

Abstract
Oxidative damage and inflammation are both implicated in the genesis of hypertension; however, the mechanisms by which these stimuli promote hypertension are not fully understood. Here, we have described a pathway in which hypertensive stimuli promote dendritic cell (DC) activation of T cells, ultimately leading to hypertension. Using multiple murine models of hypertension, we determined that proteins oxidatively modified by highly reactive γ-ketoaldehydes (isoketals) are formed in hypertension and accumulate in DCs. Isoketal accumulation was associated with DC production of IL-6, IL-1β, and IL-23 and an increase in costimulatory proteins CD80 and CD86. These activated DCs promoted T cell, particularly CD8+ T cell, proliferation; production of IFN-γ and IL-17A; and hypertension. Moreover, isoketal scavengers prevented these hypertension-associated events. Plasma F2-isoprostanes, which are formed in concert with isoketals, were found to be elevated in humans with treated hypertension and were markedly elevated in patients with resistant hypertension. Isoketal-modified proteins were also markedly elevated in circulating monocytes and DCs from humans with hypertension. Our data reveal that hypertension activates DCs, in large part by promoting the formation of isoketals, and suggest that reducing isoketals has potential as a treatment strategy for this disease.
Introduction
It is well established that inflammation is involved in the genesis of hypertension (1). T cells and macrophages accumulate in the kidneys and vasculature of animals with experimental hypertension and likely contribute to the end-organ damage that accompanies this disease (2, 3). Immunosuppression ameliorates end-organ damage resulting from hypertension, and the renin-angiotensin system has been reported to regulate immune responses (4). Previous studies from our laboratory and others have shown that cells of the adaptive immune system are involved in the genesis of hypertension (5). Mice lacking lymphocytes (Rag1–/– mice) develop blunted hypertension and are protected from vascular dysfunction and vascular oxidative stress in response to various stimuli, including angiotensin II, norepinephrine, and deoxycorticosterone acetate plus NaCl (DOCA-salt). Adoptive transfer of T cells, but not B cells, restores hypertension in these animals. In addition, mice with severe combined immunodeficiency are protected from experimental hypertension (6), as are mice lacking the T cell cytokine IL-17A (7). The triggering events that lead to T cell activation in hypertension are currently not known.
Antigen-presenting dendritic cells (DCs) capture and process antigens and present antigenic peptides to T cells, leading to T cell proliferation, cytokine production, and alterations in surface molecule expression (8). DC-derived cytokines also influence the ultimate polarization of T cells and thus have long-lasting influence on the inflammatory milieu associated with the activated T cell. We have shown previously that DCs from hypertensive mice have increased surface expression of the B7 ligands CD80 and CD86, suggestive of DC maturation and activation (9). Blockade of these costimulatory molecules prevents hypertension and T cell activation during both angiotensin II– and DOCA-salt–induced hypertension (9).
There is also ample evidence that ROS produced by the NADPH oxidases are involved in hypertension. Mice lacking components of the NADPH oxidases, including p47phox, Nox1, and Nox4, are protected against hypertension and its end-organ damage (10, 11). Overexpression of either p22phox or Nox1 sensitizes mice to hypertension (12). Isoketals (alternatively named isolevuglandins or γ-ketoaldehydes) are formed via rearrangement of H2-isoprostane intermediates in the F2-isoprostane pathway of free radical–mediated lipid peroxidation (13). Isoketals adduct to lysine residues on proteins and extensively crosslink proteins, leading to alteration of protein function. Increased formation of isoketal protein adducts has been found in several diseases associated with oxidative stress, including alcohol-induced liver damage, atherosclerosis, Alzheimer’s disease, and asthma (14–17). In the current studies, we show that hypertension causes isoketals to accumulate in DCs and that they promote DC cytokine production and immunogenicity. We further show that compounds that scavenge isoketals prevent DC activation and experimental hypertension. These studies define a new mechanism of hypertension and provide a potentially new therapeutic approach for the treatment of this disease.
Results
Angiotensin II–induced hypertension increases superoxide production in DCs.
We have shown previously that DCs in the spleens and lymph nodes undergo maturation during angiotensin II–induced hypertension (9). Because mice lacking components of the NADPH oxidase are protected against hypertension, we considered the hypothesis that the NADPH oxidase in DCs might participate in this process. In initial experiments, we measured DC superoxide (O2η–) using HPLC to monitor conversion of dihydroethidium to the specific O2η– adduct 2-hydroxyethidium. We found that angiotensin II infusion markedly increased DC O2η– production (Figure 1, A and B). This increase in O2η– was absent in Nox2–/– (gp91phox–/–) mice, indicating that it is Nox2 dependent (Figure 1B). In vitro treatment of DCs with angiotensin II (100 nM) for 24 hours also modestly increased their O2η– production (Figure 1C).
Figure 1. Angiotensin II–induced hypertension increases O2η– production in DCs and isoketal formation in tissues.

(A) WT and Nox2–/– mice received angiotensin II or sham infusions for 2 weeks, and CD11c+ cells were isolated from the spleens. O2η– was determined using HPLC to monitor conversion of dihydroethidium to the O2η– oxidation adduct 2-hydroxyethidium, as indicated by the first peak (arrows) of the HPLC tracings. (B) Quantification of O2η– in WT and Nox2–/– mice (n = 6). (C) O2η– was measured in DCs at either baseline or following 24 hours of angiotensin II treatment in vitro (100 nM) (n = 7). (D) Pathway illustrating lipid peroxidation and formation of isoketals. The isoketals react with lysine residues on proteins forming lactam adducts that cross-linked proteins. This process is prevented by scavenging with 2-HOBA. (E) Immunohistochemistry of heart and aortic sections showing accumulation of isoketals in hypertension using a single-chain antibody that recognizes isoketal-lysine adducts on all proteins (scale bar: 100 μm). (F) Quantification of isoketals in the hearts and aortas (n = 6–7, *P < 0.05).
Isoketals contribute to hypertension and kidney damage.
A potential mechanism by which O2η– and related ROS could promote T cell activation is via oxidative protein modification and formation of proteins not identified as self. One such modification is formation of isoketals via the isoprostane pathway of lipid peroxidation. Once formed, isoketals react with lysine residues on proteins forming lactam adducts and cross-linked proteins (Figure 1D). This process can be prevented using isoketal scavengers, such as 2-hydroxybenzylamine (2-HOBA) (18). To determine whether isoketals are formed in hypertension and to examine their role in this disease, we used several approaches. Immunohistochemistry analysis, using a single-chain antibody that recognizes isoketal-lysine adducts on any protein (19), showed that there was an exuberant accumulation of isoketals in the hearts and vasculatures of hypertensive mice (Figure 1, E and F). Coadministration of 2-HOBA prevented isoketal immunoreactivity in these tissues (Figure 1, E and F). We also examined the ability of 2-HOBA and several other isoketal scavengers to prevent hypertension using telemetry to monitor blood pressure. We also treated control mice with compounds with related chemical structures that are unable to scavenge isoketals (20). We found that compounds known to scavenge isoketals with high efficacy, including 2-HOBA, 5-methyl-2-HOBA, and pentylpyridoxamine, blunted the hypertensive response to angiotensin II (Figure 2A). Conversely, related compounds that exhibit low reactivity with isoketals, including N-methyl-2-HOBA and 4-HOBA, did not affect angiotensin II–induced hypertension (Figure 2A). Supplemental Table 1 (supplemental material available online with this article; doi:10.1172/JCI74084DS1) shows the relative scavenging efficacy and specificity of these compounds for isoketals and malondialdehyde (MDA). Of note, all of these compounds demonstrated slow rate constants for reactions with MDA, and these rates were unrelated to their ability to lower blood pressure. In other experiments, we showed that 2-HOBA has no scavenging activity for either O2η– or peroxynitrite (Supplemental Figure 1).
Figure 2. Isoketals contribute to angiotensin II hypertension and kidney damage.

