

Abstract
Prostate cancer is the most commonly diagnosed cancer disease in men in the Unites States and its management remains challenge in everyday oncology practice. Thus, advanced therapeutic strategies are required to treat prostate cancer patients. Curcumin (CUR) is a promising anticancer agent for various cancer types. The objective of this study was to evaluate therapeutic potential of novel poly(lactic-co-glycolic acid)- CUR nanoparticles (PLGA-CUR NPs) for prostate cancer treatment. Our results indicate that PLGA-CUR NPs efficiently internalize in prostate cancer cells and release biologically active CUR in cytosolic compartment of cells for effective therapeutic activity. Cell proliferation (MTS), clonogenic, and Western blot analyses reveal that PLGA-CUR NPs can effectively inhibit proliferation and colony formation ability of prostate cancer cells than free CUR. PLGA-CUR NPs showed superior tumor regression compared to CUR in xenograft mice. Further investigations reveal that PLGA-CUR NPs inhibit nuclear β-catenin and AR expression in cells and in tumor xenograft tissues. It also suppresses STAT3 and AKT phosphorylation and leads to apoptosis via inhibition of key anti-apoptotic proteins, MCL-1, Bcl-xL and caused induction of PARP cleavage. Additionally, PLGA-CUR NPs significant downregulation of oncogenic miR21 and up-regulation of miR-205 was observed with PLGA-CUR NPs treatment as determined by RT-PCR and in situ hybridization analyses. A superior anti-cancer potential was attained with PSMA antibody conjugated PLGA-CUR NPs in prostate cancer cells and a significant tumor targeting of 131I labelled PSMA antibody was achieved with PLGA-CUR NPs in prostate cancer xenograft mice model. In conclusion, PLGA-CUR NPs can significantly accumulate and exhibit superior anticancer activity in prostate cancer.
Keywords: Nanoparticles, curcumin, chemoprevention, prostate cancer, targeted delivery
1. Introduction
Prostate cancer is the most commonly diagnosed cancer in men. Recent statistics estimated 233,00 new cases and 29,480 deaths occurred from prostate cancer in the United States in 2014 [1]. Prostate cancer mortality has been declining but therapy and surgery expenses are still very high [2, 3]. Conventional chemotherapy results in off-target effects: anti-cancer drug(s) cause damage to healthy rapidly dividing cells and thus patients often experience normal organ toxicity [4]. Epidemiological reports indicate that natural polyphenol compounds used in daily diet reduce the risk and incidence of various types of cancers [5, 6]. These compounds benefit overall health and longevity. Use of anti-cancer natural compounds is highly suitable for prostate cancer due to its high incidence rate as well as very long latency [7]. Natural polyphenol compounds may also act as an adjunct to surgery, chemotherapy and radiation [8]. Among many natural anti-cancer agents, curcumin is a favorable phytochemical that has demonstrated remarkable therapeutic potential for prostate cancer [9]. Research studies have proven that curcumin efficiently induces apoptosis through a number of different molecular targets and inhibits metastasis, invasion and angiogenesis [10]. At this stage, there are about 40 clinical trials studying the therapeutic effects of CUR in curing cancer(s). Based on these data, we expect that curcumin formulated in the form of pills or gel capsules has the potential to either delay the onset or prevent cancer progression.
Despite curcumin’s significant therapeutic value, its translation from basic research to clinical trials and human application is hindered due to its extreme degradation and metabolization which are responsible for poor pharmacokinetics, low bioavailability and pharmacological activity [11]. It is important to note that a trace amount of curcumin was detected in the serum of human when up to 4–12 g/day of CUR was administered [12]. Such a low bioavailability profile significantly hampers its clinical utility [13]. Additionally, it is a prerequisite that curcumin molecules reach the tumor site for its effective pharmacological actions. To achieve this, curcumin must be formulated using nanotechnology approaches for better medical application. To-date a number of drug delivery strategies have been adopted to incorporate or encapsulate curcumin in polymer nanoparticles (NPs), liposomes, inclusion complex, and self-assemblies utilizing their complex molecular structures. This resulted in active binding and enhanced uptake by the cancer cells and thus exhibited enhanced anti-cancer activity [14–16]. Poly(lactic-co-glycolic acid) (PLGA) based nanoformulations are non-toxic, biodegradable and non-immunogenic and are thus valuable in medical applications [17]. Abraxane®, a paclitaxel albumin-bound nanoparticle formulation, has been approved by the FDA and is in the market for metastasis cancer treatment(s) [18]. Another successful formulation of modified PLGA based docetaxel has successfully completed Phase I clinical trials for prostate cancer [19]. The clinical proof-of-success has inspired us to encapsulate curcumin in PLGA NPs [20] that can be a suitable/better choice for prostate cancer therapeutics. Furthermore, it has been demonstrated that curcumin bioavailability can be improved by formulating with PLGA polymer or PLGA with other polymers/copolymers.
Targeted nanotherapies may improve therapeutic outcome of prostate cancer treatment through targeting, accumulation and sustained release of therapeutics. Targeted nanoformulations can target the antigens specifically present and/or overexpressed in cancer tissues such as prostate stem cell antigen (PSCA), prostate specific membrane antigen (PSMA), six transmembrane epithelial antigen of the prostate 1 (STEAP1), prostatic acid phosphatase (PAP), T cell receptor gamma alternate reading frame protein (TARP), transient receptor potential (trp)-p8 (Trp-p8), etc. PSMA is a 100 kDa prostate epithelial cell type II transmembrane glycoprotein [21, 22]. PSMA is highly expressed in almost all prostate tumors and its expression progressively increases in higher Gleason grade and hormone refractory PrCa [21, 22]. PSMA is a highly validated target. This information suggests its applicability for targeting advanced stage PrCa; therefore, we propose to develop an anti-PSMA antibody coupled PLGA-CUR nanoparticle for prostate cancer.
Inhibition of AR and β-catenin nuclear localization play important roles in prostate tumorigenesis [23]. AR binds to β-catenin directly to stimulate AR-mediated gene transcription [24] and importantly, the AR gene itself is a transcriptional target of β-catenin [25]. Furthermore, enhanced crosstalk between AR and β-catenin has been observed in in vivo models. Therefore, inhibitors of nuclear AR and β-catenin, which act as oncogenes in prostate cancer, are highly desired. In the present study, we have generated an antibody conjugation compatible curcumin loaded PLGA nanoparticle formulation for improved targeting AR/β-catenin to induce anti-cancer activity in prostate cancer.
2. Experimental design
2.1. Materials
All reagents, solvents, chemicals and cell culture plastics were purchased from Sigma-Aldrich Co. (St. Louis, MO) or Fisher (Pittsburgh, PA) unless otherwise mentioned. All chemicals were used as received without further purification. PLGA-CUR was prepared following our previously published protocol using nanoprecipitation method [20]. Blank PLGA NPs were also prepared to use as control for all our studies.
2.2. Cell culture
LNCaP sublines (including C4-2) have been generated to provide an androgen dependent (AD) state and the most clinically relevant phenomenon. C4-2, a subline of LNCaP cells (metastatic lesion of human prostatic adenocarcinoma), were procured from Dr. Jaggi’s lab. Human prostate cancer cell lines (DU-145 and PC-3 cells, androgen independent (AI) characteristic) were purchased from the American Type Culture Collection (ATCC), have the propensity of prostate cancer to metastasize to bone but these cells in bone do not fully mimic clinical human disease. These cell lines were cultured in Roswell Park Memorial Institute (RPMI)-1640/Dulbecco’s Modified Eagle Medium (DMEM)-high medium (FBS, Invitrogen) supplemented with fetal bovine serum (10%, volume percentage) and 1% antibiotics (penicillin, 100 units/mL plus streptomycin) at 37 °C in a humidified incubator containing 5% CO2.
2.3. Cellular internalization, accumulation and retention of PLGA-CUR NPs
Transmission electron microscopy (TEM) and flow cytometry (FCM) methods were employed to elucidate the internalization of PLGA-CUR NPs in C4-2, DU-145 and PC-3 cells. For the TEM internalization study, cells (1 × 107 cells in 20 mL) were seeded in 150 mm dish and were allowed to attach overnight. To observe internalization and trafficking, cells were incubated with 10 μM CUR equivalent PLGA-CUR NPs for 0–18 hrs. Cells were then washed with cold phosphate buffer saline (PBS) solution, trypsinized, and centrifuged at 3,000 rpm to obtain a cell pellet. Cells were fixed with standard ice-cold formaldehyde (4%)-glutaraldehyde (1%) fixative solution followed by osmium tetroxide fixative solution, thin sectioned, and imaged under TEM according to our previously published method [20]. PLGA-CUR NPs in cancer cells were distinguished with high electron density (due to uranyl acetate staining). For flow cytometry, cells (5 × 105 cells per well) were seeded in 6-well plates, allowed to attach and then treated with 10 μM CUR or PLGA-CUR NPs. After treatment, the cells were washed with PBS, trypsinized, and centrifuged at 3,000 rpm and the cell pellet was resuspended in PBS containing 5% FBS. The internalization of CUR or PLGA-CUR was assessed by cellular fluorescence due to curcumin using an Accuri C6 flow cytometer (BD Accuri Cytometers, Inc., Ann Arbor, MI) with FL1 channel (488 nm excitation, Blue laser, 530 ± 15 nm, FITC/GFP) [20]. The flow cytometer acquisition was performed within 90 min of cell collection, thus there was no significant leach of CUR or PLGA-CUR NPs from the cancer cells. Similarly, the uptake of PLGA-CUR NPs were tested in the presence of endocytosis inhibitors such as genistein, chlorpromazine, nocodazole, methyl-β-cyclodextrin, or at 4°C (energy deprivation) to evaluate the internalization mechanisms in C4-2 and PC-3 (1×105 in 6 well-plates) cancer cells. The treatment concentrations were 10 μM or 15 μM PLGA-CUR NPs for C4-2 and PC-3 cells, respectively. The higher concentration was chosen in the case of PC-3 cells because this cell line exhibits lower cellular uptake compared to C4-2 cells. The internalization acquisition of PLGA-CUR NPs in the presence of endocytosis inhibitors or energy deprivation method was assessed using a flow cytometer as mentioned above. To examine the cellular accumulation and retention of PLGA-CUR NPs, cells (2 × 105) were grown in 6-well plates and incubated with CUR or PLGA-CUR NPs for 1, 2 and 4 days. After treatment, cells were washed with ice-cold PBS and the media was replaced with fresh media. Cells were then collected and processed to estimate curcumin levels by flow cytometer as described above.
