

Abstract
Bone is a hierarchically structured composite material whose basic building block is the mineralized collagen fibril, where the collagen is the scaffold into which the hydroxyapatite (HA) crystals nucleate and grow. Understanding the mechanisms of hydroxyapatite formation inside the collagen is key to unravelling osteogenesis. In this work, we employed a biomimetic in vitro mineralization system to investigate the role of the amorphous precursor calcium phosphate phase in the mineralization of collagen. We observed that the rate of collagen mineralization is highly dependent on the concentration of polyaspartic acid, an inhibitor of hydroxyapatite nucleation and inducer of intrafibrillar mineralization. The lower the concentration of the polymer, the faster the mineralization and crystallization. Addition of the non-collagenous protein C-DMP1, a nucleator of hydroxyapatite, substantially accelerates mineral infiltration as well as HA nucleation. We have also demonstrated that Cu ions interfere with the mineralization process first by inhibiting the entry of the calcium phosphate into the collagen, and secondly by stabilizing the ACP, such that it does not convert into HA. Interestingly, under these conditions mineralization happens preferentially in the overlap regions of the collagen fibril. Our results show that the interactions between the amorphous precursor phase and the collagen fibril play an important role in the control over mineralization.
1. Introduction
Bone is a hierarchically structured composite material whose basic building block is the mineralized collagen fibril, where the collagen is the scaffold into which the hydroxyapatite (HA) crystals nucleate and grow.1, 2 The triple-helical collagen molecules are packed in a quasi-hexagonal manner and are axially organized into parallel arrays, with their ends separated by a 40 nm gap and neighbouring molecules staggered by 67 nm.3–5 The fibril, thus, has a periodic cross-striated structure where a densely packed 27 nm-long region - the overlap zone - alternates with the less dense 40 nm-long gap zone.
Understanding the mechanisms of HA nucleation and growth inside the collagen lies at the core of unravelling bone formation, and therefore has been widely investigated, both in in vivo and in vitro systems. Early studies on bone formation have been able to establish the spatial and crystallographic relationship between collagen and the mineral. Conventional transmission electron microscopy (TEM) studies, combined with X-ray and electron diffraction have shown that the HA crystals nucleate and grow within 40 nm gaps present in the collagen fibril, and have their c-axis aligned parallel to the long axis of collagen.1, 2, 6–11 The collagen fibrils, however, cannot induce HA mineralization on their own, and require acidic non-collagenous proteins (NCPs) to promote HA formation.
One of the major issues in bone formation has been to characterize the pathways through which the calcium and phosphate ions are translocated to the mineral deposition site within the collagen. Two main modes have been proposed: 1) the crystals are actively nucleated from solution by the NCPs associated to the collagen gap zones, without intervention of cellular processes;2, 12 and 2) matrix vesicles that bud from the plasma membrane accumulate ions extracellularly through their molecular composition.13, 14 Recent research has demonstrated that osteoblasts concentrate within intracellular vesicles calcium phosphate in a non-crystalline, disordered phase, composed of amorphous calcium phosphate (ACP).15, 16 This precursor phase is then delivered to the collagen at the bone growth front, infiltrates into the fibrils, and then crystallizes into HA. Although the presence of an amorphous precursor to HA in bone mineralization has only been recently identified, this strategy has been well known for several years, in particular in calcium carbonate-based biomineralization in invertebrates. In these organisms, amorphous calcium carbonate forms the first mineral deposits that subsequently transforms either into calcite or aragonite.17–20 The importance of using amorphous phases as precursors is that these materials are isotropic and easily moldable, and thus allow the overcoming of directional restrictions of crystals when building mineralized structures.21 Moreover, this strategy allows the transport of large enough quantities of the mineral to the growth front that are necessary to build mineralized structures.
One issue, however, that has been overlooked is whether the amorphous precursor can play an active role in helping to direct the mineralization process itself by interacting with the pre-formed organic matrix template. In in vitro studies on collagen mineralization, we have demonstrated that the charge interaction between the collagen and the ACP drives the entry of the mineral into the fibril. Subsequently, the charged residues of collagen form nucleation sites that mediate the transformation of ACP into oriented HA.22 Thus, parameters such as net charge, particle size and chemistry of the ACP particles are important in mediating the interaction with collagen and hence the mineralization process.
Here, we employed cryo-transmission electron microscopy (cryoTEM) and cryo-electron tomography (cryoET) to further investigate the role of the ACP in the mineralization of collagen. We employed a biomimetic mineralization system, where the NCPs were replaced by polyaspartic acid (pAsp), which inhibits HA formation in solution and stabilizes the ACP, directing it into the collagen.23, 24 In the first step of the work, we investigated the influence of the concentration of pAsp on the mineralization. Furthermore, we accidentally found that Cu ions derived from Cu cryoTEM grids inhibit HA formation, stabilizing the ACP for long periods of time. We then studied how the mineralization of collagen proceeded under these conditions, both in presence of pAsp and the C-terminal fragment of the NCP dentin matrix protein 1 (C-DMP1), which is known to induce HA nucleation.25–27 We show that the stabilization of the ACP phase here realized both by different concentrations of pAsp and by the presence of Cu ions is an important aspect in controlling mineralization.
