

Abstract
We report a protocol for oxidative [3+2] cycloadditions of phenols and alkenes applicable to the modular synthesis of a large family of dihydrobenzofuran natural products. Visible light-activated transition metal photocatalysis enables the use of ammonium persulfate as an easily handled, benign terminal oxidant. The broad range of organic substrates that are readily oxidized by photoredox catalysis suggests that this strategy may be applicable to a variety of useful oxidative transformations.
Keywords: cycloaddition, heterocycles, phenols, photocatalysis, photooxidation
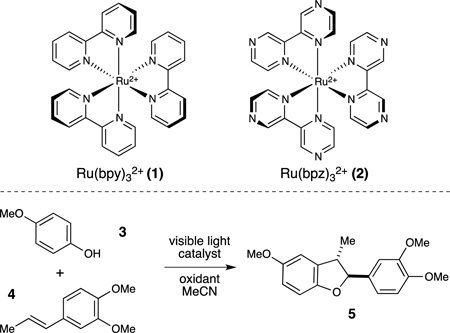
The choice of terminal oxidant is important in the design of oxidative reactions.1 Many of the most commonly used oxidants produce stoichiometric byproducts that can be practically or environmentally problematic. Our laboratory has a long-standing interest in the propensity of photoexcited Ru*(bpy)32+ to undergo redox reactions with a diverse range of quenchers,2 a feature that has been increasingly exploited in the design of synthetic reactions.3 We wondered if photoredox catalysis might offer a general strategy to employ benign, kinetically inert oxidants in oxidative transformations. Recent interest in photoredox catalysis has largely been focused on redox-neutral and net reductive reactions; the examples of net oxidative transformations published to date have generally utilized either stoichiometric halocarbon oxidants,4 which are not ideal from an environmental standpoint, or molecular oxygen,5 which is a triplet quencher of many photoexcited molecules6 that can negatively impact the overall efficiency of photocatalytic reactions. Thus, there exists a need for a more practical, general approach to the design of oxidative photoredox reactions.
We became interested in studying phenol oxidation as a starting point to explore this challenge. Phenols participate in a rich variety of oxidatively induced transformations,7 which can produce a number of complex structures common in bioactive molecules. For example, many neolignans, resveratrol oligomers, and peptide-derived natural products feature a 2,3-dihydrobenzofuran core (Figure 1),8 the biogenic origin of which presumably involves oxidative [3+2] phenol-alkene cycloaddition. Several synthetic approaches to this transformation have been reported,9 but they often suffer from low yields, limited scope, or a need for specialized equipment.10,11 The most practical methods for this reaction reported to date exploit hypervalent iodine(III) reagents,12 which generate iodoarenes as stoichiometric byproducts. We report herein an alternate photocatalytic protocol for [3+2] phenol–olefin cycloaddition that enables the use of ammonium persulfate as an inexpensive terminal oxidant with a benign bisulfate salt as the stoichiometric byproduct.13
Figure 1.

Bioactive dihydrobenzofuran-containing natural products and an oxidative [3+2] cycloaddition strategy for their synthesis..
Our initial investigations (Table 1) focused upon the photocatalytic reaction of p-methoxyphenol (3) with methylisoeugenol (4). A screen of oxidants in the presence of Ru(bpy)32+(1) revealed that while inorganic and organic hydroperoxides were ineffective (entries 1–4), the desired cycloadduct is formed slowly upon irradiation in the presence of Oxone® (entry 5). This observation led us to examine other persulfates, and K2S2O8 proved to be a more effective terminal oxidant (entry 6). Peroxydisulfates have long been known as oxidative quenchers of photoexcited ruthenium polypyridyl complexes,14 but their use as terminal oxidants for synthetic photocatalytic reactions has been limited.15 A brief screen of photocatalysts revealed that the more strongly oxidizing Ru(bpz)32+(2) chromophore provided faster rates than Ru(bpy)32+, affording good yield of the cycloadduct after 24 h. We also examined other peroxydisulfate salts and found that (NH4)2S2O8 provided optimal yields. Finally, control studies indicated that this reaction requires both catalyst and light (entries 10–11), validating the photocatalytic nature of this process.