2.4. Cell proliferation assay
The cytotoxicity of the PLGA-CUR nanoformulation was performed in prostate cancer cells using CellTiter 96® AQueous One Solution Cell Proliferation solution (Promega Corporation, Madison, WI). For this assay, cells (5,000 cells) were plated in 96-well plates and allowed to attach overnight. The cells were treated with 5–40 μM CUR or PLGA-CUR NPs for 2 days. Appropriate equivalent amounts of dimethyl sulfoxide (DMSO) or PLGA in PBS were used as controls. After completion of the treatment, the media was replaced with 100 μL fresh media containing 25 μL CellTiter 96® AQueous One Solution for 2–3 hrs. The color intensity developed by intracellular formazan was measured at 492 nm using a microplate reader (BioMate 3 UV-Vis spectrophotometer, Thermo Scientific, Waltham, MA). The percentage of cell growth was calculated as the percentage of the absorption of treated cells to the absorption of non-treated cells. Each treatment condition was replicated six times.
2.5. Clonogenic assay
Cancer cells (500 cells/well in 2 mL medium) were seeded in 6-well plates. After confirming that cells initiated colonies (day 3), cells were treated with 2–8 μM CUR or PLGA-CUR for a week, then media was replaced with fresh media without drugs and maintained until day 14. At the end of the treatment, cells were rinsed with PBS, fixed with cold methanol and stained with hematoxylin. The colonies were photographed using a Multimage™ light cabinet (Alpha Innotech Corporation, San Leandro, CA) with the help of AlphaEaseFC™ (Alpha ImagerHP AIC) software. The number of colonies was counted and quantified using the analysis tool for the auto count method at a constant density threshold as described in our previous publication [20]. Equivalent amounts of dimethyl sulfoxide (DMSO) or PLGA treated cells were considered as controls for these experiments.
2.6. Western blot
For Western blots, prostate cancer cells (1 × 106 in 10 mL medium) were seeded in a 100 mm culture dish and treated with 10 or 20 μM CUR or PLGA-CUR for 48 hrs. Cells were lysed with 2X SDS lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA). The process of extraction and quantification of proteins and the Western blotting method were followed in our established protocol [26]. In brief, equal protein amounts were loaded and separated in 4–20% SDS-PAGE acrylamide gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes. These membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.05% Tween-20 for 1 hr. The membranes were incubated with antibodies to: B-cell lymphoma-extra large (Bcl-xL), induced myeloid leukemia cell differentiation protein (Mcl-1), poly(ADP-ribose) polymerase (PARP), β-catenin, androgen receptor (AR), and β-actin (for protein loading control) (Cell Signaling, Danvers, MA). Primary antibodies were incubated overnight at 4 °C, washed and then probed with 1:2000 diluted horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit secondary antibody (Promega) for 1 hr at room temperature. The protein bands were detected and imaged with the Lumi-Light detection kit (Roche, Nutley, NJ) using a BioSpectrum® 500 Imaging System (UVP, Upland, CA, USA).
2.7. Immunofluorescence
For immunofluorescence, C4-2 cells (1 × 105/well) were grown on 4-well chamber glass slides and treated with 20 μM CUR or PLGA-CUR NPs or respective controls. Cells were then washed, fixed in 2% paraformaldehyde, permeabilized, blocked with 2% goat serum, and incubated with primary antibody (AR or β-catenin) overnight at 4 °C. The slides were washed with PBS and incubated with Alexa488 or Cy3 labeled secondary antibody for 1 hr at room temperature. The slides were washed and coverslips were mounted with aqueous antifade medium (Vector Laboratories, Burlingame, CA) for detection. Fluorescence images of stained cells were scanned and analyzed on a confocal laser scanning microscopy (Olympus FV1000 Laser Scanning Microscope, Olympus, Japan).
2.8. Reverse transcription–quantitative real-time polymerase chain reaction (QRT-PCR)
Prostate cancer cells were seeded in a 6-well plate, allowed to attach and treated with 10 μM CUR or PLGA-CUR for 2 days. Total RNA was extracted from treated cells using TRIzol reagent (Invitrogen) [27]. The integrity of the RNA was checked with an RNA 6000 Nano Assay kit and 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). For miRNA detection, 100 ng total RNA was reverse transcribed into cDNA using specific primers designed for miRNA analysis (Applied Biosystems, Foster City, CA). The miRNA expression levels were determined by QRT-PCR using Taqman PCR master mixture (no AmpErase UNG) and specific primers designed for detection of mature miRNAs (Applied Biosystems). The expression of miRNA was normalized with the expression of endogenous control, 18S rRNA.
2.9. PSMA MAb or 131I radiolabeling PSMA MAb conjugation to PLGA-CUR NPs
Prostate specific membrane antigen monoclonal antibody (PSMA MAb) (100 μg of J591, acquired from Dr. Neil Bander, Weill Cornell Medical College, New York) was labeled with 131I radio nucleotide using a Pierce iodination tube (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) for 2 min. Then radiolabelled 131I PSMA-MAb was passed through 1% BSA saturated G-20 Spandex silica gel column (GE Healthcare) to remove free radiolabel and the radiolabelled antibody was collected in 15 fractions. Radioactivity associated with 131I PSMA-MAb was determined using a CRC-25R radioisotope dose calibrator (Capintec Inc., Ramsey, NJ). The fractions containing the majority of the 131I labelled antibody were pooled together and used for PLGA-NPs and PLGA-CUR NPs conjugation reactions and for animal experiments. To formulate PSMA-MAb targeted PLGA-NPs or PLGA-CUR NPs and 131I PSMA-MAb targeted PLGA-NPs or PLGA-CUR NPs, 100 μg PSMA-MAb or 131I labelled 100 μg PSMA-MAb was conjugated to amine groups of PLL of PLGA-NPs or PLGA-CUR NPs (1 mg) through PEG–N-hydroxysuccinimide (NHS)-PEG, (PG-2-NS-5k, NANOCS, Boston, MA) coupling reaction.
2.10. Cellular binding and superior anti-cancer potential of PSMA MAb PLGA-CUR NPs
To investigate target specificity of PSMA MAb PLGA-CUR NPs (PSMA-PLGA-CUR NPs), a cellular binding experiment was performed on the C4-2 (PSMA expressing) cell line and evaluated by flow cytometry and immunofluorescence methods. In the flow cytometry experiment, C4-2 cells (1×105) were seeded in a 6 well-plate. The next day, cells were incubated with 1 μg equivalent PSMA-MAb or PSMA-PLGA-CUR NPs for 1 hr. For comparison, we also treated cells with control PLGA NPs and PLGA-CUR NPs. Then the cells were trypsinized, pelleted and re-suspended in phenol red free medium for fluorescence counts on an Accuri C6 flow cytometer (BD Accuri Cytometers, Inc.) with FL1 channel as mentioned in Section 2.3. Similarly, C4-2 cells (1×105) were treated with 1 μg equivalent PSMA-MAb or PSMA-PLGA-CUR NPs or PLGA-CUR NPs for 1 hr, fixed with 2% formaldehyde, permeabilized, blocked with 2% goat serum, and incubated with Alexa488 labeled secondary antibody for 1 hr at room temperature. These slides were washed, mounted with aqueous antifade medium and covered with coverslips to image on a confocal laser scanning microscopy (Olympus FV1000 Laser Scanning Microscope, Olympus, Japan). In order to examine the superior anti-cancer activity of PSMA-MAb-PLGA-CUR NPs over PLGA-CUR NPs and CUR, we plated C4-2 (PSMA expressing) and PC-3 (PSMA null) cancer cells (2.5×104) in a 6 well-plate, and treated with 10 μM CUR or equivalent PLGA-CUR NPs or PSMA-MAb-PLGA-CUR NPs for 6 hrs. The medium was replaced with fresh medium and cell proliferation was assessed by counting cells at day 1, 2, 3 and 4 using cell counter (Countess® Automatic Cell Counter, Life Technologies).
2.11. I131 PSMA-MAb PSMA MAb PLGA-CUR NPs binding specificity
Cellular binding experiments were performed with C4-2 (PSMA expressing) and PC-3 (PSMA null) cell lines. In brief, cells (5×105) were placed into a 15 mL falcon tube and incubated with 1 μCi equivalent I131 PSMA-MAb, PLGA NP-conjugated PSMA-MAb, or PLGA-CUR NP-conjugated PSMA-MAb in 1 mL medium. After incubation (1 hr), the cells were pelleted and the supernatant, which contained radiolabelled MAb or MAb conjugated NPs that had not bound to the cells, was measured for radioactivity using WIZARD2 Automatic Gamma Counter (Perkin Elmer, Waltham, MA). Untreated cells were used as the control in this experiment.