2. Experimental
2.1 CryoTEM and cryoET
Sample vitrification was performed using an automated vitrification robot (FEI Vitrobot™ Mark III) for plunging in liquid ethane.28 CryoTEM Au or Cu grids, R2/2 Quantifoil Jena grids (Quantifoil Micro Tools GmbH) were surface plasma treated for 40 seconds using a Cressington 208 carbon coater prior to use. For 2D imaging and tomography, samples were studied on the TU/e CryoTitan (FEI, www.cryotem.nl), equipped with a with a field emission gun (FEG) operating at 300 kV and a post column Gatan Energy Filter (GIF). Images were recorded using a post-GIF 2k × 2k Gatan CCD camera. Low-dose selected area electron diffraction (LDSAED) and energy-dispersive X-ray analysis (EDX) were performed on a FEI Tecnai 20 (Type Sphera) equipped with a LaB6 filament operating at 200 kV and a Gatan cryo holder operating at −170 °C was used. The images were recorded using a 1k × 1k Gatan CCD camera and Cryo-EDX spectra were recorded on an EDAX detector in cryo-STEM mode, spot size 3.
The alignment and 3-dimensional reconstructions of the data sets were performed in IMOD or Inspect3D v.3.0 (FEI Company). For the segmentation and visualization of the 3D volume, Amira 4.1.0 (Mercury Computer Systems) was used. Image analysis was done in Gatan Digital Micrograph and NIH ImageJ.
2.2 Assembly of collagen on TEM grids
Type I collagen extract from horse tendon was kindly provided by Dr. Giuseppe Falini (Department of Chemistry, University of Bologna, Italy) and prepared as described elsewhere.29 Briefly, 1 g of the extract was mixed overnight with 10 ml of aqueous acetic acid (50 mM, pH 2.5), centrifuged at 5000 rpm for 10 minutes and the supernatant was collected and stored at 4° C. At pH 2.5 the collagen fibrils are disassembled and remain in solution.
CryoTEM grids were laid on a 15 μl drop of collagen solution for 10 seconds. The excess of collagen solution was manually blotted and the grids were transferred to a 15 μl drop of Hepes buffer (10 mM, pH 7.4) containing NaCl (150 mM), for 30 minutes.30 The increase in pH to physiological levels triggers collagen assembly into fibrils and its subsequent precipitation.29, 31 This procedure was performed inside a glove box,28 where temperature and humidity are controlled at 22 ° C and 100 % relative humidity. The grids were then transferred to the mineralization solution (see below).
2.3 Mineralization experiments
Collagen mineralization in presence of polyaspartic acid (pAsp) was achieved by incubating the collagen-adsorbed cryoTEM grids in Hepes buffer (10 mH, pH 7.4, Sigma) containing CaCl2 (2.7 mM, Merck), K2HPO4 (1.35 mM, Merck) and polyaspartic acid (10 μg/ml, molecular weight 2000-11000 Da, Sigma) at 37 °C, as described elsewhere.22, 24
Collagen mineralization in presence of C-DMP1 was achieved by incubating collagen-adsorbed cryoTEM grids in Hepes buffer (10 mM, pH 7.4, Sigma) containing CaCl2 (2.7 mM, Merck), K2HPO4 (1.35 mM, Merck) and C-DMP1 (15 μg/ml, see below) + polyaspartic acid (1.5 μg/ml – 10 μg/ml, molecular weight 2000-11000 Da, Sigma) at 37 °C. In the control experiments collagen was mineralized without any additives.
After mineralization, grids were washed with MilliQ water for 10 minutes. The excess water was removed by manual blotting and the grids were transferred to the automated vitrification robot. Air dried samples were prepared by washing the grids in MilliQ water for 10 minutes; manual blotting with filter paper to remove the excess of water; dehydrating the samples in 100 % ethanol for 5 minutes and air drying.
2.4 Staining of collagen with uranyl acetate
Staining of collagen with uranyl acetate was performed on some samples immediately before vitrification of the grids in liquid ethane, following the procedure described by Traub et al.10, with modifications. After removal of the samples from the mineralization solution and subsequent washing with MilliQ water, the grids were incubated in 0.5 % uranyl acetate in MilliQ water for 15 seconds, then washed with MilliQ water for 1 minute, manually blotted and transferred to the automated vitrification robot.