Table 1.
Optimization of conditions for oxidative [3+2] cycloaddition
 | |||
|---|---|---|---|
| Entry[a] | Catalyst | Oxidant | Yield[b] |
| 1 | Ru(bpy)3(PF6)2 | H2O2 (30% aq) | 0% |
| 2 | Ru(bpy)3(PF6)2 | H2O2•urea | 0% |
| 3 | Ru(bpy)3(PF6)2 | t-BuOOH | 0% |
| 4 | Ru(bpy)3(PF6)2 | m-CPBA | 0% |
| 5 | Ru(bpy)3(PF6)2 | Oxone® | 7% |
| 6 | Ru(bpy)3(PF6)2 | K2S2O8 | 20% |
| 7 | Ru(bpz)3(PF6)2 | K2S2O8 | 75% |
| 8 | Ru(bpz)3(PF6)2 | Na2S2O8 | 23% |
| 9 | Ru(bpz)3(PF6)2 | (NH4)2S2O8 | 78% |
| 10c | Ru(bpz)3(PF6)2 | (NH4)2S2O8 | 0% |
| 11 | none | (NH4)2S2O8 | 0% |
All reactions were irradiated for 24 h using 0.10 mmol phenol, 0.13 mmol styrene, 0.20 mmol oxidant, and 0.005 mmol catalyst, unless noted.
Yields determined by 1H NMR spectroscopy using TMSPh as an internal standard.
Reaction performed in the dark.
A broad range of coupling partners participate readily in this oxidative [3+2] cycloaddition (Figure 2). The reaction requires electron-rich phenols bearing alkoxy substituents at the 2- or 4-position, consistent with the need to stabilize the putative phenoxonium intermediate. Within this constraint, however, the scope proved to be quite broad. Benzyl (7) and allyl (8) ethers were tolerated easily without any trace of oxidative degradation, as were unprotected alcohols (9). Other substituents are also tolerated, including aryl substituents (11), bulky alkyl groups (12) and halides (14). Unsymmetrical 3-substituted phenols undergo clean, highly regioselective [3+2] cycloadditions (15 and 16), suggesting that the reaction is susceptible to steric control. Nevertheless, 3,5-disubstituted phenols do not suffer from significantly lower reactivity (17). Condensed polycyclic phenols are also excellent substrates for this reaction (18). We also examined the scope of this reaction with respect to the styrene component. In line with the highly electrophilic nature of the phenoxonium intermediate, electron-rich styrenes bearing para or ortho alkoxy groups were the most reactive cycloaddition partners (19–21). However, styrenes lacking these activating groups still reacted smoothly (22–24). As with the phenol component, a variety of potentially sensitive functional groups could be present on the alkene substrate, including halides (25), sulfonamides (26), and acetals (27). The reaction was also tolerant of steric bulk at the α and β positions of the styrene (28–30).
Figure 2.

Scope studies for the oxidative [3+2] cycloaddition[a] All reactions conducted on 0.40 mmol phenol, 0.52 mmol alkene, 0.80 mmol (NH4)2S2O8, and 0.02 mmol 2•(PF6)2, unless otherwise noted. Yields represent the averaged isolated yields of two reproducible experiments.[b] Reactions conducted with 0.80 mmol alkene.
Several experiments were conducted to probe the mechanism of this reaction. Although both coupling partners are electron-rich, we designed this reaction as an entry into the versatile reactivity of oxidized phenol compounds. In accord with this hypothesis, reactions conducted with readily oxidized phenols such as 3 (Eox = +1.05 V vs. SCE)10b proceed even in the presence of alkenes that possess oxidation potentials outside of the working range for the photocatalyst ([Ru]3+/2+, +1.98 V; styrene 31,+2.05 V, Scheme 2).16 However, readily oxidized alkenes, such as anethole (4, +1.10 V)17 do not generate product in the presence of less electronrich phenol (32, Eox =+1.30 V).18 This suggests that phenol oxidation, rather than alkene oxidation, is the key step that initiates the cycloaddition.