2.12. In vivo evaluation of PSMA-PLGA-CUR nanoformulation
We have established a C4-2 xenograft mouse model by subcutaneous injection of 5 × 106 cells in 100 μL Matrigel (BD Biosciences, Sparks, MD). Cells were implanted in the left or right flank of male athymic nude (nu/nu) mice (Charles River Laboratories, Wilmington, MA). Animals were housed under pathogen-free conditions with free access to food and acidified water. The drinking water was supplemented with potassium sorbate to prevent bacterial or fungal contamination. Mice developed tumors at least ~100 mm3 within 3 weeks. In vivo studies of systemic toxicity, tumor uptake and tumor targeting capabilities of PLGA-CUR NPs were conducted using xenograft mice in compliance with the principles and procedures approved by the local Institutional Animal Care and Use Committee.
2.12.1. Systemic toxicity
Systemic toxicity of PLGA-CUR NPs was evaluated in mice treated intraperitonially with 2.5 mg/kg CUR or equivalent curcumin in PLGA-CUR NPs or respective vehicle controls. Red blood cells were collected after euthanization under anesthetic condition. Toxicity of this formulation on red blood cells was evaluated using an Olympus BX 41 microscope (Olympus, Center Valley, PA).
2.12.2. Anti-tumor efficacy
The anti-cancer potential of PLGA-CUR nanoparticles was determined with the C4-2 xenograft mouse model. Mice bearing tumors were randomly divided into four groups (n=6): (1) curcumin (25 μg in DMSO); (2) PLGA-CUR NPs (25 μg curcumin equivalent NPs in PBS); (3) DMSO control; (4) Blank PLGA NPs. All treatments were injected intratumorally and the tumor size was measured using a digital Vernier caliper. The tumor volume was calculated using the ellipsoid volume equation: tumor volume (mm3) = π/6×L×W×H, wherein L is length, W is width, and H is height. The mice were euthanized after 7 days following treatment. Tumors were excised, collected, and fixed with 10% formalin for immunohistochemistry to determine cellular and molecular changes that occurred within the tumors.
2.12.3. Tumor specific targeting
Tumor specific targeting was determined using the C4-2 xenograft mouse model. Mice bearing tumors were treated with 5, 10, or 25 μCi I131 of radiolabelled PSMA MAb, PLGA-CUR-conjugated I131 radiolabelled PSMA MAb, or their respective controls. All treatments were delivered by intraperitoneal injection. These concentrations were chosen based on our previous experience in radiolabelled experiments. Tumor uptake and targeting efficiency was determined with the Carestream Xtreme animal imager (Carestream Health, Rochester, NY) using a radiographic screen at 24, 48, and 72 hrs. Animals were euthanized at 72 hrs and organs were harvested for ex vivo imaging.
2.13. Immunohistochemistry
Immunohistochemistry was used to determine the expression pattern of β-catenin, AR, F4/80, and CD31 using formalin-fixed, paraffin-embedded (FFPE) tissues of control and treated xenograft mice. The slides were stained using heat-induced antigen retrieval immunohistochemistry techniques with the Vector Biocon kit (Vector Laboratories) using indicated primary antibodies and analyzed as previously described [28].
2.14. In situ hybridization for miRNAs
In situ hybridization was conducted according to the manufacturer’s protocol for FFPE tissues of control and treated xenograft mice to detect the expression of miR-21 using the Biochain kit (Biochain, San Francisco, CA). DIG-labelled LNA oligo-nucleotides (EXIQON, Woburn, MA) were used for overnight hybridization at 50°C. The staining was carried out as per Exiqon user manual instructions. Briefly, after deparaffinization, specimens were fixed in 4% paraformaldehyde in DEPC-PBS for 20 min and subjected to digestion using 2X standard saline citrate and 0.1% Triton-X for the next 25 min. The slides were prehybridized with prehybridization solution for 4 hrs at 48°C followed by hybridization of the slides with hybridization buffer and probe (Digoxigenin labelled) at 45°C overnight. After stringent washing of slides with various grades of standard saline citrate, the slides were blocked using 1X blocking solution provided with the kit. The tissues were subsequently incubated overnight with the AP-conjugated anti-digoxingenin antibody. The slides were washed twice for 5 min with 1X Alkaline Phosphatase buffer. The final visualization was carried out with NBT/BCIP overnight followed by nuclear fast red counterstaining. The slides were mounted and imaged and analyzed under ScanScope® XT/XT2 system (Aperio, Vista, CA). All the reagents used for the assay were provided with the kit from Biochain.
2.15. Statistical analysis
Values were processed using Microsoft Excel 2007 software and presented as mean ± standard error of the mean (S.E.M.). Statistical analyses were performed using an unpaired, two-tailed student t-test. The level of significance was set at 3p < 0.05. All the graphs were plotted using Origin 6.1 software.
3. Results
The aim of this study was to evaluate the anti-cancer effects of PLGA-CUR NPs in androgen independent and androgen dependent prostate cancer cell line models. Therefore, we used C4-2, PC-3, and DU-145 prostate cancer cells throughout the study. PLGA was chosen to generate a curcumin loaded nanoparticle formulation because this copolymer has been widely examined and used as drug nanocarrier(s). Our PLGA-CUR NPs were formulated with a batch of 90 mg PLGA, 10 mg poly(L-lysine) (PLL), 200 mg poly(vinyl alcohol) (PVA), and 10 mg CUR. Physico-chemical properties of PLGA-CUR NPs were consistent with our previous report [20] (Figure 1A). The effect of PLGA-CUR NPs was examined in human prostate cancer cells using in vitro and in vivo xenograft mouse models. Tumor-targeted delivery of I131 labeled anti-PSMA conjugated PLGA-CUR NPs was evaluated in C4-2 xenograft mice model.
Figure 1. PLGA-CUR NPs improves internalization, accumulation, retention and alters cellular structure in prostate cancer cells.

(A) Hypothetical structure of PLGA-CUR NPs and the particle size determined by dynamic light scattering and transmission electron microscopy (TEM) [29]. (B) PLGA-CUR NPs internalize in a time dependent manner in DU145 cancer cells, (C) PLGA-CUR NPs accumulation depends on type of prostate cancer cells. Prostate cancer cells (1 × 107) were treated with 10 μM CUR equivalent PLGA-CUR NPs, washed with PBS, tripsinized, centrifuged at 3,000 rpm, and obtained cell pellet was fixed with formaldehyde (4%)-glutaraldehyde (1%) fixative solution followed by osmium tetroxide fixative solution. The thin sections of these cells were processed and imaged under TEM. (D) PLGA-CUR NPs revealed an enhanced cellular uptake and retention compared to curcumin, in prostate cancer cells. Cells (2 × 105) were treated with 5 μM CUR or equivalent PLGA-CUR NPs, on day 1, 2 and 4, cells were washed with PBS, trypsinized, and cell suspension was injected into Acuri C6 flow cytometer (Accuri Cytometers, Inc., Ann Arbor, MI, USA) to determine the fluorescence levels in FL1 channel (488 excitation, Blue laser, 530 ± 15 nm, FITC/GFP). Data is mean ± SEM (n = 3). *p value ≤ 0.05 when compared to curcumin. (E) PLGA-CUR NPs efficiently alters cellular structures and induces vacuoles in prostate cancer cells. Prostate cancer cells (1 × 107) were treated with 20 μM CUR equivalent PLGA-CUR NPs, cellular structures and lysosomal activity (vacuoles formation) was imaged using TEM.
3.1. Interaction of NPs with prostate cancer cells
Insufficient cellular internalization of a drug, or drug-loaded NPs, leads to suboptimal intracellular drug concentration, less effective cancer therapy and the potential for drug resistance. Therefore, we sought to determine the internalization efficiency of PLGA-CUR NPs in prostate cancer cells (Figure 1). PLGA-CUR NPs were efficiently internalized in a time dependent manner. By 1 hr there was a substantial number of NPs engulfed within membrane vesicles and by 3 hr, NPs were fully internalized into the cells (blue arrows). At the 18 hr time point, NPs were located near the nucleus (Figure 1B, blue arrows). Based on the robust internalization and distribution pattern of NPs, the process is likely a combination of clathrin mediated endocytosis and phagocytosis (Supplementary Information 1) [29]. In accordance with previous studies, this observation confirms that early time points play a key role in transporting NPs into cancer cells [30]. Endocytosed nanoformulations can escape from the endosome, localize in cytoplasm/cytosol and become distributed around the nucleus. Drugs released from the NPs at this point will likely induce superior cytotoxicity/therapeutic effects [31]. The uptake of PLGA-CUR NPs was further confirmed by flow cytometry (Supplementary Information 2), which showed clear cellular localization at the 18 hr time point. The extent of endocytosis of PLGA-CUR NPs varied in different prostate cancer cells (Figure 1C, blue arrows) (Supplementary Information 2). The possible differences in the mechanism of internalization of PLGA-CUR NPs in the three tested prostate cancer cell lines may be due to the presence of various lipid membranes on their surfaces. Additional details on the uptake of PLGA-CUR NPs are presented in Section 3.2.
In addition to internalization, long term accumulation and retention is also important to improve the therapeutic efficacy of cancer drugs. Many cancer drugs fail to reach effective doses within cells due to rapid degradation or cellular export. To assess the PLGA-CUR NPs in this regard, we studied the retention of free CUR and PLGA-CUR NPs at day 1, 2 and 4 by flow cytometry (Figure 1D). As expected, PLGA-CUR NPs exhibited higher accumulation and retention at each time point compared to free CUR. The extent of retention varied amongst the 3 cell lines tested and was in concordance with the TEM data described above. DU-145 cells exhibited the highest level of accumulation, which peaked at day 2 and was significantly lower at day 4. C4-2 cells had a gradual increase from day 1 to 2 and remained relatively similar at day 4. PC-3 had much less retention of both free curcumin and PLGA-CUR NPs, with a peak accumulation at day 2.