(A) Effect of isoketal scavengers 2-HOBA, 5-methyl-2-HOBA (5-Me-2-HOBA), and pentylpyridoxamine (PnPM) and control compounds N-methyl-2-HOBA (N-Me-2-HOBA) and 4-HOBA on the hypertensive response to angiotensin II. (B) Masson’s Trichrome immunohistochemistry showing fibrosis (green arrows) in kidney section from mice with angiotensin II–induced hypertension (scale bar: 100 μm). (C) Quantification of total renal fibrosis. (D) Collagen IV staining in kidney sections (scale bar: 10 μm). (E) Anti-CD3 staining showing T cell infiltration in kidney sections from angiotensin II–infused mice (scale bar: 10 μm). (F) Quantification of collagen IV staining. (G) Nephrin and (H) albumin concentrations were measured in urine using an ELISA-based assay (n = 6–7, *P < 0.05, **P < 0.01, ***P < 0.001).
Natural killer T cells (NKT) cells respond to the MHCI-related glycoprotein CD1d and specialize in presentation of lipid antigens. Because isoketal-adducted proteins could represent lipid-modified antigens, we studied mice lacking Jα18 (Ja18–/– mice), an invariant α chain of the T cell receptor on NKT cells. Using telemetry, we found that Ja18–/– mice developed similar degrees of hypertension in response to angiotensin II as those observed in WT mice (Supplemental Figure 2, A–C). NKT cells respond to lipid antigens presented in the context of CD1d. In separate experiments, we found that the hypertensive response to angiotensin II in Cd1d–/– mice was also similar to that in WT mice (Supplemental Figure 2D). These data indicate that NKT cells do not play a role in angiotensin II–induced hypertension and that isoketals do not act via the CD1d/NKT axis.
Scavenging isoketals with 2-HOBA also prevented angiotensin II–induced renal damage. This was demonstrated by a prevention of renal fibrosis, as indicated by trichrome staining (Figure 2, B and C). Renal injury in mice and humans is characterized by accumulation of collagen IV (21), which we found to be markedly increased in the kidneys of angiotensin II–treated mice and to be prevented by 2-HOBA treatment (Figure 2, D and F). Immunostaining with anti-CD3 revealed a striking renal infiltration of T cells in response to angiotensin II, which was completely prevented by 2-HOBA (Figure 2E). Angiotensin II–induced hypertension was associated with a 10-fold increase in urinary albumin and nephrin, both indicative of glomerular damage, and these perturbations were completely normalized by treatment with 2-HOBA (Figure 2, G and H). These results suggest that isoketals contribute to the genesis of hypertension and its associated renal damage.
Isoketals accumulate and activate DCs in hypertension.
To examine isoketal adduct formation in DCs, we used flow cytometry and intracellular staining using an Alexa Fluor 488–tagged single-chain antibody specific for isoketal adduct D11 (19). A gating strategy for MHC class II I-Ab–positive splenocytes was based on the presence of surface markers CD11b and CD11c (Supplemental Figure 3A). We found that angiotensin II infusion increased intracellular accumulation of isoketals to the greatest extent in CD11b+/CD11c– (Figure 3A) and CD11b+/CD11c+ cells (Figure 3B) and to a lesser extent in CD11b–/CD11c+ cells (Figure 3C). Subsequent gating strategies using the markers MerTK and CD64 showed that these cells were not macrophages (Supplemental Figure 3B).
Figure 3. DC accumulation of isoketals in hypertension.

(A) Intracellular staining of isoketals using Alexa Fluor 488–tagged D11 in CD11b+/CD11c–, (B) CD11b+/CD11c+, and (C) CD11b–/CD11c+ cells from sham and angiotensin II–infused mice with or without 2-HOBA treatment. Mean data are shown in the bar graphs (n = 6, *P < 0.05, **P < 0.01, ***P < 0.001). (D) Analysis of isoketals in various DC subtypes from sham or angiotensin II–infused mice (shown are representative data of 6 mice per group). (E) Stable isotope dilution multiple reaction monitoring mass spectrometry analysis of isoketal-lysine-lactam adduct in DCs. Representative LC/MS chromatographs from pooled samples for each group are shown. The top row shows multiple reaction monitoring chromatographs for isoketal-lysine-lactam (IsoK-Lys) in sample, while the bottom row shows multiple reaction monitoring chromatograph for [13C615N2] internal standard for same samples.
To further characterized the specific subtypes of DCs that accumulate isoketal-modified proteins, we used a gating strategy to identify CD8+, B220+, MHCIIhi, and MHCIIint, and endothelial surface adhesion molecule high (ESAMhi) and ESAMlo CD11c+/CD11b+ cells (Supplemental Figure 4). We found that hypertension is associated with an increase in isoketal-adducted proteins in all of these, but the most prominent accumulation was present in CD11b+/CD11c+ cells, independent of ESAM status (Figure 3D). The B220+ cells, which correspond to plasmacytoid cells, had the least accumulation of isoketal protein adducts. The CD8+ DCs, which are known to cross-present antigens to CD8+ T cells, also accumulated a modest amount of isoketals in angiotensin II–treated animals.
To further confirm that DCs accumulate isoketal protein adducts in hypertension, we used mass spectrometry. DCs were pooled from 10 sham-infused mice, 10 mice treated with angiotensin II, and 10 mice treated with angiotensin II and 2-HOBA, and liquid chromatography mass spectrometry analysis was performed using methods established in our laboratories (22). These studies confirmed that hypertension increases isoketals in DCs and that 2-HOBA reduces this (Figure 3E).
Maturation of DCs upon antigen uptake and presentation is associated with an increase in surface expression of the costimulatory B7 ligands CD80 and CD86. We found that cells staining positively for isoketal protein adducts also exhibited a striking increase in surface expression of CD80 and CD86 (Figure 4). This was most significant in the CD11b+/CD11c+ DCs and was prevented by treatment with 2-HOBA (Figure 4, C and F).
Figure 4. Accumulation of isoketals in activated DCs.