Additionally, we observe an induction period of several hours (see Supporting Information, Figure S1), during which time an orange precipitate forms in the reaction mixture. Upon filtration, the supernatant is not catalytically active (Figure S2). The precipitate, however, can be used to catalyze the [3+2] cycloaddition, and we observed no induction period in this experiment (Figure S3). One reasonable interpretation of these data is that the active photocatalyst is an insoluble Ru(bpz)32+ persulfate complex that forms via a slow salt metathesis process. Consistent with this interpretation, authentic Ru(bpz)3(S2O8), precipitated from a combination of Ru(bpz)3(PF6)2 with (Bu4N)2(S2O8) in the dark, proved to be a competent catalyst, and we also observed no induction period in this experiment (Figure S4).
Based upon these observations, we have proposed the mechanistic model outlined in Scheme 2. A slow salt metathesis results in the precipitation of Ru(bpz)3(S2O8). Photoexcitation of this salt followed by oxidative quenching then generates the active oxidant, Ru(bpz)33+, along with sulfate radical anion. Oxidation of phenol generates the corresponding radical cation, which can be further oxidized to generate a resonance-stabilized phenoxonium cation that is trapped by an electron rich olefin to afford the observed dihydrobenzofuran.12a
Many bioactive natural products feature dihydrobenzofuran scaffolds, and the broad scope of the oxidative [3+2] cycloaddition makes it readily applicable to the efficient, modular assembly of compounds in this class (Scheme 3). For instance, the dihydrobenzofuran 35, isolated along with several similar benzofuranoid neolignans from Piper aequale,19 presumably arises from an oxidative [3+2] phenol cycloaddition. This coupling can be replicated using photocatalytic [3+2] cycloaddition of 33 with TBS-protected 4-propenylphenol 34. Subsequent deprotection of the silyl group affords the natural product in 81% yield over these two steps. Similarly, the antiprotozoal neolignan 37, isolated from K. ixina,20 is available in three steps from cycloadduct 7. Selective hydrogenolysis of the primary benzyl ether followed by treatment with triflic anhydride affords aryl triflate 36 in 93% yield over two steps. Suzuki coupling with trans-propenyl boronic acid produces 37 in 95% yield. Together, these syntheses demonstrate the applicability of this photocatalytic method to access this large family of bioactive benzofuranoid natural products.
In conclusion, we have developed a robust photocatalytic method for the oxidative [3+2] cycloaddition of phenols and electron-rich styrenes. Transition metal photoredox catalysis enables the use of ammonium persulfate as a terminal oxidant, which results in the formation of an innocuous and easily separated inorganic byproduct. Given the diverse range of organic substrates that participate readily in electron-transfer processes with photogenerated ruthenium polypyridyl oxidants, this strategy should be widely applicable to the design of other photocatalytically enabled oxidative transformations. This broad objective is a continuing goal of research in our laboratory.
Supplementary Material
Scheme 1.

Evidence for initial phenol oxidation.
Scheme 2.

Proposed mechanism for oxidative [3+2] cycloaddition of phenols using Ru(bpz)32+.
Scheme 3.

Modular synthesis of neolignan natural products.