Due to the different cellular levels of PLGA-CUR NPs, the therapeutic response may be different in each cancer cell type. Using TEM, large and extensive production of vacuoles was observed in both C4-2 and DU-145 cancer cells (Figure 1E, red arrows). These cells also showed excessive lysosomal activity that probably arose from destabilization of lysosomal membranes which trigger cell death/apoptosis signals [32, 33]. It is also interesting to note that all the vacuoles were located around the periphery of the nucleus where internalized NPs can release bioactive CUR [34]. The existence of PLGA-CUR NPs in prostate cancer cells resulted in protrusion of cell membrane and disrupted cytoskeleton by inducing vacuoles in the cell body [35]. Free CUR was unable to induce this kind of vacuolization in prostate cancer cells [36]. PC-3 cells exhibited relatively fewer and smaller cytoplasmic vacuoles which suggest less NPs uptake.
3.2. Internalization mechanism
Our studies suggest an efficient internalization of PLGA-CUR NPs in prostate cancer cells. Therefore, we were interested to learn about internalization mechanisms of these NPs. Table 1 demonstrates that PLGA-CUR NP uptake by C4-2 and PC-3 was decreased when cells were co-cultured with various biochemical endocytosis inhibitors (clathrin-mediated endocytosis, caveolae-mediated endocytosis, macropinocytosis, and energy dependence). Therefore, the internalization process is largely mediated by a combination of clathrin and caveolae pathways. Low temperature significantly inhibits the endocytosis uptake of PLGA-CUR NPs compared to all other endocytosis inhibitors in C4-2 and PC-3 cancer cells. The order of inhibition of PLGA-CUR NPs was found to be: 4°C > methyl-β-cyclodextrin > chlorpromazine > genestein > nocodazole in C4-2 cells and 4°C > chlorpromazine > nocodazole > methyl-β-cyclodextrin in PC-3 cells. We did not observe any inhibition of uptake of PLGA-CUR NPs when PC-3 cells were incubated with genestein.
Table 1.
The influence of endocytosis inhibitors on cellular uptake of PLGA-CUR NPs.
| Name of inhibitor or condition | Function or mechanism of inhibitor/condition | Process | % Uptake | Response | ||
|---|---|---|---|---|---|---|
|
| ||||||
| C4-2 cells | PC-3 cells | C4-2 cells | PC-3 cells | |||
| No inhibitor | Normal uptake | No inhibition of endocytosis | 100.0±1.1 | 99.0±0.8 | NA | NA |
| Genestein | Tyrosine kinase inhibitor | Inhibitor of caveolae-mediated endocytosis | 81.8±0.6 | 108.0.5± | + | − |
| Methyl-β-cyclodextrin | Cholesterol extracting agent | Disrupts caveolae associated endocytosis/Per turbs the formation of clathrin-coated endocytosis | 67.2±1.2 | 86.3±8.0 | + | + |
| Chlorpromazine | Reversible translocation of clathrin from cell membrane | Blocking agents of clathrin-coated pit formation | 75.3±0.3 | 71.7±0.1 | + | + |
| Nocadazole | Polymerization of microtubule | Inhibitor of macro pinocytosis | 88.2±0.6 | 78.0±0.7 | + | + |
| 4°C | Low temperature | Blocks endocytosis active entry | 32.1±0.1 | 44.9±0.2 | ++ | ++ |
+ indicates significant blocking the uptake of NPs by cells; ++ indicates drastically blocking the uptake of NPs by cells; − indicates no change/effect on uptake of NPs by cells; NA indicates not applicable.
3.3. Anti-cancer effect of PLGA-CUR NPs
We next sought to determine the cytotoxicity profile of the PLGA-CUR NPs. Treatment with 2.5–40 μM of free CUR or CUR equivalent PLGA-CUR NPs for 48 hrs was clearly cytotoxic at the higher doses (Figure 2A–B). Upon treatment with free CUR or PLGA-CUR NPs, cells showed morphological changes such as shrinkage, aggregation, and spherical and micro nucleation. These morphological changes and severe membrane damage are clear signs of apoptosis [37]. These cellular changes were more pronounced with increasing concentrations of PLGA-CUR NPs. To quantify the effect of CUR and PLGA-CUR NPs, cell proliferation was assessed by an MTS colorimetric method. Empty PLGA NPs did not affect cell growth, even at high concentrations. However, as expected, both free CUR and PLGA-CUR NPs exhibited a dose-dependent effect in cell proliferation (Figure 2B). As expected, the IC50 value varied amongst the cell lines and between free CUR and PLGA-CUR NPs. The IC50 of PLGA-CUR NPs was 9.5, 4.9, and 20.4 μM while free CUR ranged from 11.2, 7.6, and > 40 μM for C4-2, DU-145 and PC-3 cancer cell lines, respectively. This suggests that C4-2 and DU-145 cancer cells are highly sensitive to PLGA-CUR NPs compared to PC-3. This may be due to a lower level of internalization of NPs.
Figure 2. PLGA-CUR NPs attenuate proliferation of prostate cancer cells.

(A) Representative phase contrast microscopic cell images demonstrates PLGA-CUR NPs induces significant cell death over curcumin. Phase contrast images of prostate cancer cells treated with vehicle (DMSO/PLGA-NPs, control), 10, 20, and 40 μM CUR or PLGA-CUR NPs for 48 hrs. Bar equals 20 microns. (B) MTS assay reveals PLGA-CUR NPs significantly inhibits proliferation of prostate cancer cells. Prostate cancer cells (5000) were treated with 2.5–40 μM curcumin or equivalent PLGA-CUR NPs or respective controls (DMSO/PLGA-NPs). After 48 hrs, cells were washed with PBS, incubated with 20 μL MTS reagent, and color density was measured using UV-Vis spectrophotometer at 492 nm. Data is mean ± SEM (n = 6). *p value ≤ 0.05 when compared to curcumin.
We also determined the anti-cancer activity of PLGA-CUR NPs through growth inhibition as determined by clonogenic assay. Cells treated with controls produced large colonies while CUR treatment resulted in a dose dependent inhibition of colony formation. Our PLGA-CUR NP formulation had a more pronounced inhibition on colony formation, even at 2 and 4 μM concentrations, compared to free CUR (Figure 3). Although 2 μM CUR treatment exhibited mild inhibitory effects on colony formation in C4-2, DU-145 and PC-3 cancer cells, a similar concentration of PLGA-CUR NPs showed a prominent reduction in colony formation assay. In all 3 cell lines, PLGA-CUR was also more effective at the 4 μM and 6 μM concentrations compared to CUR. This suggests a slow and sustained release of active CUR over time. Another interesting observation is that PLGA-CUR NPs exhibited a superior effect on PC-3 cells compared to its inhibitory effect in MTS assay (Figure 2B and 3B), suggesting that sustained exposure to CUR is effective in inhibiting growth of relatively resistance cell lines, such as PC-3. Overall, the slow and sustained release of CUR from internalized PLGA-CUR NPs may substantially improve anti-cancer effects of curcumin.
Figure 3. PLGA-CUR NPs inhibits clonogenic potential of prostate cancer cells.
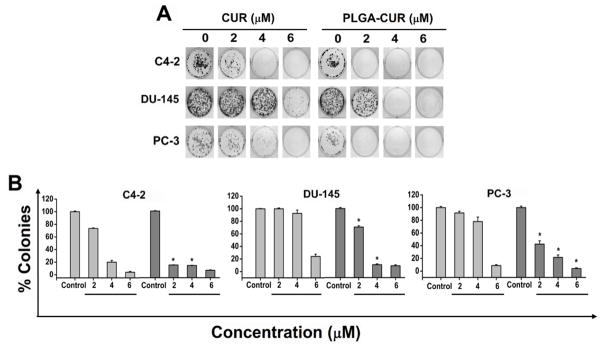
Prostate cancer cells (500) were incubated with 0, 2, 4, and 6 μM CUR or equivalent PLGA-CUR NPs or respective controls (DMSO/PLGA-NPs). After a week, cells were replaced with fresh medium without CUR or PLGA-CUR NPs. Day 14, cells were rinsed with PBS, fixed with cold methanol and stained with hematoxylin. (A) Photographs of clonogenic pattern in Multimage™ light cabinet (Alpha Innotech Corporation, San Leandro, CA) represent superior inhibition of clonogenic formulation with PLGA-CUR NPs. (B) A comparative quantification of clonogenic potential of CUR or PLGA-CUR NPs in prostate cancer cells. The number of colonies was quantified using AlphaEaseFC™ (Alpha ImagerHP AIC) software analysis tool. Data is mean ± SEM (n = 6). *p value ≤ 0.05 when compared to curcumin.
3.4. Anti-tumor effects of PLGA-CUR NPs in xenograft mice models
The anti-tumor efficacy of PLGA-CUR NPs was determined in a pre-clinical mouse xenograft model of prostate cancer. Mice with C4-2 xenograft tumors were treated with intratumoral injection of CUR or PLGA-CUR NPs (25 μg of CUR or PLGA-CUR NPs per mouse). Both CUR and PLGA-CUR NPs resulted in a lower tumor volume compared to control vehicles (DMSO or PLGA NPs) (Figure 4A). However, a superior anti-tumor effect was observed with PLGA-CUR NPs compared to curcumin. Additionally, no systemic toxicity associated with the PLGA-CUR formulation was observed as determined by analysis of red blood cells (Figure 4B).
Figure 4. Effect of PLGA-CUR NPs in tumor growth and cytocompatibility in xenograft male nude mouse model.