Mice were made hypertensive with angiotensin II, as in Figure 1. (A) Gating strategy to identify CD11c+/CD11b+ cells. (B and E) Intracellular staining of isoketals in CD11b+/CD11c–, (C and F) CD11b+/CD11c+, and (D and G) CD11b+/CD11c– cells expressing CD80 and CD86 (n = 6, *P < 0.05, **P < 0.01 ***P < 0.001).
Another aspect of DC activation is production of cytokines that influence the local inflammatory milieu and guide T cell polarization. We isolated DCs from spleens of sham or angiotensin II–infused mice and measured a panel of cytokines using a Luminex-based assay (Table 1). Angiotensin II–induced hypertension was associated with a 2-fold increase in IL-6 and IL-1β and a 3-fold increase in IL-23 production by DCs. These responses were completely normalized by cotreatment with 2-HOBA. Other cytokines that released into the media but were not produced at significantly different rates when compared across treatment groups included GM-CSF, TGF-β1, and TGF-β2. Other cytokines were produced by DCs in only trace amounts (Table 1). Taken together, these results suggest that hypertension and the formation of isoketals increase maturation and cytokine production by DCs.
Table 1.
Effect of hypertension on cytokine production in DCs

Isoketals contribute to DOCA-salt hypertension.
To determine whether isoketals contribute to hypertension caused by stimuli other than angiotensin II, we produced DOCA-salt hypertension in mice. DOCA-salt hypertension was associated with formation of isoketal protein adducts in DCs, and this was prevented by 2-HOBA treatment (Figure 5, A and B). As with angiotensin II–induced hypertension, 2-HOBA significantly reduced the hypertensive response to DOCA-salt challenge (Figure 5C).
Figure 5. Isoketals contribute to DOCA-salt hypertension.

(A and B) Intracellular staining of isoketal adducts in CD11c+ cells from sham mice and mice with DOCA-salt hypertension, treated or not with 2-HOBA. (C) Systolic pressures from radiotelemetry recordings showing the effect of 2-HOBA on DOCA-salt hypertension (n = 6, **P < 0.01).
DCs from angiotensin II–hypertensive mice promote T cell survival, proliferation, and cytokine production.
In additional experiments, we sought to determine whether isoketal-modified proteins in DCs cause T cell activation in hypertension. To accomplish this, we developed an assay to specifically examine the ability of DCs from hypertensive, angiotensin II–treated mice to activate T cells from normotensive mice not treated with angiotensin II (Figure 6A). DCs from sham-treated mice failed to support survival (Figure 6, B–F) or proliferation of either CD4+ or CD8+ T cells (Figure 6, G and H), as reflected by CSFE dilution assays. In contrast, DCs from angiotensin II–treated mice supported both survival and proliferation of CD8+ T cells. Cotreatment of the DC donor mice with 2-HOBA prevented both survival and proliferation of the recipient T cells in this assay (Figure 6, F–H). In additional experiments, we measured cytokine release by T cells into the media. T cells exposed to DCs from angiotensin II–treated mice released increased amounts of IFN-γ, IL-17A, and TNF-α compared with T cells exposed to DCs of sham-treated mice. Cotreatment of donor mice with 2-HOBA during angiotensin II infusion eliminated T cells production of these cytokines (Figure 6I).
Figure 6. DCs from angiotensin II–hypertensive mice promote T cell survival, proliferation, and cytokine production.

(A) Experimental approach for the immunization assay. Flow cytometry was used to determine percentages of live cells using (B) 7-AAD, (C) CD45+ cells, and (D) CD3+ cells. (E) Total number of live cells, CD45+ cells, and CD3+ cells. (F–H) Representative flow cytometry profiles and graph showing percentages and numbers of CD4+ and CD8+ T cells. Proliferation of (G) CD8+ T cells and (H) CD4+ T cells, as reflected by CSFE dilution assays. (I) Cytokines IFN-γ, IL-17a, and TNF-α released by T cells stimulated with sham, angiotensin II, or angiotensin II and 2-HOBA DCs (n = 6–12, *P < 0.05, **P < 0.01, ***P < 0.001).
Oxidant stress and isoketals alter DC gene expression in hypertension.
In additional experiments, we demonstrated that hypertension modulates DC gene expression (Figure 7A). Microarray analysis of DC gene expression indicated that 505 genes were altered in mice that were made hypertensive by angiotensin II for 2 weeks. Of these, 150 genes were not altered in Nox2–/– mice and 141 were normalized by 2-HOBA treatment. Fifty-five genes altered by angiotensin II infusion were commonly normalized in both Nox2–/– and 2-HOBA–treated groups (Figure 7B). A comprehensive list of the top genes altered by angiotensin II infusion and returned to baseline both by absence of Nox2 and by isoketal scavenging is shown in Supplemental Table 2. To determine whether this change in DC gene expression was simply mediated by blood pressure lowering, we normalized blood pressure during angiotensin II infusion in a group of mice by cotreatment with a combination of hydralazine and hydrochlorothiazide (Figure 7C). While lowering blood pressure reduced the alteration in DC gene expression in response to angiotensin II, this effect was not as great as that observed in Nox2–/– mice or upon treatment with 2-HOBA (Figure 7, D–G). These data demonstrate that angiotensin II–induced hypertension alters DC gene expression and that many of these gene changes are dependent on oxidative stress and subsequent isoketal protein modification.
Figure 7. Oxidant stress and isoketals alter DC gene expression in hypertension.

