

Abstract
Background/Objective
Progeria is a rare segmental premature aging disease with significant skeletal abnormalities. Defining the full scope of radiologic abnormalities requires examination of a large proportion of the world’s Progeria population (estimated at 1 in 4 million). There has been no comprehensive prospective study describing the skeletal abnormalities associated with Progeria. We define characteristic radiographic features of this syndrome.
Materials and Methods
Thirty-nine children with classic Progeria, ages 2–17 years, from 29 countries were studied at a single site. Comprehensive radiographic imaging studies were performed.
Results
Sample included 23 females/16 males, the largest number of prospectively evaluated patients with Progeria to date. Eight new and two little known Progeria-associated radiologic findings were identified (frequencies of 3–36%). Additionally, 23 commonly reported findings were evaluated. Of these, 2 were not encountered, 21 were present and ranked according to their frequency. Nine abnormalities were associated with increasing patient age (P=0.02–0.0001).
Conclusion
This study considerably expands the radiographic morphologic spectrum of Progeria. A better understanding of the radiologic abnormalities associated with Progeria and improved understanding of the biology of progerin (the molecule responsible for this disease), will improve our ability to treat the spectrum of bony abnormalities.
Keywords: Progeria, phenotype, skeletal findings, frequency
Introduction
Hutchinson-Gilford Progeria Syndrome (HGPS or Progeria) is a sporadic, rare autosomal dominant disorder that presents as a segmental model of premature aging affecting multiple organ systems. The classic Progeria mutation is a de novo heterozygous c.1824 C>T base substitution within the LMNA gene, located on chromosome 1q[1, 2]. The LMNA gene normally encodes for the inner nuclear membrane protein, lamin A, serving both structural and regulatory functions in most cell types [3]. The silent mutation creates a cryptic splice site, resulting in a shortened and aberrant protein product called progerin [2]. The cellular defects in Progeria stem from accumulation of progerin leading to nuclear membrane distortion and a decreased cellular life span [4, 5]. Clinical manifestations likely stem from accumulation of progerin within tissues [6, 7] and abnormal transcription of hundreds of genes that lie downstream of the LMNA defect [8–10].
HGPS was first identified nearly 126 years ago, with a single case report by Sir Jonathan Hutchinson in 1886, and further described by his colleague Hastings Gilford in a series of case reports starting in 1897 in which he coined the term “progeria” to describe the syndrome. Subsequently, in 1928, an article in French by Waldorp and del Castillo used the conjoined eponym “Hutchinson-Gilford syndrome” for the first time (11). Gene mutation identification came nearly 117 years later in 2003 (1,2).
Patients with the classic gene mutation for HGPS have a normal appearance at birth. By one year of age, there is progressive development of characteristic signs and symptoms [12]. These include generalized growth failure, alopecia, significant loss of subcutaneous fat, joint contractures and extreme short stature, along with multiple additional features. Later in childhood, atherosclerosis is apparent and children die of myocardial infarction or cerebral vascular accidents at an average age of 13 years [11].
Skeletal involvement results in a characteristic facial appearance with retrognathia, crowded dentition, narrowed nasal bridge, as well as short stature. The largest retrospective skeletal survey of patients with HGPS evaluated by conventional radiography reported osteopenia, narrowed bones with accentuated demineralization at the ends of long bones comprising the appendicular skeleton, coxa valga, hip dysplasia, avascular necrosis, acro-osteolysis, narrow chest apices, small clavicles, resorption of the distal clavicles, thin ribs, resorption of the anterior ribs, rib fractures, ovoid vertebral bodies, cardiomegaly, diastasis of the cranial sutures and Wormian bones [13]. Others have noted kyphoscoliosis [14, 15]. Recently using peripheral quantitative computed tomography (pQCT) and dual energy x-ray analysis (DXA) we demonstrated in a subgroup of the cohort of HGPS patients investigated here that the bone structure of the appendicular skeleton was highly dysmorphic [16]. Analysis of the DXA data indicated that these children had moderately low Z-scores for the projected area bone mineral density (aBMD) at the lumbar spine and proximal femur, which improved after adjustment for height age, while the overall actual volumetric BMD measured at the radius using pQCT was not different for children with HGPS compared to normal age-matched controls. Further cross-sectional pQCT images revealed distinct abnormalities of the structural geometry at the diaphysis and metaphysis of the radius in children with HGPS compared with healthy controls. Taken together, these findings suggested that the phenotype of HGPS represents a unique skeletal dysplasia.