(A) PLGA-CUR NPs significantly reduce the tumor growth in C4-2 xenograft mice. Following tumor development, the mice were treated intratumorally one time with 25 μg CUR or PLGA-CUR NPs or respective controls (DMSO or PLGA-NPS). Tumor growth was measured after 7 days using a digital Vernier caliper. Data is mean ± SEM (n = 6). *p value ≤ 0.05 when compared to respective control. (B) PLGA-CUR NPs represent cyto-compatibility in mice. Following tumor development, the mice were treated intraperitonially with 2.5 mg/kg CUR or equivalent PLGA-CUR NPs or respective vehicle controls. Red blood cells were collected from blood after euthanization under anesthetic condition. Cyto-compatibility of the formulation on red blood cells was evaluated by Olympus BX 41 microscope (Olympus, Center Valley, PA). Bar equals 100 microns.
3.5. Modulation of key oncogenic signaling molecules and tumor microenvironment
As we had evidence that PLGA-CUR NPs were capable of reducing both in vitro and in vivo prostate cancer cell growth, we examined molecular pathways involved in apoptosis, oncogenic signaling pathways, and the tumor microenvironment. Prostate cancer cells treated with CUR or PLGA-CUR NPs exhibited PARP cleavage and inhibited the expression of anti-apoptotic proteins, Bcl-XL and Mcl-1 (Figure 5A). The presence of cleaved PARP is a hallmark for apoptosis and high expression levels of Bcl-xL are involved in cancer cell survival and chemo/radio-resistance [38]. We also observed that PLGA-CUR treatment inhibited the expression of STAT3 and phosphorylation of AKT at even the lowest concentration (Figure 5B). STAT3 promotes metastatic progression of prostate cancer [39] and PLGA-CUR treatment was able to inhibit STAT3 expression significantly as compared to control, whereas CUR alone showed no effects even at 20 μM. The PI3K/AKT pathway is also aberrantly expressed in prostate cancer and increased AKT phosphorylation accounts for prostate cancer progression [40].
Figure 5. PLGA-CUR NPs inhibits key oncogenic proteins and induces apoptosis in C4-2 prostate cancer cells.

(A) Representative Western blot of the PARP, Bcl-xL, Mcl-1, (B) STAT-3, p-Akt (Ser 473), and (C) AR protein expression. C4-2 prostate cancer cells (1 × 106) were treated with 10, 20, and 30 μM CUR or equivalent PLGA-CUR NPs or respective controls (DMSO) or PLGA-NPs) for 48 hrs, collected lysates in 2X SDS lysis buffer for Western blotting.
Further, we evaluated the impact of PLGA-CUR on AR and β-catenin. As a transcription factor, nuclear localization of AR is critical for its function and enhanced crosstalk between AR and β-catenin [41]. Therefore, inhibitors of nuclear AR and β-catenin are desired to inhibit prostate cancer and reduce its invasiveness. CUR is already known to down-regulate AR gene expression/activation in prostate cancer cells. Treatment with PLGA-CUR NPs drastically decreases the AR expression level (Figure 5C) compared to free curcumin. To further confirm the alterations in AR and β-catenin expression levels and their subcellular localization in response to PLGA-CUR NPs, C4-2 cells were treated and processed for immunocytochemical staining. Treatment with PLGA-CUR NPs substantially decreased the expression of AR in the nucleus and enhanced β-catenin localization on the cell membrane (Figure 6A and B). Further, treatment with PLGA-CUR NPs also resulted in increased expression of the tumor suppressor PKD1 (Figure 6B), which is important for inhibiting the oncogenic signals induced by nuclear β-catenin in prostate cancer cells [42].
Figure 6. Effect of PLGA-CUR NPs on the AR and β-catenin.
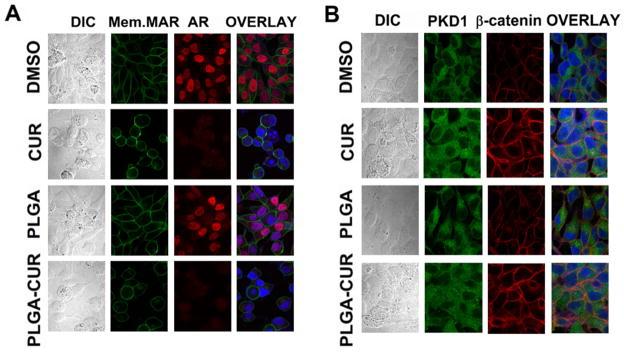
(A) PLGA-CUR NPs lowers the AR expression in C4-2 cells. C4-2 cells were treated with 20 μM curcumin or equivalent PLGA-CUR NPs, or respective control (DMSO or PLGA-NPs) for 24 hrs, immunostained for membrane marker (green), AR (red) and counter-stained with DAPI (blue). Curcumin and PLGA-CUR NPs treated cells showed significantly lower AR staining compared to control C4-2 cells. (B) PLGA-CUR NPs treatment improves the cellular localization of β-catenin and PKD1. C4-2 cells were treated with 20 μM curcumin or equivalent PLGA-CUR NPs, or respective control (DMSO or PLGA-NPs) for 1 hr, immunostained for PKD1 (green), β-catenin (red) and counter-stained with DAPI (blue). CUR and PLGA-CUR NPs treated cells exhibited higher membrane β-catenin staining and localized with PKD1, compared to control C4-2 cells. Original Magnifications 600X.
As described above and shown in Figure 4, mice treated with PLGA-CUR NPs by intratumoral injection had smaller tumors than mice treated with PLGA control. To further explore the molecular effects of CUR and PLGA-CUR NPs, the tumors from these mice were processed for immunohistochemical analysis. Both CUR and PLGA-CUR NP treatment dramatically reduced the expression of Bcl-xL and nuclear AR, in concordance with the in vitro data. Additionally, CUR and PLGA-CUR NP treatment dramatically enhanced membrane staining of β-catenin compared to mice treated with controls (DMSO and PLGA) (Figure 7). Thus, these data suggest that PLGA-CUR NPs can inhibit Bcl-xL, AR and nuclear β-catenin activity that are often dysregulated in prostate cancer. Additionally, treatment with PLGA-CUR NPs significantly reduced the tumor blood vessels density as indicated by fewer CD31+ vessels when compared to vehicle controls (Figure 7). The tumors from both CUR and PLGA-CUR NP-treated mice showed significantly less developed vasculature as compared to well-developed blood vessels in vehicle control-treated tumors. This indicates that PLGA-CUR NPs limit blood and nutrient supply to the tumors. Interestingly, a larger F4/80 + macrophage population was also found in the tumor tissues of mice that were treated with PLGA-CUR NPs (Figure 7). Tumor-suppressing macrophages act as a double-edged sword and their role has been largely described as linked to orchestration of T cell anti-tumor immunity [43].
Figure 7. Effects of CUR and PLGA-CUR NPs treatment on the expression of key oncoproteins.

The expression of Bcl-xL, β-catenin, AR, F4/80, and CD31 in C4-2 prostate tumor xenografts after treatment with an intratumoral injection of CUR or PLGA-CUR NPs (25 μg/mice). Representative images were obtained using an Olympus BX 41 Microscope. Original Magnifications 400X.
3.6. Expression miR-21 and miR-205
The role of miRNAs in cancer development and progression has recently been recognized. We investigated the expression of miRNAs which are known to be deregulated in prostate cancer. Of interest, real-time PCR showed at least a 9-fold reduction in expression of the oncomir, miR-21, in prostate cancer cells (C4-2 and DU-145) treated with PLGA-CUR NPs as compared to the PLGA control treatment (Figure 8A). Up-regulation of miR-21 has been detected in prostate cancer and suggested as a potential therapeutic target [44]. Additionally, we observed a 10-fold increase in miR-205 levels compared to PLGA (Supporting Information 3). miR-205 is down-regulated in prostate cancer and exerts tumor suppressive functions [45]. Of note, free CUR also affected the expression of miR-21 and miR-205, but not to the same extent as PLGA-CUR NPs.The decrease in miR-21 was also confirmed through in situ hybridization in tumor tissues obtained from the xenograft mouse model. We found a substantial decrease in miR-21 expression in the tumor tissues obtained from mice that were treated with both CUR and PLGA-CUR (Figure 8B).
Figure 8. Effects of CUR and PLGA-CUR on miR-21.

(A) PLGA-CUR NPs significantly lowers miR-21 expression in C4-2 and DU-145 cells. Cells were treated with 10 μM CUR or equivalent PLGA-CUR NPs, or respective control (DMSO or PLGA-NPs) for 48 hrs, the total RNA was extracted from these treated cells, and miRNA expression levels were determined by QRT-PCR using Taqman PCR master mixture (no AmpErase UNG) and specific primers designed for detection of mature miRNAs (Applied Biosystems). The expression of miRNA was normalized with the expression of endogenous control, 18S rRNA. *p value ≤ 0.05 when compared respective control. (B) PLGA-CUR NPs attenuates the expression of miR-21 in C4-2 prostate tumor xenografts. In situ hybridization of paraffin embedded tissues of control and treated xenograft mice was employed to detect the expression of miR-21 using DIG-labelled LNA oligo-nucleotides and the visualization step was achieved with NBT/BCIP. Representative images were obtained using under ScanScope® XT/XT2 system. Original Magnifications 400X.