(A) Microarray analysis of CD11c+ cells from spleens of WT mice infused with either sham or angiotensin II with or without 2-HOBA treatment or cotreatment with a combination of hydralazine and hydrochlorothiazide as well as cells from spleens of Nox2–/– mice. The gene expression profiles for the various treatment groups. (B) Genes altered by angiotensin II (blue), genes reversed by Nox2–/– (pink), genes reversed by 2-HOBA treatment (green), and a Venn diagram summarizing altered genes, including those normalized in both Nox2–/– and 2-HOBA–treated groups. (C) Normalization of angiotensin II–induced hypertension by cotreatment with a combination of hydralazine and hydrochlorothiazide. (D) Volcano plot showing the 505 genes altered by angiotensin II compared with sham and colored by P value. Red represents high and blue represents low P values in WT angiotensin II vs. WT sham. Color codes for each gene in D were used in E–G. The genes altered by angiotensin II in C but plotted based on how these genes behaved (E) after 2-HOBA treatment, (F) in Nox2–/– mice, and (G) after treatment with a combination of hydralazine and hydrochlorothiazide, when compared with WT sham (n = 6, **P < 0.01)
Additional experiments were performed to determine how isoketals alter DC immunogenicity. To determine whether MHCI is directly modified by isoketals, we immunoprecipitated MHCI from DCs of sham and angiotensin II–treated mice and performed Western blots using D11 antibody. Neither the heavy nor the light chain of MHCI demonstrated D11 reactivity, suggesting that MHCI was not directly modified by isoketals either at baseline or in response to angiotensin II (Figure 8A). To determine whether lipid-modified proteins could act as neoantigens that drive T cell activation, we exposed mouse kidney homogenates (100 μg total protein) to 100 μmol/liter of isoketals, hydroxynonenal, MDA, or methylglyoxal for 30 minutes. One million DCs were then pulsed with these modified proteins for 1 hour, washed, and exposed to T cells prelabeled with CFSE at a ratio of 1 DC to 10 T cells for 7 days. DCs pulsed with isoketal-modified proteins caused robust proliferation of CD8+ T cells (Figure 8B, left panel). In striking contrast, responses to homogenates modified by hydroxynonenal, methylglyoxal, and MDA were minimal. The effect of isoketal-modified proteins was greater for T cells from angiotensin II–treated mice than for those from sham-treated mice (Figure 8B, middle panel), suggesting that in vivo exposure to hypertension primes responsiveness of T cells to isoketal-modified proteins. Direct exposure of DCs to lipid adducts alone had no effect (Figure 8B, right panel). CD4+ T cells demonstrated similar albeit less robust responses (Figure 8C). In additional experiments, we found that CD8+ memory T cells isolated from the spleens of angiotensin II–treated mice robustly proliferated in response to DCs pulsed with isoketal-modified proteins. CD4+ memory T cells also proliferated, albeit to a lesser extent. Naive T cells from angiotensin II–treated mice did not respond in this assay (Figure 8D).
Figure 8. Effect of isoketal protein modification on DC immunogenicity.

(A) MHCI was immunoprecipitated from DCs of sham and angiotensin II–treated mice, and Western blot using D11 antibody was performed. Mouse kidney homogenates (100 μg total protein) were exposed to 100 μmol/liter of isoketal, hydroxynonenal (HNE), MDA, or methylglyoxal (MGO) for 30 minutes. One million DCs were then pulsed with these modified proteins for 1 hour, washed, and exposed to T cells prelabeled with CFSE at a ratio of 1 DC to 10 T cells for 7 days. (B) Effect of DCs pulsed with lipid-modified proteins on CD8+ T cells. (C) Effect of DCs pulsed with lipid-modified proteins on CD4+ T cells. The left and middle panels of B and C show proliferation of T cells that were obtained from sham or angiotensin II–treated mice, respectively. The right panels of B and C show the effects of the lipid adducts directly added to DCs. (D) Proliferation of memory and naive T cells from angiotensin II–treated mice in response to DCs pulsed with isoketal-adducted proteins.
Since DCs from angiotensin II–infused mice predominantly support proliferation of CD8+ T cells, we examined surface expression of MHCI. We found increased expression of both H-2Db and H-2Kb in hypertensive mice (Supplemental Figure 5A). The fluorescence intensity of H-2Kb increased by 15.7% while that of H-2Db increased by 20.3% among CD11c+ cells from angiotensin II–infused mice when compared with sham mice, and these changes were prevented by treatment with 2-HOBA. These data suggest that the MHCI surface repertoire is altered by hypertension.
Because DCs from hypertensive mice seem to present isoketal-modified antigens more efficiently, we sought to determine whether this extended to irrelevant peptides. DCs were pulsed with the ovalbumin peptide OVA257-264 (SIINFEKL) and cocultured with T cells from OT1 transgenic mice for 3 days. Surprisingly, DCs from angiotensin II–infused mice were less efficient in presenting OVA257-264 peptide to OT1 cells, and this was prevented by 2-HOBA treatment (Supplemental Figure 5B and C). We also used flow cytometry and the SIINFEKL/H-2Kb antibody, which identifies SIINFEKL bound to MHCI. Following incubation of DCs with 10 μg/ml SIINFEKL for 1 hour, we found that DCs from angiotensin II–infused mice were less efficiently loaded with the peptide, and 2-HOBA normalized this (Supplemental Figure 5D). These data show that alterations of DC immunogenicity in response to hypertension do not extend to irrelevant antigens like the OT1 peptide.
Transfer of hypertension by DCs.
In additional studies, we examined the ability of DCs to confer hypertension to recipient mice. One million DCs obtained from either sham or angiotensin II–infused mice were adoptively transferred to recipient WT mice. Ten days later, recipient mice were implanted with minipumps for infusion of angiotensin II at what is generally a subpressor rate. This experimental design is shown in Figure 9A. Telemetry was used to monitor systolic (Figure 9B), diastolic (Supplemental Figure 6A), and mean arterial pressure (Supplemental Figure 6B). This normally subpressor dose of angiotensin II had no effect on blood pressure in mice that received DCs from sham mice but caused severe hypertension in mice that received DCs from angiotensin II–infused mice. Cotreatment of donor mice with the isoketal scavenger 2-HOBA prevented DCs from angiotensin II–infused mice from priming hypertension (Figure 9B). In additional experiments, we adoptively transferred DCs from angiotensin II–infused mice into Rag1–/– mice. Unlike the response in WT mice, low-dose angiotensin II failed to increase blood pressure in Rag1–/– mice, indicating that T cells are required for the DC-mediated priming of hypertension. Additional studies were performed in which DCs from mice transgenic for enhanced green fluorescent protein were used for adoptive transfer. Using flow cytometry of various tissues in the recipient mice 10 days later, we found that these cells predominantly accumulated in the spleens of recipient mice, and to a lesser extent in the kidneys and aortas, and this is increased if the donor mouse was treated with angiotensin II (Supplemental Figure 6C).
Figure 9. Transfer of hypertension by DCs.