The objective of the current study is to provide a comprehensive survey of the skeletal dysmorphisms observed in patients with HGPS using conventional radiography. We identified radiographic findings from 39 children with the classic HGPS genotype, representing approximately 15–20% of the world’s population, the largest number of patients prospectively reviewed with this syndrome. To date, only one prospective study has reported a limited set of X-ray findings for children with HGPS [17]. The results of this current study add information for characterizing the skeletal abnormalities associated with HGPS and provides reliable data on which to base treatment study outcomes.
Materials and Methods
Study Population
Participants were flown to the central study site, from 29 countries speaking 14 different languages. Ages ranged from 2.2–17.5 years. There were 23 males and 16 females. All had the classic clinical phenotype and genetically confirmed LMNA c.1824C>T (p.Gly608Gly) mutation [12]. Most pre-trial clinical information was obtained from The Progeria Research Foundation Medical and Research Database (Brown University Center for Gerontology and Healthcare Research, Providence, RI) with parental consent. For some patients, pre-trial clinical information was provided directly from the referring physicians.
Children were enrolled into either of two clinical trials for Progeria (see clinicaltrials.gov study numbers NCT00425607 and NCT00916747). As such, the patients included in this present study are included in databases providing information for other investigations, including one published earlier [16]. However the conventional imaging characteristics have not previously been categorized or reported as a part of these. Only the baseline, pre-treatment X-ray studies are reported in this study. They have not been reported elsewhere.
Consenting and Oversight
The central study site’s Committee on Clinical Investigation approved both study protocols. Written informed consent was obtained from the parents of all participants and study assent was obtained from children old enough to partake in the consent process. Consent was provided in written and oral form in the language of the parents and interpreters were provided during all testing periods for non-English speaking participants. The study was HIPAA compliant and overseen by a data and safety monitoring committee.
Radiographic Imaging
Images included AP of the hands, PA and lateral of the chest, AP and frog views of the pelvis (N=39) with dental images including an AP of much of the skull (N=30). Subsequently, AP and lateral images of each humerus, forearm and lower leg were added to the protocol. A subgroup of the 39 patients received these additional images (N = 13). All images were interpreted by the lead author (RHC), an American Board of Radiology subspecialty certified Pediatric Radiologist with 36 years experience as a pediatric radiologist.
Statistical Analyses
Comparison of age-related incidences of observations were performed using Fisher’s exact test with statistical significance defined as a two-sided p ≤ 0.05.
Results
Thirty-one skeletal abnormalities were detected on radiographic images. Table 1 lists the observations and the number and percentage of children who exhibited the observation in descending order of frequency. Eight of these observations have not previously been reported as associated with HGPS. Two have been rarely mentioned as associated with HGPS. These ten observations are shown in figures 1–4. Those findings with age specificity are listed in Table 2.
Table 1.
Rank order of observations
| Observation | Patients Affected (N=39) | % Affected |
|---|---|---|
| Small clavicles | 39 | 100 |
| Coxa valga | 37 | 95 |
| Acroosteolysis | 36 | 92 |
| Resorption of the distal clavicles | 32 | 82 |
| Narrow apices | 32 | 82 |
| Hip dysplasia | 27 | 69 |
| Thin ribs | 23 | 59 |
| Resorption of the anterior ribs | 17 | 44 |
| Closed sagittal suture | 13 (N = 30)(a) | 43 |
| Generalized osteopenia | 15 | 38 |
| Focal cortical defect* | 14(b) | 36 |
| Flexed fingers | 14 | 36 |
| Ulnar minus variant* | 12 | 31 |
| Enlarged heart | 12 | 31 |
| Dystrophic calcification** | 11 | 28 |
| Sagittal suture diastasis | 8 (N = 30)(a) | 27 |
| Enlarged femoral head*** | 10 | 26 |
| Pseudoarthrosis** | 9 | 23 |
| Enlarged femoral greater trochanter* | 9 | 23 |
| Avascular necrosis of the proximal femur | 7 | 18 |
| Kyphoscoliosis | 6 | 15 |
| Enlarged humoral head*** | 5 | 13 |
| Narrowed humoral diaphysis | 5 | 13 |
| Prominent pulmonary vessels | 5 | 13 |
| Wormian bones | 4 (N = 35)(a) | 11 |
| Accentuated osteopenia of proximal humoral/femoral epiphysis | 4 | 10 |
| Rib fracture | 4 | 10 |
| Madelung’s deformity* | 3 | 8 |
| Ivory epiphyses* | 1 | 3 |
| Bifid rib* | 1 | 3 |
| Congenitally fused ribs* | 1 | 3 |
| Narrowed femoral diaphysis**** | 0(c) | 0 |
| Oval vertebrae**** | 0 | 0 |
New observation
Observation previously rarely noted in HGPS
When femoral and humeral heads are enlarged and the distal aspect of the long bone was imaged, it was always comparably enlarged. The distal enlargement is a newly noted observation (the distal femur and humerus were only imaged in 9 of the patients).
