS, et al. Independent confirmation of a prognostic gene-expression signature in adult acute myeloid leukemia with a normal karyotype: a Cancer and Leukemia Group B study. Blood. 2006;108:1677–83. doi: 10.1182/blood-2006-02-005538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Advani AS. C-kit as a target in the treatment of acute myelogenous leukemia. Curr Hematol Rep. 2005;4:51–8. [PubMed] [Google Scholar]
- 25.Lu C, Hassan HT. Human stem cell factor-antibody [anti-SCF] enhances chemotherapy cytotoxicity in human CD34+ resistant myeloid leukaemia cells. Leuk Res. 2006;30:296–302. doi: 10.1016/j.leukres.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 26.Heidel F, Cortes J, Rücker FG, et al. Results of a multicenter phase II trial for older patients with c-Kit-positive acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (HR-MDS) using low-dose ara-C and imatinib. Cancer. 2007;109:907–14. doi: 10.1002/cncr.22471. [DOI] [PubMed] [Google Scholar]
- 27.Cairoli R, Beghini A, Morello E, et al. Imatinib mesylate in the treatment of Core Binding Factor leukemias with KIT mutations: a report of three cases. Leuk Res. 2005;29:397–400. doi: 10.1016/j.leukres.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 28.Nanri T, Matsuno N, Kawakita T, et al. Imatinib mesylate for refractory acute myeloblastic leukemia harboring inv(16) and a C-KIT exon 8 mutation. Leukemia. 2005;19:1673–5. doi: 10.1038/sj.leu.2403889. [DOI] [PubMed] [Google Scholar]
- 29.Schnittenhelm MM, Shiraga S, Schroeder A, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66:473–81. doi: 10.1158/0008-5472.CAN-05-2050. [DOI] [PubMed] [Google Scholar]
- 30.Fabbro D, Ruetz S, Bodis S, et al. PKC412: a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 2000;15:17–28. [PubMed] [Google Scholar]
- 31.Gotlib J, Berubé C, Growney JD, et al. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816 KIT mutation. Blood. 2005;106:2865–70. doi: 10.1182/blood-2005-04-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]