(A) DCs were obtained from either sham or angiotensin II–infused mice, and 1 × 106 cells were adoptively transferred to recipient WT or Rag1–/– mice, and blood pressure was monitored using telemetry. (B) Systolic blood pressure in response to low-dose angiotensin II infusion (140 ng/kg/min) 10 days after DC adoptive transfer (n = 5–7, **P < 0.01). DCs were treated with 1 mM t-BHP, and isoketals were measured by flow cytometry using Alexa Fluor 488–tagged D11 antibody in (C) CD11b+/CD11c–, (D) CD11b+/CD11c+, and (E) CD11b–/CD11c+ cells. (F) Expression of CD86 in DCs. (G) Coculture of t-BHP–treated DCs with T cells promoted survival of CD8+ T cell. (H) Adoptive transfer of t-BHP–treated DCs in mice increased systolic blood pressure (n = 4–7, *P < 0.05, **P < 0.01, ***P < 0.001).
Induction of oxidative stress in DCs promotes isoketal formation, activation of T cells, and hypertension.
To confirm the role of oxidative stress and isoketals in T cell activation and hypertension, we treated DCs ex vivo with 1 mM tert-butyl hydroperoxide (t-BHP) for 30 minutes. We found that this brief exposure of DCs to t-BHP led to intracellular isoketal formation, particularly in CD11b+/CD11c+ cells (Figure 9, C–E), and surface expression of CD86 (Figure 9F). Coculture of t-BHP–treated DCs with T cells promoted survival and proliferation of CD8+ T cells (Figure 9G). The formation of isoketal-modified proteins in DCs and the enhanced capacity of these cells to drive T cell proliferation was prevented by cotreatment with 2-HOBA (Supplemental Figure 7, A–C). In additional experiments, we found that adoptive transfer of t-BHP–treated DCs augmented the systolic (Figure 9H), diastolic (Supplemental Figure 7D, and mean arterial pressure (Supplemental Figure 7E) elevation in response to low-dose angiotensin II in recipient mice. These results further support the concept that oxidative stress in DCs is sufficient to activate T cells and promote hypertension.
Increased F2-isoprostanes and isoketals in hypertensive humans.
Isoketals and F2-isoprostanes are coproduced in the isoprostane pathway of lipid peroxidation. To determine whether isoketals play a role in human hypertension, we studied 2 cohorts of patients. In the first, we measured levels of F2-isoprostanes levels in the plasma of normotensive subjects, patients with controlled hypertension, and patients with resistant hypertension. The general characteristics of these groups are shown in Table 2. BMI, glucose, and HbA1C were higher in the 2 hypertensive groups compared with the normotensive volunteers. Subjects with resistant hypertension were taking a mean of 4.3 classes of antihypertensive drugs, while well-controlled patients were taking an average of 2.3 drugs daily. There were differences in proportion of diuretics, calcium channel blockers, and beta-blockers between hypertension and resistant hypertension subjects. As expected, systolic, diastolic, and pulse pressures were highest in patients with resistant hypertension. The patients with resistant hypertension had increased levels of plasma F2-isoprostanes compared with well-controlled and normotensive subjects (23.2 ± 11.5 vs. 18.3 ± 10.3 vs. 11.6 ± 7.0 pg/ml, respectively, P < 0.05; Figure 10A). The association between F2-isoprostane levels and hypertension grade was confirmed by multiple linear regression adjusted for BMI, HbA1C, LDL, and statin use (Table 3).
Table 2.
General characteristics of study groups for analysis of plasma isoprostanes

Figure 10. Increased isoprostanes and isoketals in hypertensive humans.

(A) Plasma levels of F2-isoprostanes in normotensive subjects (NT) as well as well-controlled hypertension (HT) and resistant hypertensive (RH) patients (*P < 0.05 vs. NT; #P < 0.05 vs. HT). (B) Gating strategy for flow cytometric analysis of isoketals in mononuclear cells obtained from the buffy coats of humans with or without hypertension. (C) Isoketal adducts in human monocytes, as detected by surface staining for CD14. (D) Quantification of isoketals in CD14+ cells (***P < 0.001). (E) Isoketal adducts in human CD83+ cells. (F) Quantification of isoketals in CD83+ cells(*P < 0.05). (G) Proposed pathway for DC activation. Hypertensive stimuli increase ROS production and formation of isoketals and protein isoketal adducts in DCs. These adducts enhance DC immunogenicity and promote DC cytokine production. These activated DCs promote T cell proliferation and cytokine production. Activated T cells infiltrate target tissues, including the kidney and vasculature, and release cytokines, such as IL-17, TNF-α, and IFN-γ, that promote hypertension.
Table 3.
Multiple linear regression for log-transformed plasma 8-isoprostane levels

In a second cohort of patients, mononuclear cells were obtained from the buffy coats of 12 humans with hypertension and 8 normotensive subjects and analyzed for isoketal protein adduct content using flow cytometry. Demographics of these individuals are presented in Table 4. Because the hypertensive patients were generally well treated, there was no significant difference in blood pressures obtained at the time of blood donation. The gating strategy for these experiments is shown in Figure 10B. The presence of isoketal adducts in monocytes of humans with hypertension, as detected by surface staining for CD14, was 3-fold greater than in humans without hypertension (Figure 10, C and D). We also observed a small population (<0.5%) of circulating CD83+ cells in these subjects and found a similar increase in isoketal protein adducts in these cells among those with hypertension (Figure 10, E and F). While CD83 is considered a marker of human DCs, subsequent analysis indicated that about one-half of these cells were positive for the B cell marker CD19. The percentage of isoketal-positive CD14+ and CD83+ cells correlated with their respective systolic blood pressures (Table 5). After adjusting the levels of BMI, glucose, and age in the multivariable linear model, systolic blood pressure remained a statistically significant predictor of isoketals in both CD14+ and CD83+ cells (Tables 6 and 7).
Table 4.
General characteristics of study groups for detection of intracellular isoketals

Table 5.
Spearman correlations between isoketal content of CD14+ and CD83+ cells and predictors

Table 6.
Adjusted model of the Spearman correlations between isoketal content of CD14+ cells and predictors
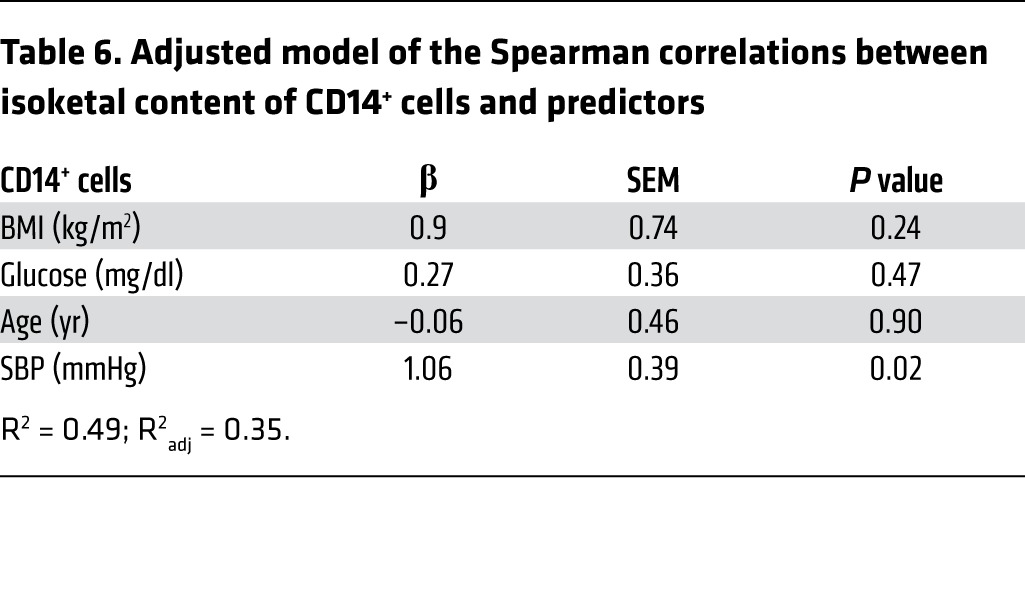
Table 7.
Adjusted model of the Spearman correlations between isoketal content of CD83+ cells and predictors