Previously published findings which we did not observe
Imaging did not provide adequate assessment for this observation in some of the patients accounting for the N of less than 39.
Focal cortical defects were noted in the metadiaphyses of multiple long bones in 11 patients and in the greater trochanter of the femur in 4 patients. Five patients had these in multiple bones.
The femoral diaphysis was not completely imaged in these patients, therefore this observation is inconclusive.
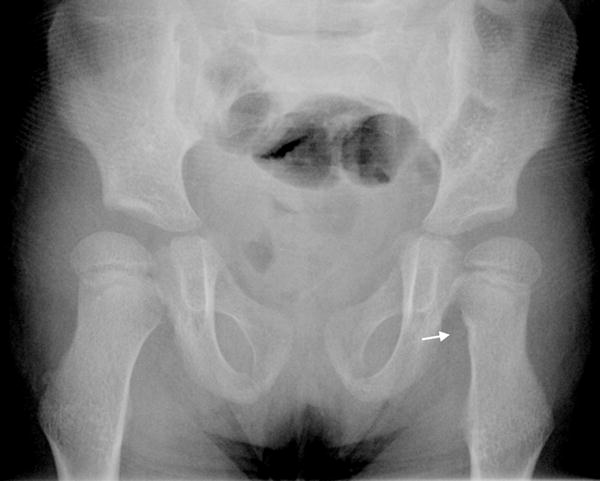
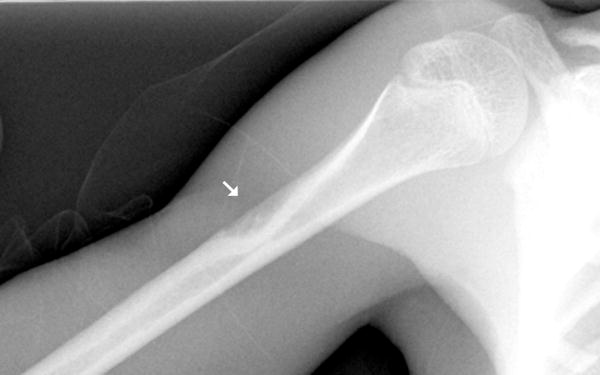
Fig 1.
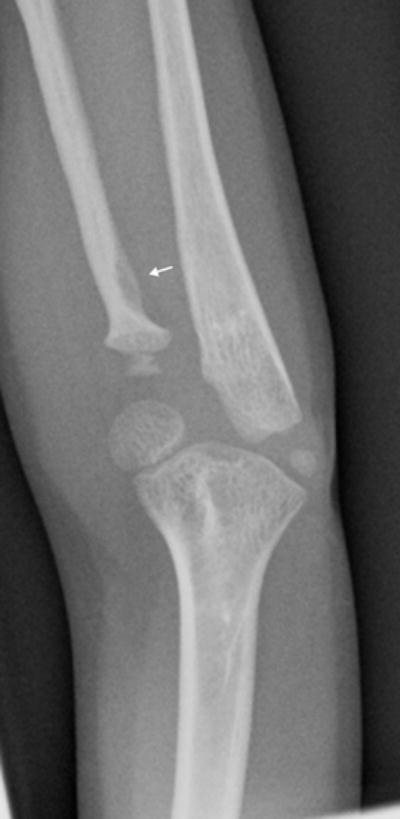
a: 5 year old girl with Progeria and a cortical notch defect: Cortical notch defects when present in the radius were always situated in the proximal diaphysis along the ulnar aspect, essentially at the site of the radial tuberosity (arrow).