3.7. Targeted Anti-cancer activity
To address the applicability of targeted PLGA-CUR NPs in prostate cancer treatment, we first examined its cellular targeting using C4-2 (PSMA expressing cells). As revealed by flow cytometry, greater fluorescence levels of CUR exist after treatment with PSMA-MAb PLGA-CUR NPs than PLGA-CUR NPs and CUR (Fig. 9A). This indicates that internalization of PSMA-MAb PLGA-CUR NPs is greater which may have significant implications for cancer treatments. Further, targeting efficacy was significantly greater with PSMA-MAb PLGA-CUR NPs (red fluorescence dots) over PSMA Mab (Fig. 9B). We did not observe such specific targeting on PC-3 (PSMA non-expressing cells). Due to such efficient targeting, PSMA-MAb PLGA-CUR NPs were capable of slowing the cellular growth on day 2, 3, and 4 compared to PLGA-CUR NPs or CUR in C4-2 cells (Fig 9C). PSMA-MAb PLGA-CUR NPs and PLGA-CUR NPs exhibited very similar cell growth arrest in PC-3 cells and was greater than CUR alone (Fig 9C). This indicates PSMA-MAb conjugated PLGA-CUR NPs are more effective in PSMA expressing cells than in PSMA null cells.
Figure 9. PSMA-MAb PLGA-CUR NPs efficiently target PSMA positive cells and effective in killing prostate cancer cells.

(A) PSMA-MAb PLGA-CUR NPs internalize significantly in PSMA+ (C4-2) over PSMA− (PC-3) cancer cells. Prostate cancer cells (1 × 106) were treated with 10 μM CUR or equivalent PLGA-CUR NPs or equivalent PSMA-MAb PLGA-CUR NPs, or with respective controls (DMSO/PLGA-NPs), washed with PBS, tripsinized, centrifuged at 3,000 rpm, the obtained cell pellet was re-suspended and cell suspension fluorescence levels in FL1 channel were analyzed suing an Acuri C6 flow cytometer Data is mean ± SEM (n = 3). *p value ≤ 0.05 when compared to curcumin vs DMSO, PLGA-CUR vs PLGA NPs, and PSMA-MAb PLGA-CUR NPs vs PLGA-CUR NPs. (B) PSMA-MAb PLGA-CUR NPs efficiently target PSMA+ (C4-2) over PSMA− (PC-3) cancer cells. Prostate cancer cells (1 × 106) were seeded in 4-well chamber slides, blocked with goat serum, and incubated with 2 μg equivalent PSMA-MAb of PSMA-MAb PLGA-CUR NPs. Then cells were washed with PBS, and probed with secondary antibody for red florescence detection on confocal microscope. (C) PSMA-MAb PLGA-CUR NPs efficiently target and significantly inhibits the growth of prostate cancer cells. Prostate cancer cells (2.5 × 105) in 12 well-plate were incubated with 10 μM CUR or equivalent PLGA-CUR NPs or equivalent PSMA-MAb PLGA-CUR NPs, or with respective controls (DMSO/PLGA-NPs). After treatment at 24, 48, 72, and 96 hrs, cells were tripsinized, centrifuged at 3,000 rpm, the obtained cell pellet was re-suspended and cell number was counted using a Countess™ automated cell counter. Data is mean ± SEM (n = 3). *p value ≤ 0.05 when compared to curcumin vs DMSO, PLGA-CUR vs PLGA NPs, and PSMA-MAb PLGA-CUR NPs vs PLGA-CUR NPs.
3.8. In vivo radio-immuno targeting
Despite significant challenges, immunotherapy with radio-labeled antibodies continues to be a promising strategy for effective cancer treatment. To explore the utility of PLGA-CUR NPs in targeted radio-immunotherapy applications, we generated 131I radiolabeled PSMA-MAb antibodies which were used as is or conjugated to PLGA NPs or PLGA-CUR NPs. After radio-labeling the PSMA MAb, fractions 3–6 were collected (Figure 10A) and the 131I-PSMA was conjugated to PLGA-NPs or PLGA-CUR NPs (Figure 10B). In cell binding experiments, 131 I -PSMA bound 2.2–2.8-fold higher to cells expressing PSMA (C4-2 cell lines) compared with PSMA null cells (PC-3 cell line) (Figure 10C). Importantly, conjugation of 131I-PSMA to PLGA-NPs or PLGA-CUR NPs did not inhibit cellular binding of PSMA to the prostate cancer cells. In fact, conjugation to NPs enhanced binding of 131I-PSMA to PSMA expressing cells.
Figure 10. 131I-labbled PSMA MAbs (J591) conjugated PLGA-CUR NPs specifically targets prostate cancer.

(A) 87.8% 131I-labeling achieved with PSMA MAbs (J591). Collection of radiolabeled PSMA MAbs at different fractions used for targeting experiments. (B) Represented illustrative structure of 131I-labeled PSMA MAbs (J591) conjugated PLGA-CUR NPs. (C) Binding efficiency of PSMA conjugated PLGA-NPs or PLGA-CUR NPs in prostate cancer cells (C4-2, PSMA expressing cells and PC-3 non-PSMA expressing cells). Data is mean ± SEM (n = 6). *p value ≤ 0.05 when compared PC-3 cells. (D) 131I-PSMA-PLGA-CUR NPs accumulate in a dose response at the tumor site 72 hrs after IP injection. The 131I was detected with the Carestream Xtreme animal imager using a radiographic screen. (E) 131I-PSMA accumulates more efficiently in the tumor when conjugated to PLGA-CUR NPs. 131I-PSMA-PLGA-CUR accumulates specifically in the tumor as shown by ex vivo imaging of organs harvested 72 hrs after IP injection (left image shows organs only). The image shows the radiographic intensity as an overlay in accordance with the scale at the bottom of the image. L: Lung, K: Kidney, H: Heart, M: Muscle, B: Bone, Lu: Lung, S: Spleen, T: Tumor.
We also examined the ability of 131I-PSMA-PLGA-CUR NPs to target prostate tumor tissue in the pre-clinical C4-2 xenograft mouse model. For these experiments, 131I PSMA-MAb and 131 I-PSMA-PLGA-CUR NPs were delivered via intraperitoneal injection into mice bearing a C4-2 xenograft tumor. The distribution of 131I was examined at 24, 48 and 72 hrs (data shown for 72 hrs). 131 I-PSMA-PLGA-CUR NPs specifically accumulated at the tumor site in a dose dependent manner (Figure 10D). As shown with whole-body and ex vivo imaging of multiple organs, 131I-PSMA-PLGA-CUR NPs accumulated and were retained within the tumor tissue at a much higher level than 131I-PSMA (Figure 10E). Interestingly, in contrast to the tumor, other organs exhibited very minimal 131I by 72 hrs. This data suggests a potential new targeted therapeutic approach for prostate cancer treatment.
4. Discussion
Prostate cancer can be treated with surgery, chemotherapy, and radiation therapy; however, 20–30% of patients experience recurrence after 5 years. An additional concern is that prostate cancer occurs mostly in older men who may be sensitive to side effects from first line chemotherapeutic agents currently used in treating prostate cancer. To lower recurrence and reduce negative side effects, alternative treatment strategies are needed and natural compounds may be beneficial. Preclinical, clinical and epidemiological data suggest that curcumin can effectively suppress prostate cancer cell proliferation, invasion, angiogenesis, and metastasis [9, 46, 47]. However, clinical translation of CUR is substantially hindered due to poor pharmacokinetics and insufficient bioavailability at the primary tumor as well as metastatic sites. Incorporating CUR into a nanoformulation may be very useful for patient treatment and is a viable option [14, 16]. Curcumin nanoformulations may target tumor tissue through either passive or active mechanisms that will improve its activity. Another advantage of a curcumin nanoformulation is its ability to bypass the efflux action of P-glycoprotein, which is commonly observed in resistant cancer cells. To date, curcumin formulations have not been designed to specifically target prostate cancer (Supplementary information 4). Therefore, in this study we begin development of a targeted CUR nanoparticle formulation (i.e., PLGA-CUR NPs) for prostate cancer treatment.
Herein, we explored the feasibility of targeting prostate cancer cells by conjugating our previously characterized PLGA-CUR nanoformulation [20] to PSMA MAb. PLGA drug NP formulations are known to improve the therapeutic index in cancer and reduce morbidity and mortality. The localization and accumulation of drug loaded NPs within tumors as well as within cancer cells can significantly improve drug activity [48]. Our data showed that our PLGA-CUR NPs can efficiently internalize and maintain CUR levels in prostate cancer cells (Figure 1). The internalization of PLGA-CUR NPs was evident by 1 hr and by 18 hrs PLGA-CUR NPs were located close to the nucleus, as evident from TEM analysis. The level of internalization varies depending on cancer cell type, but each of the three prostate cancer cell lines studied internalized the PLGA-CUR NPs. It is evident from the internalization experiment that endocytosis is the main pathway by which these PLGA-CUR NPs are taken up by the prostate cancer cells. This observation is elucidated in Table 1 that shows that nanoparticle internalization is significantly inhibited in the presence of endocytosis inhibitors. In this study, we demonstrate improved anti-cancer efficacy of PLGA-CUR NPs with in vitro and in vivo tumorigenic assays (Figure 2–4). The improved anti-cancer efficacy of our PLGA-CUR NP formulation is in accordance with previous studies [16, 49]. It is thought that NP formulations are more effective due to the sustained drug release over time compared to the short exposure to free CUR.
Our PLGA-CUR NPs potently induced cell death in prostate cancer cells through inducing PARP cleavage and down-regulation of anti-apoptotic proteins such as Bcl-xL and Mcl-1 (Figure 5A). Cleaved PARP is a key arbitrator of apoptosis which usually is caused by activation of Caspase-3/7 [50]. Mcl-1/Bcl-xL down-regulation is associated with suppression of platelet-derived growth factors (PDGF) and β-catenin transcription factors. We also revealed that PLGA-CUR NPs inhibit nuclear β-catenin and AR expression (Figures 5–7). β-catenin is a multifunctional protein that plays an important role in ontogenesis and oncogenesis. Dysregulation of β-catenin has been associated with increased AR activation [41] and development of many types of cancers, including prostate cancer. Additionally, PLGA-CUR up-regulates PKD1 (Figures 6B and 7), which is known to inhibit nuclear β-catenin and AR expression [51, 52].