Discussion
The present studies describe a new pathway that contributes to hypertension (Figure 10G). We demonstrate for the first time that DCs play a causal role, likely related to increased formation of ROS and ultimately isoketal protein adducts in DCs. Our findings indicate that DCs from angiotensin II–infused mice produce increased amounts of O2η–, accumulate isoketal protein adducts, and release cytokines, such as IL-6, IL-1β, and IL-23. DCs from angiotensin II–infused mice promote T cell proliferation and polarize T cells to an inflammatory phenotype involving production of IL-17, IFN-γ, and TNF-α. DCs from hypertensive mice also prime a hypertensive response in recipient mice to low-dose angiotensin II. Scavenging isoketals prevents these parameters of DC activation and ameliorates hypertension. Exposure of DCs to the prooxidant t-BHP promotes their ability to support CD8+ T cell proliferation and hypertension, thus mimicking the effect of angiotensin II in vivo. Likewise, DCs pulsed with isoketal-modified proteins from renal homogenates potently stimulate T cell proliferation, while DCs pulsed with other oxidized lipid products do not. Finally, we have shown that humans with hypertension have elevated plasma F2-isoprostanes and increased isoketal adducts in their mononuclear cells and that these increase with the severity of hypertension. Our data strongly suggest that isoketal-modified proteins serve as neoantigens that promote an immune response in hypertension.
DCs and similar antigen-presenting cells are essential for T cell activation by presenting antigenic peptides in MHCs and by providing costimulation. In a prior study, we showed that hypertension increases expression of the costimulatory ligands CD80 and CD86 on DCs and that inhibition of costimulation either pharmacologically or genetically prevents hypertension caused by either angiotensin II or DOCA-salt (9). Our current study provides insight into mechanisms responsible for DC activation and immunogenicity. Our data strongly suggest that isoketal-modified proteins act as neoantigens in hypertension. These modified proteins accumulate in DCs and promote DC maturation. Removal of these by isoketal scavengers prevents DC immunogenicity and maturation. Increasing DC isoketals either by a strong oxidant or by addition of isoketal-modified proteins promotes the ability of DCs to activate T cells. Thus, protein modification by isoketals likely leads to formation of neoepitopes that are not recognized as self and promote an autoimmune-like reaction, leading to hypertension.
The definition of DCs is evolving, and it is now clear that subsets of these cells have unique roles and functions. Importantly, we found that hypertension induced formation of isoketal-modified proteins in all subsets, except for the plasmacytoid subset. Of particular interest, both ESAMlo and ESAMhi CD11b+/CD11c+ cells exhibited the most striking modification. CD11b+/ESAMhi cells are known to present antigens to CD4+ T cells, while the role of CD11b+/ESAMlo cells is less well defined. We also observed isoketal protein adducts in CD8+ DCs, which cross-present to CD8+ T cells. In keeping with these findings, we observed that DCs of hypertensive mice promoted proliferation of both CD4+ and CD8+ T cells, although the response was greatest for the latter.
An important finding of the present study is that O2η– production by DCs is absent in Nox2-deficient mice. We and others have shown that the NADPH oxidase is critical for many forms of hypertension. Importantly, O2η– serves as a progenitor for many biologically important ROS, including hydrogen peroxide, lipid peroxides, peroxynitrite, and hydroxyl and hypohalous acids. The electron potential of these indicate that they are more likely to react with arachidonic acid than O2η– (23). Thus, while we demonstrated an increase in DC production of O2η–, it is likely that other ROS, derived from O2η–, are responsible for isoketal formation.
We found that exposing DCs to isoketal-modified proteins promoted their ability to drive CD8+ and, to a lesser extent, CD4+ T cell proliferation. This was most pronounced if T cells were obtained from angiotensin II–treated mice, suggesting that these cells had been primed or enriched to respond to such antigens. This indicates that oxidative injury of other cells, aside from DCs, might lead to formation of immunogenic isoketal protein adducts that can be phagocytosed by DCs and cross-presented to CD8+ T cells. Thus, activation of the NADPH oxidase in vascular or renal cells might initiate this immune-like reaction. Of note, NADPH oxidase–derived O2η– alters antigen cross-presentation in the context of MHCI (24). Redox regulation has also been found to facilitate optimal class I peptide selection during antigen processing (25). DCs increase H2O2 production during antigen presentation, and scavenging of H2O2 using ebselen reduces DC cytokine production and T cell activation (26). Thus, a complex interplay may exist between ROS formed in DCs and that formed in other cells to promote T cell activation and inflammation in hypertension.
Oxidative injury and inflammation are intimately linked. Redox-sensitive transcription factors stimulate vascular cell adhesion molecule expression and chemotaxis of inflammatory cells into the subendothelial space. Oxidation promotes endothelial permeability and injury, critical features of inflammation. Our current results provide a direct link between lipid oxidation and immune activation. It is possible that a similar cascade of events contributes to other diseases in which oxidant stress has been implicated. T cells are often present in sites of inflammation for unexplained reasons. It is possible that their activation involves the presence of isoketal adducts. In keeping with these considerations, oxidatively modified macromolecules have been proposed to serve as neoantigens that participate in recognized autoimmune diseases (27).
In addition to antigen presentation, an important role of DCs is the release of mediators that promote T cell polarization. Indeed, hypertension augmented the capacity of DCs to produce IL-1β, IL-6, and IL-23, which are known to polarize T cells to produce IL-17. There has been substantial interest in the role of this cytokine in hypertension. We found that mice lacking IL-17A exhibit blunted hypertensive responses to angiotensin II (7), and others have found that infusion of IL-17A raises blood pressure, in part by altering activation of the endothelial nitric oxide synthase (28). In keeping with this, we found that DCs from hypertensive mice stimulate T cell production of IL-17A as well as TNF-α and IFN-γ, which have also been implicated in hypertension (5, 29, 30). In keeping with a critical role of DCs in this disease, we also found that hypertension causes a striking alteration of DC gene expression and that these alterations were in large part mediated by ROS derived from the NOX2 containing NADPH oxidase and by isoketal formation. These data further support a rather diverse effect of isoketal formation on DC function.