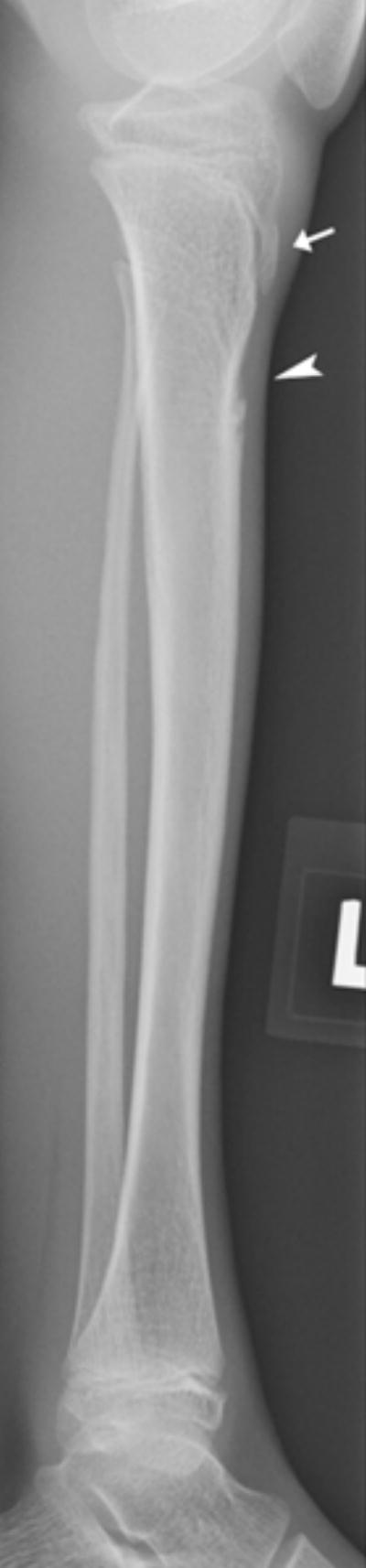
b: 10 year old girl with Progeria and a cortical notch defect: When present in the tibia, the cortical notch defects were in the anterior cortex of the proximal diaphysis somewhat below the level of the normal concavity associated with the tibial tubercle apophysis (arrow indicates the tibial tubercle; arrowhead indicates the abnormal cortical notch).
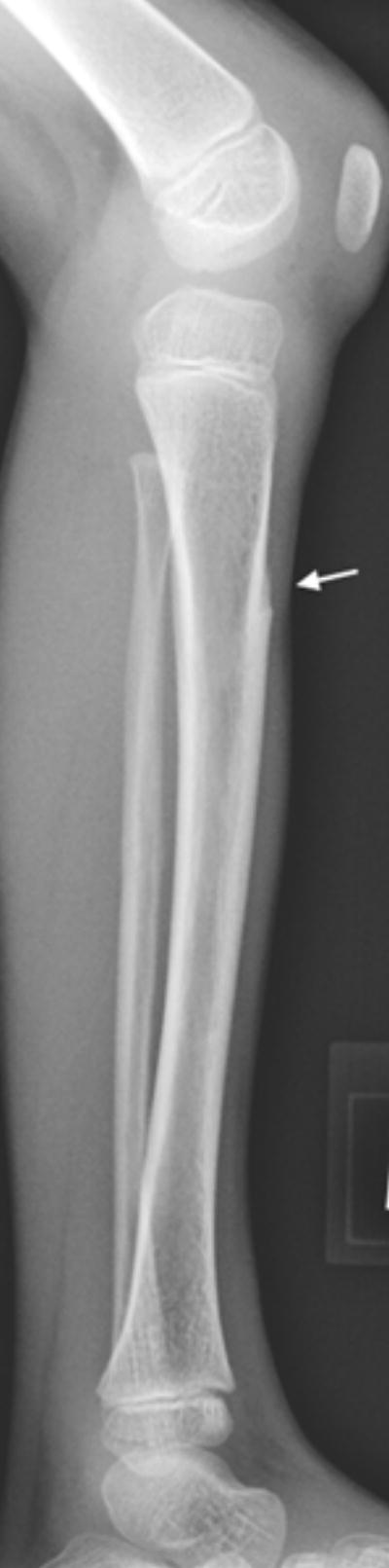
c: 5 year old girl with Progeria and a cortical notch defect: The defect was always similarly placed in the anterior proximal diaphysial cortex when present in the tibia (arrow).