3. Results
In order to investigate the role of the amorphous phase in the mineralization of collagen, we first investigated the effect of pAsp concentration on the infiltration of ACP into the fibrils and its subsequent transformation into oriented hydroxyapatite (HA) crystals. When pAsp was present at a concentration of 1.5 μg/ml in the mineralization solution, the collagen fibrils were completely impregnated with amorphous calcium phosphate (ACP) after 24 h of reaction (figure 1A). At this stage, the ACP had already started to convert into oriented HA crystals, as shown by the presence of needle-like, dense objects within the amorphous phase (figure 1A, arrows), as reported.22 Higher concentrations of pAsp resulted in considerably slower mineralization rates. Between 3 and 10 μg/ml we could only observe aggregates of ACP infiltrating into the collagen through the region at the border between the gap and overlap regions (figure 1B–D).22 These results show that by increasing the amount of pAsp in the solution and hence making the ACP phase more stable, the rate of infiltration of the ACP into the collagen significantly slows down. At a concentration of 10 μg/ml of pAsp, 72 h were necessary for the complete mineralization of collagen with HA crystals (figure 2). In this case, infiltration of the fibril with ACP occurred within 24–48 h (figures 2A and B), followed by the crystallization of ACP into HA (figure 2C). Interestingly, ACP and oriented HA crystals were found only on well organized regions of the collagen (figure 2D, black circle area). On poorly organized areas, very little mineral was present, consisting mostly of few HA crystals randomly oriented (figure 2D, white circle area and white dashed circle). These observations suggest a role for the collagen not only in directing the infiltration of the mineral phase, but also in templating the orientation and growth of the HA crystals. In addition, with the progression of the mineralization, the collagen fibrils expanded in the direction perpendicular to their long axis, becoming deformed by the developing mineral (figure 2E). The cross-sectional area of the fibril at the point with the least amount of mineral (dashed-line 1) increased from 53000 nm2 to 102000 nm2 at the point with the highest mineral content (dashed-line 2). Although in both cases the collagen contained a mixture of ACP and HA, the expansion of the fibril is proportional to the amount of calcium phosphate that is inside. Therefore, it is likely that it was the infiltration of the ACP into the collagen that caused the fibril to expand, rather than the transformation of ACP into HA. Regarding the presence of HA crystals randomly oriented on poorly ordered regions of collagen (figure 2D), we must point out that these images are 2-dimensional projections of a 3-dimensional object. Therefore, we cannot distinguish whether the crystals are actually present inside or on the surface of the collagen.
Fig. 1.

CryoTEM images of collagen mineralized with calcium phosphate for 24 h in presence of different concentrations of pAsp, on cryoTEM grids made of Au. A. 1.5 μg/ml of pAsp. White arrows: apatite crystals nucleating and growing within the amorphous calcium phosphate phase. B. 3 μg/ml of pAsp. C. 6 μg/ml of pAsp. D. 10 μg/ml of pAsp. White arrows: amorphous calcium phosphate particles infiltrating into the collagen. Panel D was adapted from Nudelman, F. et al.,22 Nature Mater., 2010, 9, 1004–1009. Reprinted with permission from Macmillan Publishers Ltd. Nature Materials, www.nature.com/nmat, Copyright 2010.
Fig. 2.

CryoTEM images of collagen at different stages of mineralization in presence of 10 μg/ml of pAsp, on cryoTEM grids made of Au. A. Mineralization for 24 h. B. Mineralization for 48 h. C. Mineralization for 72 h. D. Collagen fibril containing two regions, one well organized (black circle area) and one poorly organized (black circle area). The well organized area is partially mineralized with amorphous calcium phosphate and apatite, while the poorly organized one contains only few, randomly oriented apatite crystals (white dashed circle). Dashed black circle: 10 nm gold marker. E. A partially mineralized collagen fibril, where deformation caused by the presence of the mineral can be observed. Dashed-lines: Region with least (dashed-line 1) and most amount of mineral (dashed-line 2) from where the cross-sectional area of the fibril was calculated. Panel E was adapted from Nudelman, F. et al.,22 Nature Mater., 2010, 9, 1004–1009. Reprinted with permission from Macmillan Publishers Ltd. Nature Materials, www.nature.com/nmat, Copyright 2010.
All the above experiments were performed with collagen absorbed on TEM grids that were made of Au. We accidentally found that when grids made of Cu were used instead, the mineralization of collagen changed drastically. Using 10 μg/ml of pAsp, after 24 h of reaction there was substantially less ACP aggregates infiltrating the collagen when compared to experiments performed on Au grids (figure 3A). After 48 h, most of the mineral was associated to the overlap region (figure 3B), becoming denser after 72 h but still with no signs of crystallinity (figures 3C and D). Cryo-energy dispersive X-ray spectroscopy (cryoEDX) confirmed that the precipitates found on the collagen were indeed composed of calcium phosphate (figure 3E). Our results indicate that Cu ions were being released from the grid into the solution, and were interfering with the crystallization process. Indeed, using atomic absorption spectroscopy, we found that after 72 h of reaction Cu ions were present in the solution at a concentration of 5 μg/ml, and after 1 week their concentration increased to 11 μg/ml.
Fig. 3.

CryoTEM images of collagen at different stages of mineralization in presence of 10 μg/ml of pAsp, on cryoTEM grids made of Cu. A. Mineralization for 24 h. White circle: amorphous calcium phosphate particles associated to the collagen. B. Mineralization for 48 h. White circle: amorphous calcium phosphate particles associated to the overlap region of collagen. Black circle: 10 nm gold markers. C. Mineralization for 72 h. White circle: amorphous calcium phosphate particles associated to the overlap region of collagen. D. Low-dose selected-area electron diffraction of C, showing that the mineral is still amorphous. E. Cryo-energy dispersive X-ray spectroscopy (cryoEDX) measurement of C, confirming that the precipitates are indeed composed of calcium phosphate.