We also found that humans with hypertension have increased levels of circulating isoprostanes and increased isoketal protein adducts in monocytes, which can give rise to DCs similar to the CD11b+/CD11c+ subset we observed in mice. We also observed an increase in isoketal protein adducts in CD83+ cells, which represent both DCs and activated B cells, both of which can present antigens. Of note, the level of both plasma F2-isoprostanes, which are formed in the same pathway as isoketals, and cellular isoketals was increased as the severity of hypertension increased. These findings indicate that oxidative stress and lipid peroxidation occurs in humans with hypertension in a fashion similar to that observed in mice with experimental hypertension. It is therefore attractive to speculate that drugs like 2-HOBA might be effective for treatment of hypertension and prevention of end-organ damage in humans.
For the past two decades, it has become clear that ROS are central to the pathogenesis of hypertension, likely via several mechanisms (31). Unfortunately, treatment with common antioxidants, such as vitamins E and C, has proven ineffective for many cardiovascular diseases and in some instances has proven deleterious (32). In prior work, we have established that very high doses of vitamin E are necessary to suppress isoprostane production in humans (33). Such high doses paradoxically increase cardiovascular events (32). It is therefore possible that specific scavenging of isoketals will be more effective than traditional antioxidants for treatment of hypertension. It is of interest that we recently found that 2-HOBA also prevents the development of working memory defects in hApoE4 mice (16). Given the emerging recognition of the association between blood pressure and cognitive decline, it is possible that isoketal scavenging will be useful for the constellation of illnesses linked to hypertension.
Methods
Please see the Supplemental Methods for additional details.
Animals.
WT, Nox2–/– mice, and mice transgenic for enhanced green fluorescent protein were obtained from The Jackson Laboratory. All experiments were performed in male animals at approximately 3 months of age. Ja18–/–, and Cd1d–/– mice were gifts of Luc Van Kaer at Vanderbilt University. Osmotic minipumps (Alzet, Model 2002) were implanted for infusion of angiotensin II (490 or 140 ng/kg/min) for 2 weeks. DOCA-salt hypertension was produced as previously described (34). In some experiments, hydrochlorothiazide and hydralazine were administered by drinking water to prevent the elevation of blood pressure caused by angiotensin II, as previously described (35). Blood pressure was monitored noninvasively using tail cuff and invasively using radiotelemetry, as previously described (5, 36, 37). After telemetry implantation, mice were allowed to recover for 10 days before the osmotic minipumps were placed. Adoptive transfer of DCs was accomplished by injection 1 × 106 DCs in 100 μl physiological buffered saline via the lateral tail veins in mice anesthetized with xylazine/ketamine. Angiotensin II infusion was started 10 days after adoptive transfer of DCs.
Gene expression analysis.
Data were uploaded into Partek Genomics Suite version 6.6 (Partek Incorporated) and RMA normalized. Gene functions were determined using NCBI Entrez Gene, Stanford SOURCE, Aceview, and PubMed databases. The online Gene Ontology Enrichment Analysis and Visualization (GOrilla) tool (38, 39) was also used to explore gene ontology process, function, and component enrichment within each gene list, using a flexible threshold of 0.05. Final data were submitted to GEO (accession no. GSE59422).
Studies of F2-isoprostanes in humans.
A cross-sectional study was performed with eligible patients from Outpatient Resistant Hypertension Clinic of the University of Campinas. Clinical follow-up was conducted for a 6-month period with ambulatory BP monitoring, screening for secondary causes of hypertension (pheochromocytoma, coarctation of the aorta, Conn’s or Cushing’s syndrome, and renal artery stenosis), and pseudoresistance hypertension (white coat hypertension and adherence assessment). Inclusion criteria were as follows: male or female, over 35 years of age, and diagnosis with true resistant hypertension according to American Heart Association Statement (40). We excluded patients with symptomatic ischemic heart disease, liver disease, impaired renal function, and history of stroke and peripheral vascular disease. Eighty-six patients classified as resistant hypertension, 44 mild-to-moderate (stages I and II) hypertension subjects, and 16 normotensive volunteers were included in this study. Plasma F2-isoprostane levels were determined using the Enzyme Immunoassay EIA Kit (Cayman Chemical Company) according to the manufacturer’s instructions.
An additional cohort of 8 normotensive and 12 hypertensive humans was recruited at Vanderbilt for detection of isoketal adducts in blood mononuclear cells. We defined patients as hypertensive if they had a systolic blood pressure greater than 140 mmHg or were currently being treated for hypertension. Buffy coat samples were labeled with anti-CD45, anti-CD83, anti-CD14, and D11, as shown in Figure 10.
Statistics.
All data are expressed as mean ± SEM. Comparisons made between 2 variables were performed using 1- and 2-tailed Student’s t tests. Comparisons among more than 2 variables were performed using 1-way ANOVA with a Tukey’s post-hoc test. To compare differences in O2η– production in WT and Nox2–/– DCs, we used 2-way ANOVA. Comparisons of blood pressure over time were made using a mixed-effects model. For the human studies, normality of distribution was assessed by the Kolmogorov-Smirnov test. According to data distribution, we used ANOVA or Kruskal-Wallis test followed by post-hoc test. The χ2 test was used for categorical variables. P values of less than 0.05 were considered statistically significant. To model the correlation between isoketals in both CD14+ and CD83+ cells and systolic blood pressure, LDL, BMI, glucose, sex, and age, the general nonlinear model based on the restricted cubic splines method was used. The multiple imputation method was used to deal with missing values, and bootstrap sampling was used to assess internal validity and refine the model. Each test of the model parameters was 2-sided at the 0.05 level of significance. All statistical analysis was performed using R package with version 3.1.0 for Windows. For gene microarray analysis, data were uploaded into Partek Genomics Suite version 6.6 (Partek Incorporated) and RMA normalized before further analysis. Partek was used to perform pairwise comparisons of average group values, and 1-way ANOVA was used for the 4 groups. Only probes that resulted in a fold change of at least 1.5 and P value of less than 0.05 were considered significantly altered.
Study approval.
All animal procedures were approved by Vanderbilt University’s Institutional Animal Care and Use Committee, and the mice were housed and cared for in accordance with the Guide for the Care and Use of Laboratory Animals, US Department of Health and Human Services. The institutional review boards of Vanderbilt University and the University of Campinas approved the human studies. The patients signed the written informed consent form before enrolling in the study.
Supplementary Material
Acknowledgments
This work was supported by a postdoctoral fellowship from the American Heart Association (13POST14440041) and NIH grants R01HL039006, P01HL058000, P01HL095070, P01GM015431, R01HL110353, R01HL108701, K08HL121671, and R01HL105294.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(10):4642–4656. doi:10.1172/JCI74084.
Address for correspondence: David G. Harrison, Room 536, Robinson Research Building, Vanderbilt University, Nashville, Tennessee 37232-6602, USA. Phone: 615.875.3049; E-mail: david.g.harrison@vanderbilt.edu.
See the related Commentary beginning on page 4234.
References