Footnotes
This research was conducted using funds from the NIH (GM095666), Sloan Foundation, Beckman Foundation, and Research Corporation. The NMR facilities at UW-Madison are funded by the NSF (CHE-9208463, CHE-9629688) and NIH (RR08389-01, RR13866-01). T.R.B. acknowledges support from an NIH Chemical Biology Interface training Grant (5 T32 GM850520).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- 1.(a) Desai LV, Malik HA, Sanford MS. Org. Lett. 2006;8:1141–1144. doi: 10.1021/ol0530272. [DOI] [PubMed] [Google Scholar]; (b) Campbell AN, Stahl SS. Acc. Chem. Res. 2012;45:851–863. doi: 10.1021/ar2002045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalyanasundaram K. Coord. Chem. Rev. 1982;46:159–244. [Google Scholar]
- 3.(a) Zeitler K. Angew. Chem. 2009;121:9969–9974. Angew. Chem. Int. Ed.2009, 48, 9785–9789. [Google Scholar]; (b) Yoon TP, Ischay MA, Du J. Nat. Chem. 2010;2:527–532. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]; (c) Narayanam JMR, Stephenson CRJ. Chem. Soc. Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; (d) Xuan J, Xiao W-J. Angew. Chem. 2012;124:6934–6944. Angew. Chem. Int. Ed.2012, 51, 68282013;6838. [Google Scholar]; (e) Prier CK, Rankic DA, MacMillan DWC. Chem. Rev. 2013:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Bergonzini G, Schindler CS, Wallentin C-J, Jacobsen EN, Stephenson CRJ. Chem. Sci. 2014;5:112–116. doi: 10.1039/C3SC52265B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Konieczynska MD, Dai C, Stephenson CRJ. Org. Biomol. Chem. 2012;10:4509–4511. doi: 10.1039/c2ob25463h. [DOI] [PubMed] [Google Scholar]; (c) Freeman DB, Furst L, Condie AG, Stephenson CRJ. Org. Lett. 2012;14:94–97. doi: 10.1021/ol202883v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhao Y, Li Z, Yang C, Lin R, Xia W. Beilstein J. Org. Chem. 2014;10:622–627. doi: 10.3762/bjoc.10.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Su Y, Zhang L, Jiao N. Org. Lett. 2011;13:2168–2171. doi: 10.1021/ol2002013. [DOI] [PubMed] [Google Scholar]; (b) Zou Y-Q, Chen J-R, Liu X-P, Lu J-Q, Davis RL, Jorgensen KA, Xiao W-J. Angew. Chem. 2012;124:808–812. Angew. Chem. Int. Ed.2012, 51, 784–788. [Google Scholar]; (d) Cheng Y, Yang J, Qu Y, Li P. Org. Lett. 2012;14:98–101. doi: 10.1021/ol2028866. [DOI] [PubMed] [Google Scholar]; (e) Cai S, Zhao X, Wang X, Liu Q, Li Z, Wang DZ. Angew. Chem. 2012;124:8174–8177. Angew. Chem., Int. Ed.2012, 51, 8050–8053. [Google Scholar]; (h) Rueping M, Vila C, Koenigs RM, Poscharny K, Fabry DC. Chem. Commun. 2011;47:2360–2362. doi: 10.1039/c0cc04539j. [DOI] [PubMed] [Google Scholar]; (m) Hari DP, König B. Org. Lett. 2011;13:3852–3855. doi: 10.1021/ol201376v. [DOI] [PubMed] [Google Scholar]; (t) Zou Y-Q, Lu L-Q, Fu L, Chang N-J, Rong J, Chen J-R, Xiao W-J. Angew. Chem. 2011;123:7309–7313. doi: 10.1002/anie.201102306. Angew. Chem., Int. Ed.2011, 50, 7171–7175. [DOI] [PubMed] [Google Scholar]
- 6.Mulazzani QG, Sun H, Hoffman MZ, Ford WE, Rodgers MAJ. J. Phys. Chem. 1994;98:1145–1150. [Google Scholar]
- 7.(a) Quideau S, Pouységu L, Deffieux D. Curr. Org. Chem. 2004;8:113–148. [Google Scholar]; (b) Yamamura S, Nishiyama S. Synlett. 2002:533–543. [Google Scholar]
- 8.(a) Coy ED, Cuca LE, Sefkow M. Bioorg. Med. Chem. Lett. 2009;19:6922–6925. doi: 10.1016/j.bmcl.2009.10.069. [DOI] [PubMed] [Google Scholar]; (b) Barrera EDC, Suarez LEC. Chem. Pharm. Bull. 2009;57:639–642. doi: 10.1248/cpb.57.639. [DOI] [PubMed] [Google Scholar]; (c) Shen T, Wang X-N, Lou H-X. Nat. Prod. Rep. 2009;26:916–935. doi: 10.1039/b905960a. [DOI] [PubMed] [Google Scholar]; (d) Zhang H, Qiu S, Tamez P, Tan GT, Aydogmus Z, Hung NV, Cuong NM, Angerhofer C, Soejarto DD, Pezzuto JM, Fong HHS. Pharm. Biol. 2002;40:221–224. [Google Scholar]; (e) Lachia M, Moody CJ. Nat. Prod. Rep. 2008;25:227–253. doi: 10.1039/b705663j. [DOI] [PubMed] [Google Scholar]
- 9.For recent reviews of dihydrobenzofuran synthesis, see: Bertolini F, Pineschi M. Org. Prep. Proc. Int. 2009;41:385–418. Sheppard TD. J. Chem. Res. 2011:377–385. Sefkow M. Synthesis. 2003;17:2595–2625.