In order to verify whether ACP was still entering the collagen or was accumulating only on the surface of the fibril, we performed cryo-electron tomography (cryoET)32 on samples that were mineralized for 48 h (figure 4). Indeed, cryoET showed the presence of mineral particles inside the collagen, although in smaller amounts than found when Au grids were used (figure 2). In fact, while in experiments performed without Cu (i. e. on Au grids) the ACP phase was found as a continuous matrix permeating the collagen, in the presence of Cu the calcium phosphate was found as discrete particles with a broad size distribution, ranging from 10 to 60 nm in size. Further analysis of the reconstruction data confirmed that most of the mineral particles were found indeed in the overlap region, while the gap zone was the least mineralized area (figures 4B–D).
Fig. 4.

Cryo-electron tomography of a collagen fibril mineralized for 48 h in the presence of 10 μg/ml of pAsp, on a Cu grid. A. Two-dimensional cryoTEM image. B. Slice from a section of the reconstructed 3-dimensional volume, showing calcium phosphate particles inside the collagen (white circles). C. Computer-generated 3-dimensional visualization of the reconstruction, where the collagen fibril is depicted in white and the calcium phosphate precipitates in red. Inset: only half of the collagen fibril is shown, revealing the calcium phosphate precipitates inside the fibril, predominantly in the overlap region. D. Graph showing the distribution of calcium phosphate within a 67 nm repeat.
In the next step, we combined chemical staining with cryoTEM22 to map if the infiltration of the ACP into the fibrils occurs through the same mechanism when Cu grids are used as compared to experiments done with Au grids. Uranyl acetate is a staining agent that binds to the charged amino acids of collagen, increasing the local mass density, thus generating a pattern of bands that mark the location of clusters of positively and negatively charged amino acids.33, 34 This staining has been widely used to visualize collagen in conventional (reviewed in33) and in cryo-TEM,10, 22 and the resulting staining pattern can be directly correlated to the amino acid sequence of collagen and to the crystal structure. When the mineralization of collagen was performed on Au grids, we observed the infiltration of the mineral into the fibrils through the a-band region, as previously reported (figure 5A and B, black arrows).22 This region corresponds to the C-terminal end of the collagen molecules, and it has been described as being highly positively charged and thus the most favourable for the negatively charged pAsp-mineral complex to interact with. When the experiments were performed on Cu grids, we still observed the ACP associated with the same areas of the collagen, namely the a-band region (figure 5C, black arrows). Analysis of the mass-density profile of the non-mineralized (figure 5D) and mineralized on Au and Cu grids (figures 5E and F, respectively) confirmed the location of the mineral phase on the collagen. The peaks corresponding to the a-bands significantly increased in intensity in both cases when compared to the non-mineralized collagen. The peaks corresponding to the other staining bands, on the other hand, did not change, suggesting little or no interaction between the ACP and the other regions of the collagen fibril, even in the presence of Cu ions.
Fig. 5.
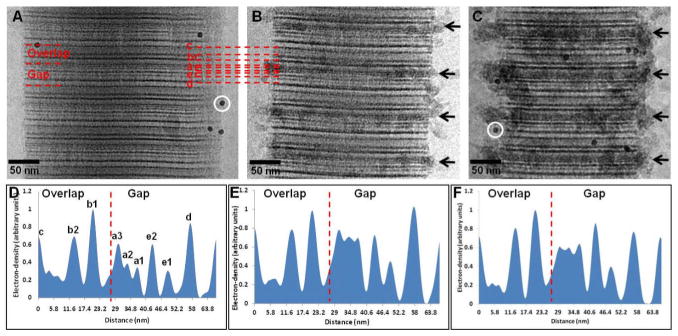
Uranyl acetate map of collagen during the early stages of mineralization. A. Non-mineralized collagen. White circle: 10 nm gold marker. B. Collagen mineralized for 24 h in presence of 10 μg/ml of pAsp, on Au grids. C. Collagen mineralized for 24 h in presence of 10 μg/ml of pAsp, on Cu grids. White circle: 10 nm gold marker. D. Intensity profile of A, non-mineralized collagen. Dashed line: border between gap and overlap regions (C-terminus). E. Intensity profile of B, collagen mineralized for 24 h on Au grids. Dashed line: border between gap and overlap regions (C-terminus). F. Intensity profile of C, collagen mineralized for 24 h on Cu grids. Dashed line: border between gap and overlap regions (C-terminus). Panels A and D were adapted from Nudelman, F. et al.,22 Nature Mater., 2010, 9, 1004–1009. Reprinted with permission from Macmillan Publishers Ltd. Nature Materials, www.nature.com/nmat, Copyright 2010.
We investigated the stability of the amorphous phase by letting the mineralization reaction proceed for 4 weeks (figure 6A and B). At this stage, the collagen fibrils were completely calcified, with mineral present both inside and on the surface of the collagen. However, even after such long incubation time the mineral phase consisted predominantly of ACP, as shown by LDSAED (figure 6A, inset). The amorphous phase did not convert into HA even when the samples were air-dried (figure 6B, inset). We performed control experiments where no additives were present in the mineralization solution. After 72 h, we observed the precipitation of few HA crystals in the solution and on the surface of the collagen (not shown). When incubated for 1 week and air dried, the samples contained large amounts of HA crystals, covering the whole surface of the collagen fibrils and of the carbon support film on the cryoTEM grid (figure 6C). In this case, LDSAED demonstrated that the mineral is composed of HA, as shown by the (002) and (211) reflections. Interestingly, when pAsp was present but collagen was absent, we could also obtain HA on air-dried samples (figure 6D).