- 1.Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. 2011;17(11):1402–1409. doi: 10.1038/nm.2541. [DOI] [PubMed] [Google Scholar]
- 2.Luft FC, et al. Hypertension-induced end-organ damage: A new transgenic approach to an old problem. Hypertension. 1999;33(1 pt 2):212–218. doi: 10.1161/01.hyp.33.1.212. [DOI] [PubMed] [Google Scholar]
- 3.Muller DN, et al. Effect of bosentan on NF-κB, inflammation, and tissue factor in angiotensin II-induced end-organ damage. Hypertension. 2000;36(2):282–290. doi: 10.1161/01.HYP.36.2.282. [DOI] [PubMed] [Google Scholar]
- 4.Nataraj C, et al. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest. 1999;104(12):1693–1701. doi: 10.1172/JCI7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guzik TJ, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10):2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298(4):R1089–R1097. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madhur MS, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- 9.Vinh A, et al. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122(24):2529–2537. doi: 10.1161/CIRCULATIONAHA.109.930446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landmesser U, et al. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40(4):511–515. doi: 10.1161/01.HYP.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuno K, et al. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112(17):2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 12.Weber DS, et al. Angiotensin II-induced hypertrophy is potentiated in mice overexpressing p22phox in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2005;288(1):37–42. doi: 10.1152/ajpheart.00638.2004. [DOI] [PubMed] [Google Scholar]
- 13.Davies SS, Amarnath V, Roberts LJ., 2nd Isoketals: highly reactive γ-ketoaldehydes formed from the H2-isoprostane pathway. Chem Phys Lipids. 2004;128(1–2):85–99. doi: 10.1016/j.chemphyslip.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 14.Roychowdhury S, McMullen MR, Pritchard MT, Li W, Salomon RG, Nagy LE. Formation of γ-ketoaldehyde-protein adducts during ethanol-induced liver injury in mice. Free Radic Biol Med. 2009;47(11):1526–1538. doi: 10.1016/j.freeradbiomed.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salomon RG, et al. Isolevuglandin-protein adducts in humans: products of free radical-induced lipid oxidation through the isoprostane pathway. Biochim Biophys Acta. 2000;1485(2–3):225–235. doi: 10.1016/s1388-1981(00)00038-x. [DOI] [PubMed] [Google Scholar]
- 16.Davies SS, et al. Treatment with a γ-ketoaldehyde scavenger prevents working memory deficits in hApoE4 mice. J Alzheimers Dis. 2011;27(1):49–59. doi: 10.3233/JAD-2011-102118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Talati M, et al. Oxidant stress modulates murine allergic airway responses. Free Radic Biol Med. 2006;40(7):1210–1219. doi: 10.1016/j.freeradbiomed.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 18.Davies SS, et al. Pyridoxamine analogues scavenge lipid-derived γ-ketoaldehydes and protect against H2O2-mediated cytotoxicity. Biochemistry. 2006;45(51):15756–15767. doi: 10.1021/bi061860g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies SS, et al. Localization of isoketal adducts in vivo using a single-chain antibody. Free Radic Biol Med. 2004;36(9):1163–1174. doi: 10.1016/j.freeradbiomed.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 20.Zagol-Ikapitte I, et al. Characterization of scavengers of γ-ketoaldehydes that do not inhibit prostaglandin biosynthesis. Chem Res Toxicol. 2010;23(1):240–250. doi: 10.1021/tx900407a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishimitsu T, et al. Urinary excretions of albumin and type IV collagen in normotensive and hypertensive subjects. Hypertens Res. 2000;23(5):459–466. doi: 10.1291/hypres.23.459. [DOI] [PubMed] [Google Scholar]
- 22.Davies SS, Amarnath V, Brame CJ, Boutaud O, Roberts LJ., 2nd Measurement of chronic oxidative and inflammatory stress by quantification of isoketal/levuglandin γ-ketoaldehyde protein adducts using liquid chromatography tandem mass spectrometry. Nat Protoc. 2007;2(9):2079–2091. doi: 10.1038/nprot.2007.298. [DOI] [PubMed] [Google Scholar]
- 23.Buettner GR. The pecking order of free radicals and antioxidants: lipid peroxidation, α-tocopherol, and ascorbate. Arch Biochem Biophys. 1993;300(2):535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 24.Lam GY, Huang J, Brumell JH. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin Immunopathol. 2010;32(4):415–430. doi: 10.1007/s00281-010-0221-0. [DOI] [PubMed] [Google Scholar]
- 25.Park B, et al. Redox regulation facilitates optimal peptide selection by MHC class I during antigen processing. Cell. 2006;127(2):369–382. doi: 10.1016/j.cell.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 26.Matsue H, et al. Generation and function of reactive oxygen species in dendritic cells during antigen presentation. J Immunol. 2003;171(6):3010–3018. doi: 10.4049/jimmunol.171.6.3010. [DOI] [PubMed] [Google Scholar]
- 27.Griffiths HR. Is the generation of neo-antigenic determinants by free radicals central to the development of autoimmune rheumatoid disease? Autoimmun Rev. 2008;7(7):544–549. doi: 10.1016/j.autrev.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97(4):696–704. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elmarakby AA, Quigley JE, Imig JD, Pollock JS, Pollock DM. TNF-α inhibition reduces renal injury in DOCA-salt hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2008;294(1):R76–R83. doi: 10.1152/ajpregu.00466.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marko L, et al. Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension. 2012;60(6):1430–1436. doi: 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 31.Harrison DG, Gongora MC. Oxidative stress and hypertension. Med Clin North Am. 2009;93(3):621–635. doi: 10.1016/j.mcna.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 32.Widder JD, Harrison DG. Can vitamin E prevent cardiovascular events and cancer? Nat Clin Pract Cardiovasc Med. 2005;2(10):510–511. doi: 10.1038/ncpcardio0291. [DOI] [PubMed] [Google Scholar]
- 33.Roberts LJ, 2nd, et al. The relationship between dose of vitamin E and suppression of oxidative stress in humans. Free Radic Biol Med. 2007;43(10):1388–1393. doi: 10.1016/j.freeradbiomed.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Landmesser U, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111(8):1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu J, et al. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res. 2014;114(4):616–625. doi: 10.1161/CIRCRESAHA.114.302157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirabo A, Oh SP, Kasahara H, Wagner KU, Sayeski PP. Vascular smooth muscle Jak2 deletion prevents angiotensin II-mediated neointima formation following injury in mice. J Mol Cell Cardiol. 2011;50(6):1026–1034. doi: 10.1016/j.yjmcc.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirabo A, et al. Vascular smooth muscle Jak2 mediates angiotensin II-induced hypertension via increased levels of reactive oxygen species. Cardiovasc Res. 2011;91(1):171–179. doi: 10.1093/cvr/cvr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eden E, Lipson D, Yogev S, Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol. 2007;3(3):e39. doi: 10.1371/journal.pcbi.0030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calhoun DA, et al. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation. 2008;117(25):e510–e512. doi: 10.1161/CIRCULATIONAHA.108.189141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.