Previous studies have shown that CUR is also involved in regulating STAT3 and AKT pathways in prostate cancer [53]. Thus, it is important to note that further decreasing the dose of CUR required to inhibit STAT3 and AKT phosphorylation activity without altering its function would benefit the cancer treatments. These improved specificities were achieved with structurally modified CUR analogs which effectively induced apoptosis. A similar significant down-regulation of activated or phosphorylation of these protein levels (Figure 5A) suggest that PLGA-CUR NPs can play a major role in suppression of tumorigenicity, chemoresistance, and resistance to hormone therapies. Additionally, our study shows that a PLGA-CUR formulation was also highly effective in suppressing angiogenesis as indicated by a decrease in CD31(+) microvessel density (Figure 7). The PLGA-CUR induced effects were accompanied by enhanced recruitment of macrophages (Figure 7) that infiltrate the tumor, induce T cell anti-tumor immunity [43] and facilitate the depletion of the tumor stroma [54]. All together, our in vitro results demonstrate that PLGA-CUR NPs exhibit greater anti-cancer potential over free CUR by inhibiting AKT and STAT3, and inducing apoptosis, which enhance its translational value as therapeutic agents. Additionally, PLGA-CUR NPs significantly down-regulate miR-21 and up-regulate miR-205 (Figure 8), which are known to be involved in prostate cancer progression and metastasis.
In this study, xenograft PrCa mice models were used to study the effect of PLGA-CUR NP treatment on in vivo tumorigenesis. A marked inhibition of tumor growth upon PLGA-CUR treatment further validates its potent anti-cancer efficacy for prostate cancer (Figure 4A). Additionally, the PLGA-NPs were evaluated for in vivo toxicity. Hemolytic toxicity evaluation of drug or drug nanoformulation is a critical parameter for clinical translation. The extensive binding, interaction or penetration of NPs may cause membranes to rupture, especially in red blood cells (RBCs), and cause severe systemic toxicity [55]. In the examination of PLGA-CUR for this toxicity in mice models, it was found that PLGA-CUR NPs in mice neither bind to RBCs nor get internalize (Figure 4B). PLGA-CUR NP treatment did not cause any detectable damage to the cell membrane of RBCs (Figure 4B). This hemo/cytocompatibility behavior of PLGA-CUR is in agreement with the evaluation of our prior ex vivo experiments with human RBCs [56]. This property encourages further development of a NP formulation for pre-clinical application of tumor specific targeted delivery. Overall, our in vitro and in vivo results suggest that PLGA-CUR NPs are effective and superior to free CUR in targeting oncogenic β-catenin and AR and suppressing cellular proliferation and inducing apoptotic cell death in prostate cancer cells.
PLGA based CUR NPs demonstrated up to 9-fold improved pharmacological and biological activity, indicating its efficient clinical utility for cancer treatment [15]. Targeted delivery of CUR to prostate cancer cells has the potential to be an effective treatment strategy. Targeting of tumor cells using antigens present on their surfaces can be the basis of a drug delivery strategy that will avoid side effects associated with nonspecificity of drug formulations. Such a formulation could be achieved by conjugating tumor specific targeting molecules (i.e. monoclonal antibody, peptides or nucleic acid/aptamers) to the surface of the NPs. To date, a specific targeted approach delivering CUR to prostate cancer cells had not been reported. Herein, we conjugated PSMA MAb to PLGA-CUR NPs. Our data clearly demonstrates that PSMA MAb PLGA-CUR NPs are specifically bound to C4-2 (PSMA expressing) and induce significant anti-cancer activity compared to PLGA-CUR and CUR (Figure 9C). This behavior is not observed in PC-3 cells because these cells do not express PSMA. Taken together, this experimental finding provides proof for targeted NPs-based therapy for prostate cancer. Because 94% of prostate cancers highly overexpress PSMA, our findings promote development of a PSMA targeted NPs approach in nano-medicine.
With this approach we can specifically target tumor cells in vivo as shown in the C4-2 xenograft mouse model (Figure 10D–E). This could be a promising nano-therapeutic strategy for prostate cancer treatment. In designing this formulation, we intentionally chose components that are already in individual clinical trials (PLGA, PSMA MAb and CUR). These components are well-known for their biodegradability and biocompatibility which will improve clinical translation. Such targeted nanoparticle approaches could be a useful strategy to improve chemo/radiation utility advancements in drug delivery technology which can co-deliver drug and radiation specifically to tumors. This proof-of-concept study delineates numerous favorable characteristics of targeted biodegradable NPs and the feasibility of developing a targeted NP platform that is capable of delivering both chemo- and radio-therapy, simultaneously.
Conclusions
Our results demonstrate that PLGA-CUR NPs efficiently inhibit growth of prostate cancer cells both in vitro and in vivo. This was achieved through lysosomal activity, apoptosis, and inhibition of AR and nuclear β-catenin activity. PLGA-CUR NPs significantly modulate the expression of miR-21 and miR-205. We show significant prostate tumor specific targeting in a xenograft mouse model using PSMA-PLGA-CUR NPs. This approach provides a proof-of-concept for the use of targeted nanotechnology-based CUR for effective and targeted killing of prostate cancer cells. Additional studies, however, are required to further confirm the improved therapeutic efficacy of this prostate cancer treatment approach.
Supplementary Material
Acknowledgments
This work was partially supported by grants from the Department of Defense (PC073887 to SCC and PC073643 to MJ), the National Institutes of Health (RO1 CA142736 to SCC and U01 CA162106 to SCC and MJ), and the College of Pharmacy 2013 Dean’s Seed Grant of the University of Tennessee Health Science Center (to MJ and MMY). Assistance from the Imaging, Molecular Pathology, Flow Cytometry and Tumor Biology Cores (supported by P20 GM103548-02 grant awarded to Dr. KM Miskimins) is gratefully acknowledged. Authors thank Cathy Christopherson (Sanford Research) for editorial assistance. Authors also thank Ashley Ezekiel Yacoubian (Department of Pathology, Tissue Services Core, University of Tennessee Health Science Center) for tissue staining and imaging.
Footnotes
Competing interests
The authors declare that they have no competing interests in this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Gianino MM, Galzerano M, Minniti D, Di NC, Martin B, Davini O, et al. A comparative costs analysis of brachytherapy and radical retropubic prostatectomy therapies for clinically localized prostate cancer. Int J Technol Assess Health Care. 2009;25:411–4. doi: 10.1017/S0266462309990043. [DOI] [PubMed] [Google Scholar]
- 3.Snyder CF, Frick KD, Blackford AL, Herbert RJ, Neville BA, Carducci MA, et al. How does initial treatment choice affect short-term and long-term costs for clinically localized prostate cancer? Cancer. 2010;116:5391–9. doi: 10.1002/cncr.25517. [DOI] [PubMed] [Google Scholar]
- 4.Sowery RD, So AI, Gleave ME. Therapeutic options in advanced prostate cancer: present and future. Curr Urol Rep. 2007;8:53–9. doi: 10.1007/s11934-007-0021-9. [DOI] [PubMed] [Google Scholar]
- 5.Sinha R, Anderson DE, McDonald SS, Greenwald P. Cancer risk and diet in india. J Postgrad Med. 2003;49:222–8. [PubMed] [Google Scholar]
- 6.Stan SD, Singh SV, Brand RE. Chemoprevention strategies for pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2010;7:347–56. doi: 10.1038/nrgastro.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cimino S, Sortino G, Favilla V, Castelli T, Madonia M, Sansalone S, et al. Polyphenols: key issues involved in chemoprevention of prostate cancer. Oxid Med Cell Longev. 2012;2012:632959. doi: 10.1155/2012/632959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anand P, Sundaram C, Jhurani S, Kunnumakkara AB, Aggarwal BB. Curcumin and cancer: an “old-age” disease with an “age-old” solution. Cancer Lett. 2008;267:133–64. doi: 10.1016/j.canlet.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 9.Aggarwal BB. Prostate cancer and curcumin: add spice to your life. Cancer Biol Ther. 2008;7:1436–40. doi: 10.4161/cbt.7.9.6659. [DOI] [PubMed] [Google Scholar]
- 10.Singh RP, Agarwal R. Mechanisms of action of novel agents for prostate cancer chemoprevention. Endocr Relat Cancer. 2006;13:751–78. doi: 10.1677/erc.1.01126. [DOI] [PubMed] [Google Scholar]
- 11.Burgos-Moron E, Calderon-Montano JM, Salvador J, Robles A, Lopez-Lazaro M. The dark side of curcumin. Int J Cancer. 2010;126:1771–5. doi: 10.1002/ijc.24967. [DOI] [PubMed] [Google Scholar]
- 12.Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4:807–18. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- 13.Epelbaum R, Schaffer M, Vizel B, Badmaev V, Bar-Sela G. Curcumin and gemcitabine in patients with advanced pancreatic cancer. Nutr Cancer. 2010;62:1137–41. doi: 10.1080/01635581.2010.513802. [DOI] [PubMed] [Google Scholar]
- 14.Ji JL, Huang XF, Zhu HL. Curcumin and its formulations: potential anti-cancer agents. Anticancer Agents Med Chem. 2012;12:210–8. doi: 10.2174/187152012800228733. [DOI] [PubMed] [Google Scholar]