Fig. 6.

A. CryoTEM image of collagen mineralized for 4 weeks in presence of 10 μg/ml of pAsp on Cu grids. The calcium phosphate is still amorphous, as shown by the LDSAED (inset). B. TEM image of dried collagen mineralized for 4 weeks in presence of 10 μg/ml of pAsp on Cu grids. Even after freeze-drying, the mineral shows no signs if crystallinity, as shown by the LDSAED (inset). C. TEM image of dried collagen mineralized for 2 weeks without additives, on a Cu grid. The surface of the collagen and of the carbon support film on the grid are completely covered with apatite crystals. Inset: LDSAED, showing the (002) and (211) reflections of apatite. D. TEM image of dried HA crystals precipitated in presence of 10 μg/ml of pAsp on a Cu grid after 2 weeks of reaction, without collagen. Inset: LDSAED, showing the (002) and (211) reflections of apatite.
With the aim of understanding how the Cu ions could be influencing mineralization, we performed experiments in the presence of the C-terminal fragment of the dentin matrix protein 1 (C-DMP1). This protein is an acidic non-collagenous protein that is expressed during the initial stages of bone and dentin mineralization.25, 26 In vitro studies have shown that in the presence of collagen, it promoted HA nucleation on the surface of the fibrils.27 Moreover, the C-terminal fragment of this protein was identified as the domain containing HA nucleation sites. Furthermore, C-DMP1 was demonstrated to induce the biomimetic mineralization of collagen when used in combination with pAsp.22 Thus, we decided to investigate whether this protein could induce the formation of HA crystals inside the collagen also in the presence of Cu ions. In control experiments performed on Au grids, C-DMP1 was used at a concentration of 15 μg/ml and in the absence of 5 pAsp. Under these conditions, we observed that after 24 h this protein induced the formation HA crystals mainly on the surface of the collagen fibrils (figure 7A). When C-DMP1 was used in combination with 10 μg/ml of pAsp, also on Au grids, the collagen fibrils were mineralized with oriented HA crystals, formed through an ACP precursor, as described (figure 7B). Interestingly, when the experiments were performed on Cu grids, C-DMP1 lost its ability to nucleate HA. In the absence of pAsp, globular structures consisting of calcium phosphate formed on the surface of the collagen (figure 7C, black arrows). Although LDSAED still needs to be performed, it is very lilely that these structures were composed of ACP. When used in combination with pAsp on Cu grids, the fibrils were mineralized with ACP that formed predominantly on the overlap region, in a similar way as when pAsp was used alone (figure 7D). However, the presence of C-DMP1 significantly accelerated mineralization, such that after 24 h the fibrils contained large amounts of ACP when compared to mineralization with only pAsp. These results indicate that the Cu ions are interacting with the protein and changing its activity. Further studies need to be conducted to clarify the effect on the Cu ions on the protein.
Fig. 7.

CryoTEM images of collagen mineralized in presence of 15 μg/ml of C-DMP1 on Au and Cu grids. A. Collagen mineralized for 24 h in presence of 15 μg/ml of C-DMP1 on Au grids. Black circle: 10 nm gold markers. B. Collagen mineralized for 24 h in presence of 15 μg/ml of C-DMP1 and 10 μg/ml of pAsp on Au grids. Black circle: 10 nm gold markers. C. Collagen mineralized for 24 h in presence of 15 μg/ml of C-DMP1 on Cu grids. Black arrows: globular structures of calcium phosphate on the surface of collagen. Black circle: 10 nm gold markers. D. Collagen mineralized for 24 h in presence of 15 μg/ml of C-DMP1 and 10 μg/ml of pAsp on Cu grids. Black circle: 10 nm gold markers.
4. Discussion
4.1 Effect of the concentration of pAsp
We have shown that the rate of collagen mineralization is highly dependent on the concentration of the HA nucleation inhibitor, namely pAsp. At low concentration of pAsp (1.5 μg/ml, figure 1A), the infiltration of the calcium phosphate into the collagen and its subsequent transformation into HA happens within 24 h. At higher concentration of pAsp (above 3 μg/ml, figures 1B–D), this process is substantially slowed down. Interestingly, when pAsp at higher concentrations is combined with C-DMP1, an HA nucleator, the mineralization of collagen is substantially accelerated, reaching the same rate as with pAsp at low concentration. Considering that the main function of pAsp is to transiently stabilize ACP and allow it to enter the collagen,24, 35 these results suggest that there must be a balance between the stabilization of ACP in solution and the promotion of HA nucleation. Interestingly, we observed that the size of the ACP aggregates infiltrating into the collagen increased at higher concentrations of pAsp (figures 1B–D). Previously, we have observed that pAsp at 10 μg/ml stabilizes the formation of loosely packed assemblies of calcium phosphate complexes36 that are 1 nm in size.22 These assemblies further aggregate, forming larger and denser structures that are similar to the ones observed entering the collagen fibril (figure 1D). It is likely that the size and the stability of these aggregates of calcium phosphate particles are dependent on the concentration of pAsp. At lower concentrations of pAsp, these aggregates will tend to be smaller and less stable, while at higher concentrations they will grow larger in size and become more stable. We note, however, that at higher concentration of pAsp (10 μg/ml) the calcium phosphate precipitates from the solution within 24 h, while infiltration of the ACP into the collagen is still ongoing and takes 24 – 48 h. Thus, the speed of the mineralization reaction is not dependent on the stabilization of the calcium phosphate into the solution, but on aggregate size. It is possible that at 1.5 μg/ml of pAsp, the assemblies of calcium phosphate complexes will be small enough to quickly diffuse through the collagen fibril, while at higher concentrations of the polymer (above 3 μg/ml), the diffusion of the particles will take substantially longer and thus the mineralization will be slower.