- 10.For examples using preparative electrochemistry, see: Shizuri Y, Nakamura K, Yamamura S. J. Chem. Soc. Chem. Comm. 1985:530–531. Gates BD, Dalidowicz P, Tebben A, Wang S, Swenton JS. J. Org. Chem. 1992;57:2135–2143. Kerns ML, Conroy SM, Swenton JS. Tet. Lett. 1994;35:7529–7532. Chiba K, Fukuda M, Kim S, Kitano Y, Tada M. J. Org. Chem. 1999;64:7654–7656. El-Seedi HR, Yamamura S, Nishiyama S. Tetrahedron. 2002;43:3301–3304.
- 11.Oxidative [3+2] cycloadditions using Mn(OAc)3: Snider BB, Han L, Xie C. J. Org. Chem. 1997;62:6978–6984.
- 12.Oxidative [3+2] cycloadditions using hypervalent iodide: Wang S, Gates BD, Swenton JS. J. Org. Chem. 1991;56:1979–1981. Bérard D, Jean A, Canesi S. Tet. Lett. 2007;48:8238–8241. Bérard D, Giroux M, Racicot L, Sabot C, Canesi S. Tetrahedron. 2008;64:7537–7544. Bérard D, Racicot L, Sabot C, Canesi S. Synlett. 2008:1076–1080. Mohr AL, Lombardo VM, Arisco TM, Morrow GW. Syn. Comm. 2009;39:3845–3855.
- 13.A very recent report describes the oxidative dimerization of resveratrol oligomers using a mesoporous carbon ntiride photocatalyst: Song T, Zhou B, Peng G-W, Zhang Q-B, Wu Li-Zhu, Liu Qiang, Wang Y. Chem. Eur. J. 2014;20:678–682. doi: 10.1002/chem.201303587.
- 14.For leading references, see: Bolletta F, Juris A, Maestri M, Sandrini D. Inorg. Chim. Acta. 1980;44:L175–L176. White HS, Becker WG, Bard AJ. J. Phys. Chem. 1984;88:1840–846.
- 15.(a) Dai C, Meschini F, Narayanam JMR, Stephenson CRJ. J. Org. Chem. 2012;77:4425–4431. doi: 10.1021/jo300162c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fancy DA, Denison C, Kim K, Xie Y, Holdeman T, Amini F, Kodadek T. Chemistry &Biology. 2000;7:697–708. doi: 10.1016/s1074-5521(00)00020-x. [DOI] [PubMed] [Google Scholar]; (c) Nickel U, Chen Y-H, Schneider S, Silva MI, Burrows HD, Formosinho SJ. J. Phys. Chem. 1994;98:2883–2888. [Google Scholar]; (d) Minisci T, Citterio A, Giordano C. Acc. Chem. Res. 1983;16:27–32. [Google Scholar]
- 16.(a) Rillema DP, Allen G, Meyer TJ, Conrad D. Inorg. Chem. 1983;22:1617–1622. [Google Scholar]; (b) Schepp NP, Johnston LJ. J. Am. Chem. Soc. 1996;118:2872–2881. [Google Scholar]
- 17.Yueh W, Bauld NL. J. Phys. Org. Chem. 1996;9:529–538. [Google Scholar]
- 18.Pratt DA, Pesavento RP, van der Donk WA. Org. Lett. 2005;7:2735–2738. doi: 10.1021/ol050916g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maxwell A, Dabideen D, Reynolds WF, McLean S. Phytochemistry. 1999;50:499–504. [Google Scholar]
- 20.Achenbach H, Utz W, Usubillaga A, Rodriguez HA. Phytochemistry. 1991;30:3753–3757. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.