- 15.Bansal SS, Goel M, Aqil F, Vadhanam MV, Gupta RC. Advanced drug delivery systems of curcumin for cancer chemoprevention. Cancer Prev Res. 2011;4:1158–71. doi: 10.1158/1940-6207.CAPR-10-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yallapu MM, Othman SF, Curtis ET, Bauer NA, Chauhan N, Kumar D, et al. Curcumin-loaded magnetic nanoparticles for breast cancer therapeutics and imaging applications. Int J Nanomedicine. 2012;7:1761–79. doi: 10.2147/IJN.S29290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161:505–22. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 18.Miele E, Spinelli GP, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int J Nanomedicine. 2009;4:99–105. doi: 10.2147/ijn.s3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4:128ra39. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 20.Yallapu MM, Gupta BK, Jaggi M, Chauhan SC. Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells. J Colloid Interface Sci. 2010;351:19–29. doi: 10.1016/j.jcis.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 21.Perner S, Hofer MD, Kim R, Shah RB, Li H, Moller P, et al. Prostate-specific membrane antigen expression as a predictor of prostate cancer progression. Hum Pathol. 2007;38:696–701. doi: 10.1016/j.humpath.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 22.Tricoli JV, Schoenfeldt M, Conley BA. Detection of prostate cancer and predicting progression: current and future diagnostic markers. Clin Cancer Res. 2004;10:3943–53. doi: 10.1158/1078-0432.CCR-03-0200. [DOI] [PubMed] [Google Scholar]
- 23.Kypta RM, Waxman J. Wnt/beta-catenin signalling in prostate cancer. Nat Rev Urol. 2012;9:418–28. doi: 10.1038/nrurol.2012.116. [DOI] [PubMed] [Google Scholar]
- 24.Song LN, Gelmann EP. Interaction of beta-catenin and TIF2/GRIP1 in transcriptional activation by the androgen receptor. J Biol Chem. 2005;280:37853–67. doi: 10.1074/jbc.M503850200. [DOI] [PubMed] [Google Scholar]
- 25.Yang X, Chen MW, Terry S, Vacherot F, Bemis DL, Capodice J, et al. Complex regulation of human androgen receptor expression by wnt signaling in prostate cancer cells. Oncogene. 2006;25:3436–44. doi: 10.1038/sj.onc.1209366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yallapu MM, Maher DM, Sundram V, Bell MC, Jaggi M, Chauhan SC. Curcumin induces chemo/radio-sensitization in ovarian cancer cells and curcumin nanoparticles inhibit ovarian cancer cell growth. J Ovarian Res. 2010;3:11. doi: 10.1186/1757-2215-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan S, Chib R, Shah BA, Wani ZA, Dhar N, Mondhe DM, et al. A cyano analogue of boswellic acid induces crosstalk between p53/puma/bax and telomerase that stages the human papillomavirus type 18 positive HeLa cells to apoptotic death. Eur J Pharmacol. 2011;660:241–8. doi: 10.1016/j.ejphar.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 28.Chauhan SC, Vannatta K, Ebeling MC, Vinayek N, Watanabe A, Pandey KK, et al. Expression and functions of transmembrane mucin MUC13 in ovarian cancer. Cancer Res. 2009;69:765–74. doi: 10.1158/0008-5472.CAN-08-0587. [DOI] [PubMed] [Google Scholar]
- 29.Panariti A, Miserocchi G, Rivolta I. The effect of nanoparticle uptake on cellular behavior: disrupting or enabling functions? Nanotech Sci Appl. 2012;5:87–100. doi: 10.2147/NSA.S25515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iversen T, Skotland TKS. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today. 2011;6:176–85. [Google Scholar]
- 31.Mo L, Hou L, Guo D, Xiao X, Mao P, Yang X. Preparation and characterization of teniposide PLGA nanoparticles and their uptake in human glioblastoma U87MG cells. Int J Pharm. 2012;436:815–24. doi: 10.1016/j.ijpharm.2012.07.050. [DOI] [PubMed] [Google Scholar]
- 32.Thomas TP, Majoros I, Kotlyar A, Mullen D, Holl MM, Baker JR., Jr Cationic poly(amidoamine) dendrimer induces lysosomal apoptotic pathway at therapeutically relevant concentrations. Biomacromolecules. 2009;10:3207–14. doi: 10.1021/bm900683r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tahara Y, Nakamura M, Yang M, Zhang M, Iijima S, Yudasaka M. Lysosomal membrane destabilization induced by high accumulation of single-walled carbon nanohorns in murine macrophage RAW 264.7. Biomaterials. 2012;33:2762–9. doi: 10.1016/j.biomaterials.2011.12.023. [DOI] [PubMed] [Google Scholar]
- 34.Mukerjee A, Vishwanatha JK. Formulation, characterization and evaluation of curcumin-loaded PLGA nanospheres for cancer therapy. Anticancer Res. 2009;29:3867–75. [PubMed] [Google Scholar]
- 35.Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol. 2007;72:29–39. doi: 10.1124/mol.106.033167. [DOI] [PubMed] [Google Scholar]
- 36.Yallapu MM, Dobberpuhl MR, Maher DM, Jaggi M, Chauhan SC. Design of curcumin loaded cellulose nanoparticles for prostate cancer. Curr Drug Metab. 2012;13:120–8. doi: 10.2174/138920012798356952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lane JD, Allan VJ, Woodman PG. Active relocation of chromatin and endoplasmic reticulum into blebs in late apoptotic cells. J Cell Sci. 2005;118:4059–71. doi: 10.1242/jcs.02529. [DOI] [PubMed] [Google Scholar]
- 38.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–15. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 39.Abdulghani J, Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, et al. Stat3 promotes metastatic progression of prostate cancer. Am J Pathol. 2008;172:1717–28. doi: 10.2353/ajpath.2008.071054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shukla S, Maclennan GT, Hartman DJ, Fu P, Resnick MI, Gupta S. Activation of PI3K-Akt signaling pathway promotes prostate cancer cell invasion. Int J Cancer. 2007;121:1424–32. doi: 10.1002/ijc.22862. [DOI] [PubMed] [Google Scholar]
- 41.Wang G, Wang J, Sadar MD. Crosstalk between the androgen receptor and beta-catenin in castrate-resistant prostate cancer. Cancer Res. 2008;68:9918–27. doi: 10.1158/0008-5472.CAN-08-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sundram V, Chauhan SC, Ebeling M, Jaggi M. Curcumin attenuates beta-catenin signaling in prostate cancer cells through activation of protein kinase D1. PloS one. 2012;7:e35368. doi: 10.1371/journal.pone.0035368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheth S, Jajoo S, Kaur T, Mukherjea D, Sheehan K, Rybak LP, et al. Resveratrol reduces prostate cancer growth and metastasis by inhibiting the Akt/MicroRNA-21 pathway. PloS one. 2012;7:e51655. doi: 10.1371/journal.pone.0051655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gandellini P, Folini M, Longoni N, Pennati M, Binda M, Colecchia M, et al. miR-205 Exerts tumor-suppressive functions in human prostate through down-regulation of protein kinase Cepsilon. Cancer Res. 2009;69:2287–95. doi: 10.1158/0008-5472.CAN-08-2894. [DOI] [PubMed] [Google Scholar]
- 46.Teiten MH, Gaascht F, Eifes S, Dicato M, Diederich M. Chemopreventive potential of curcumin in prostate cancer. Genes Nutr. 2010;5:61–74. doi: 10.1007/s12263-009-0152-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269:199–225. doi: 10.1016/j.canlet.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 48.Shekhar C. Lean and mean: nanoparticle-based delivery improves performance of cancer drugs. Chem Biol. 2009;16:349–50. doi: 10.1016/j.chembiol.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Yallapu MM, Othman SF, Curtis ET, Gupta BK, Jaggi M, Chauhan SC. Multi-functional magnetic nanoparticles for magnetic resonance imaging and cancer therapy. Biomaterials. 2011;32:1890–905. doi: 10.1016/j.biomaterials.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Algeciras-Schimnich A, Barnhart BC, Peter ME. Apoptosis dependent and independent functions of caspases. In: Marek L, Walczak H, editors. Caspases—their role in cell death and cell survival. New York, USA: Kluwer Academic/Plenum Publishers; 2002. [Google Scholar]
- 51.Mak P, Jaggi M, Syed V, Chauhan SC, Hassan S, Biswas H, et al. Protein kinase D1 (PKD1) influences androgen receptor (AR) function in prostate cancer cells. Biochem Biophys Res Commun. 2008;373:618–23. doi: 10.1016/j.bbrc.2008.06.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jaggi M, Chauhan SC, Du C, Balaji KC. Bryostatin 1 modulates beta-catenin subcellular localization and transcription activity through protein kinase D1 activation. Mol Cancer Ther. 2008;7:2703–12. doi: 10.1158/1535-7163.MCT-08-0119. [DOI] [PubMed] [Google Scholar]
- 53.Ravindran J, Prasad S, Aggarwal BB. Curcumin and cancer cells: how many ways can curry kill tumor cells selectively? AAPS J. 2009;11:495–510. doi: 10.1208/s12248-009-9128-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Merika EE, Syrigos KN, Saif MW. Desmoplasia in pancreatic cancer. Can we fight it? Gastroenterol Res Pract. 2012;2012:781765. doi: 10.1155/2012/781765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ungaro F, Conte C, Ostacolo L, Maglio G, Barbieri A, Arra C, et al. Core-shell biodegradable nanoassemblies for the passive targeting of docetaxel: features, antiproliferative activity and in vivo toxicity. Nanomedicine. 2012;8:637–46. doi: 10.1016/j.nano.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 56.Yallapu MM, Ebeling MC, Chauhan N, Jaggi M, Chauhan SC. Interaction of curcumin nanoformulations with human plasma proteins and erythrocytes. Int J Nanomedicine. 2011;6:2779–90. doi: 10.2147/IJN.S25534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.