4.2 The role of the 3-dimensional structure of collagen in controlling mineralization
It is noteworthy that intrafibrillar mineralization occurred only when the collagen fibril was well organized, and not when it was poorly ordered (figure 2D). This result is significant since it highlights that the 3-dimensional structure of collagen is essential for its function in controlling mineralization. Since the fibril is composed of individual collagen molecules organized in a parallel, staggered array, this organization results in the proper alignment of the positively charged amino acids of neighbouring molecules into domains that in turn are able to interact with the ACP and mediate its infiltration into the fibril. Furthermore, the lateral and axial organizations of the collagen molecules also creates HA nucleation sites by organizing the charged amino acids of adjacent molecules into a 3-dimensional structural template that induces oriented HA nucleation.37–39 Thus, on disorganized regions of the fibril such structural template is absent and diffusion of ACP into the collagen and its transformation into oriented HA crystals cannot occur. These findings bear important consequences for studies that use single collagen molecules to investigate mineralization.38
4.3 Expansion of collagen during mineralization
The expansion of the fibril during mineralization shows that, contrary to what has been previously assumed, availability of space does not limit crystal growth. The liquid-crystalline nature of collagen allows enough flexibility so that the molecules can be pushed aside during mineralization.6 In addition, during HA formation the intermolecular spacing between the collagen molecules in fact decreases, leading to regions with a close-packed arrangement of molecules.6 Moreover, it must be considered 1) that the increase in the cross-sectional area of the collagen is proportional to the amount of mineral inside the fibril; and 2) that mineralization starts with filling the intermolecular spaces with ACP. It is likely, then, that it was during the infiltration stage that the molecules were pushed aside and the fibril expanded. We further propose that the crystallization of ACP into HA will cause the shrinkage of the fibrils due to extrusion of water and the decrease of the intermolecular spacing, as observed in the turkey tendon.6
4.4 The effect of Cu ions on collagen mineralization
We have also demonstrated that Cu ions interfere with the mineralization process first by inhibiting the entry of the calcium phosphate into the collagen, and second by stabilizing the ACP, such that it does not convert into HA. Interestingly, HA crystals still form either in the absence of additives, or in the presence of pAsp but without the collagen fibril. In other words, the Cu ions are preventing crystal formation inside the collagen, but not in solution. Although Cu ions are known to inhibit the transformation of ACP into HA40, our observations mean that stabilization of the ACP by the Cu in our experiments occurs not only by the direct interaction of the ion with the calcium phosphate. Cu must be also complexing with the pAsp, the C-DMP1, and possibly with the collagen as well, and therefore affecting their function, both in mediating the infiltration of the ACP into collagen and subsequent HA nucleation.
At present, we do not have further information on the possible mechanism through which Cu ions interfere with the mineralization process. Regarding the infiltration of the ACP into the collagen, we speculate that Cu may associate to the sites in the collagen through which the calcium phosphate goes in, and therefore slowing the diffusion of the ACP into the fibril. Alternatively, it is also possible that in the presence of Cu, larger complexes of ACP are formed in solution which cannot easily diffuse into the collagen.41 Nevertheless, the mineral infiltration site remains the same, namely the C-terminal end of the collagen molecule. It is interesting to note that when the mineralization experiments were performed employing both pAsp and C-DMP1, mineral infiltration into the collagen was significantly faster (figure 7D), either in the presence of Cu ions (using Cu grids) or in their absence (using Au grids). The fast infiltration of ACP into the collagen, without nucleation of HA, means that one of the functions of C-DMP1 is to help drive the ACP into the fibril. More experiments on the activity of the C-DMP1 need to be performed to understand its precise mechanism of action during collagen mineralization.
Considering the inhibition of the transformation of ACP into HA inside the collagen, we hypothesize that the Cu ions may bind to the nucleation sites in the collagen,37, 39 thus inhibiting their activity. Interestingly, in the presence of Cu ions the C-DMP1 also lost the ability to nucleate HA, leading to the formation of globular mineral structures instead, which are likely composed of ACP (figure 7C). Furthermore, Cu ions may also induce protein aggregation, which could explain the formation of globular mineral structures of calcium phosphate and C-DMP1.
It is interesting to note that when Cu ions are present, the ACP accumulates inside the collagen mainly in the overlap region of the fibril, both induced by pAsp alone (figures 3 and 4) and by pAsp combined with C-DMP1 (figure 7D). In situ TEM observations of HA formation on the mineralizing turkey tendon have demonstrated that it is the gap region of collagen the site where the HA crystals nucleate and grow.2, 8 More specifically, the region in the gap zone that corresponds to the e-band of uranyl acetate staining, has been implicated as the site where the HA crystals nucleate and grows.10 In this respect, we also note that also the biomimetic mineralization system that we employ lacks this specificity, since HA crystals form everywhere within a 67 nm repeat. Although collagen has nucleation sites both in the gap and overlap regions,37, 39 it is possible that part of the function of the NCPs is to target HA formation to specific sites. It must be pointed out, however, that there are studies that described HA nucleation and growth occurring concomitantly in the gap and overlap regions in the mineralizing turkey tendon,6, 42–44 which is in line with our observations.
4.5 Outlook
The present work raises several questions on the possible interactions between the ACP and the collagen. Nevertheless, our results show that the interactions between the amorphous precursor phase and the collagen fibril play an important role in the control over mineralization. Further experiments need to be conducted to fully understand the importance of the particle size and stability of the ACP for its infiltration into the collagen. In addition, the stabilization of ACP by Cu ions is also an interesting topic that demands further research to unravel how this ion interacts with the collagen fibrils, the pAsp and the C-DMP1, and thus affecting the mineralization process. Understanding these mechanisms can lead to further insights on the role of the amorphous precursor phase on collagen mineralization and hence on bone formation.
Acknowledgments
We thank G. Falini (University of Bologna, Italy) for kindly providing the horse tendon collagen; Laura J. Brylka (Eindhoven University of Technology) for her help with the initial mineralization experiments; J. van Roosmalen (Eindhoven University of Technology, The Netherlands) for his help with the tomography reconstructions. Supported by the Dutch Science Foundation, NWO, The Netherlands; by the European Community (FP6, project code NMP4-CT-2006-033277 TEM-PLANT) and by NIH Grant DE 11657 (A.G).
Contributor Information
Anne George, Email: anneg@uic.edu.
Nico A. J. M. Sommerdijk, Email: n.sommerdijk@tue.nl.
References
- 1.Weiner S, Wagner HD. Annu Rev Mater Sci. 1998;28:271–298. [Google Scholar]
- 2.Glimcher MJ, Muir H. Philosophical Transactions of the Royal Society of London Series B, Biological Sciences. 1984;304:479–508. doi: 10.1098/rstb.1984.0041. [DOI] [PubMed] [Google Scholar]
- 3.Hodge AJ, Petruska JA. In: Aspects of Protein Structure. Ramachandran GN, editor. Academic Press; New York: 1963. pp. 289–300. [Google Scholar]
- 4.Miller A. Philosophical Transactions of the Royal Society of London Series B, Biological Sciences. 1984;304:455–477. doi: 10.1098/rstb.1984.0040. [DOI] [PubMed] [Google Scholar]
- 5.Orgel JPRO, Irving TC, Miller A, Wess TJ. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9001–9005. doi: 10.1073/pnas.0502718103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fratzl P, Fratzl-Zelman N, Klaushofer K. Biophysical Journal. 1993;64:260–266. doi: 10.1016/S0006-3495(93)81362-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hulmes DJS, Wess TJ, Prockop DJ, Fratzl P. Biophysical Journal. 1995;68:1661–1670. doi: 10.1016/S0006-3495(95)80391-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landis WJ, Song MJ, Leith A, Mcewen L, Mcewen BF. Journal of Structural Biology. 1993;110:39–54. doi: 10.1006/jsbi.1993.1003. [DOI] [PubMed] [Google Scholar]
- 9.Traub W, Arad T, Weiner S. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:9822–9826. doi: 10.1073/pnas.86.24.9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Traub W, Arad T, Weiner S. Matrix. 1992;12:251–255. doi: 10.1016/s0934-8832(11)80076-4. [DOI] [PubMed] [Google Scholar]
- 11.Traub W, Arad T, Weiner S. Connective Tissue Research. 1992;28:99–111. doi: 10.3109/03008209209014230. [DOI] [PubMed] [Google Scholar]
- 12.Veis A, Perry A. Biochemistry-Us. 1967;6:2409. doi: 10.1021/bi00860a017. [DOI] [PubMed] [Google Scholar]
- 13.Ali SY, Sajdera SW, Anderson HC. Proceedings of the National Academy of Sciences of the United States of America. 1970;67:1513. doi: 10.1073/pnas.67.3.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson HC, Garimella R, Tague SE. Front Biosci. 2005;10:822–837. doi: 10.2741/1576. [DOI] [PubMed] [Google Scholar]
- 15.Mahamid J, Aichmayer B, Shimoni E, Ziblat R, Li C, Siegel S, Paris O, Fratzl P, Weiner S, Addadi L. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:6316–6321. doi: 10.1073/pnas.0914218107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahamid J, Sharir A, Addadi L, Weiner S. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12748–12753. doi: 10.1073/pnas.0803354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Addadi L, Politi Y, Nudelman F, Weiner S. 39th Course of the International School of Crystallography on Engineering of Crystalline Materials Properties; Erice, Italy. 2007. [Google Scholar]
- 18.Beniash E, Aizenberg J, Addadi L, Weiner S. P Roy Soc Lond B Bio. 1997;264:461–465. [Google Scholar]
- 19.Politi Y, Arad T, Klein E, Weiner S, Addadi L. Science. 2004;306:1161–1164. doi: 10.1126/science.1102289. [DOI] [PubMed] [Google Scholar]
- 20.Weiss IM, Tuross N, Addadi L, Weiner S. J Exp Zool. 2002;293:478–491. doi: 10.1002/jez.90004. [DOI] [PubMed] [Google Scholar]
- 21.Addadi L, Raz S, Weiner S. Adv Mater. 2003;15:959–970. [Google Scholar]
- 22.Nudelman F, Pieterse K, George A, Bomans PHH, Friedrich H, Brylka LJ, Hilbers PAJ, de With G, Sommerdijk NAJM. Nat Mater. 2010;9:1004–1009. doi: 10.1038/nmat2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deshpande AS, Beniash E. Crystal Growth & Design. 2008;8:3084–3090. doi: 10.1021/cg800252f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olszta MJ, Cheng XG, Jee SS, Kumar R, Kim YY, Kaufman MJ, Douglas EP, Gower LB. Materials Science & Engineering R-Reports. 2007;58:77–116. [Google Scholar]
- 25.George A, Veis A. Chemical Reviews. 2008;108:4670–4693. doi: 10.1021/cr0782729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He G, Dahl T, Veis A, George A. Nat Mater. 2003;2:552–558. doi: 10.1038/nmat945. [DOI] [PubMed] [Google Scholar]
- 27.He G, George A. Journal of Biological Chemistry. 2004;279:11649–11656. doi: 10.1074/jbc.M309296200. [DOI] [PubMed] [Google Scholar]
- 28.Vos MR, Bomans PHH, Frederik PM, Sommerdijk NAJM. Ultramicroscopy. 2008;108:1478–1483. doi: 10.1016/j.ultramic.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 29.Tampieri A, Celotti G, Landi E, Sandri M, Roveri N, Falini G. Journal of Biomedical Materials Research Part A. 2003;67A:618–625. doi: 10.1002/jbm.a.10039. [DOI] [PubMed] [Google Scholar]
- 30.Sanders HMHF, Iafisco M, Pouget EM, Bomans PHH, Nudelman F, Falini G, de With G, Merkx M, Strijkers GJ, Nicolay K, Sommerdijk NAJM. Chem Commun. 2011;47:1503–1505. doi: 10.1039/c0cc02901g. [DOI] [PubMed] [Google Scholar]
- 31.Jiang FZ, Horber H, Howard J, Muller DJ. Journal of Structural Biology. 2004;148:268–278. doi: 10.1016/j.jsb.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 32.Nudelman F, de With G, Sommerdijk NAJM. Soft Matter. 2011;7:17–24. [Google Scholar]
- 33.Chapman JA, Tzaphlidou M, Meek KM, Kadler KE. Electron Microscopy Reviews. 1990;3:143–182. doi: 10.1016/0892-0354(90)90018-n. [DOI] [PubMed] [Google Scholar]
- 34.Hodge AJ, Schmitt FO. Proceedings of the National Academy of Sciences of the United States of America. 1960;46:186–197. doi: 10.1073/pnas.46.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gower LB. Chemical Reviews. 2008;108:4551–4627. doi: 10.1021/cr800443h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Posner AS, Betts F. Accounts Chem Res. 1975;8:273–281. [Google Scholar]
- 37.Katz EP, Li S. Journal of Molecular Biology. 1973;80:1–15. doi: 10.1016/0022-2836(73)90230-1. [DOI] [PubMed] [Google Scholar]
- 38.Kawska A, Hochrein O, Brickmann A, Kniep R, Zahn D. Angewandte Chemie-International Edition. 2008;47:4982–4985. doi: 10.1002/anie.200800908. [DOI] [PubMed] [Google Scholar]
- 39.Landis WJ, Silver FH. Cells Tissues Organs. 2009;189:20–24. doi: 10.1159/000151454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okamoto Y, Hidaka S. J Biomed Mater Res. 1994;28:1403–1410. doi: 10.1002/jbm.820281204. [DOI] [PubMed] [Google Scholar]
- 41.Toroian D, Lim JE, Price PA. Journal of Biological Chemistry. 2007;282:22437–22447. doi: 10.1074/jbc.M700591200. [DOI] [PubMed] [Google Scholar]
- 42.Maitland ME, Arsenault AL. Calcified Tissue International. 1991;48:341–352. doi: 10.1007/BF02556154. [DOI] [PubMed] [Google Scholar]
- 43.Arsenault AL. Calcified Tissue International. 1988;43:202–212. doi: 10.1007/BF02555136. [DOI] [PubMed] [Google Scholar]
- 44.Arsenault AL. Calcified Tissue International. 1991;48:56–62. doi: 10.1007/BF02555796. [DOI] [PubMed] [Google Scholar]