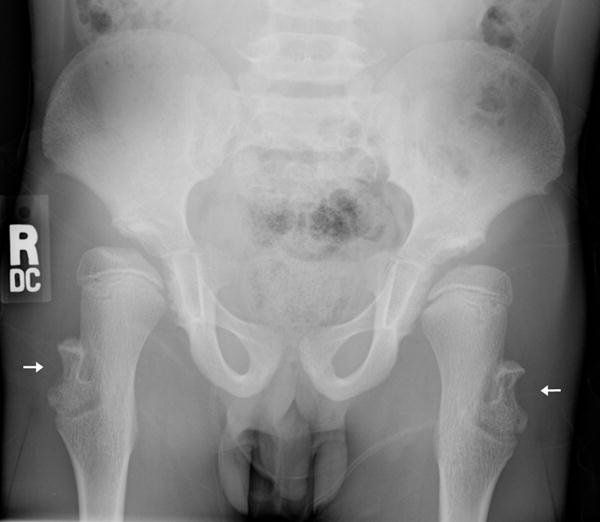
d: 9 year old boy with Progeria and a cortical notch defect: When the greater trochanter was enlarged and elongated, it often had a laterally placed notch defect (arrows). (Also note the presence of hip dysplasia, coxa valga and moderately enlarged proximal femurs.)
e: 5 year old girl with Progeria and a cortical notch defect: When present in the femoral neck, cortical notch defects were situated in the mid aspect of the medial margin, as noted on the left in this child (arrow). (Also note the presence of hip dysplasia, coxa valga and moderately enlarged proximal femurs.)
f: 10 year old girl with Progeria and a cortical notch defect: One patient had a cortical notch defect in the proximal anterior diaphysis of the right humerus (arrow). There is questionable partial internal ossification.
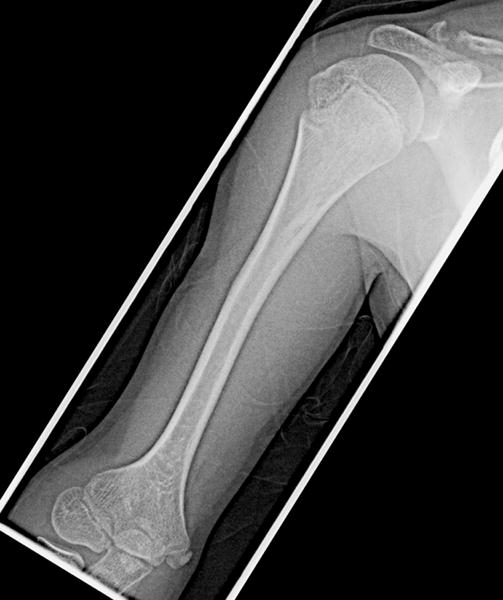
Fig 4.
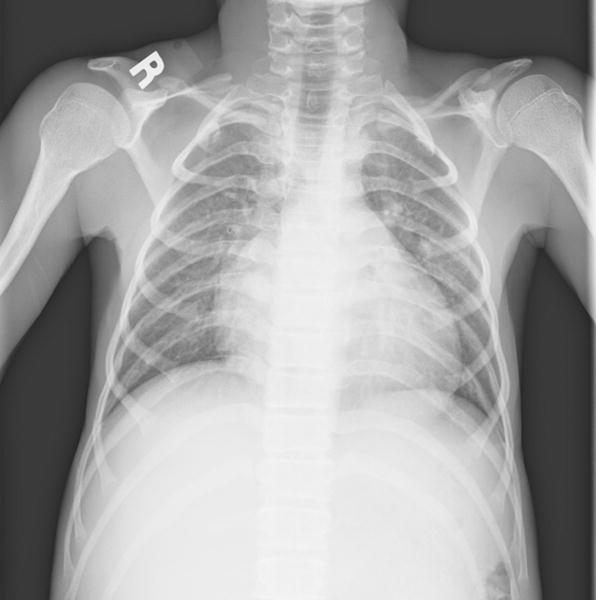
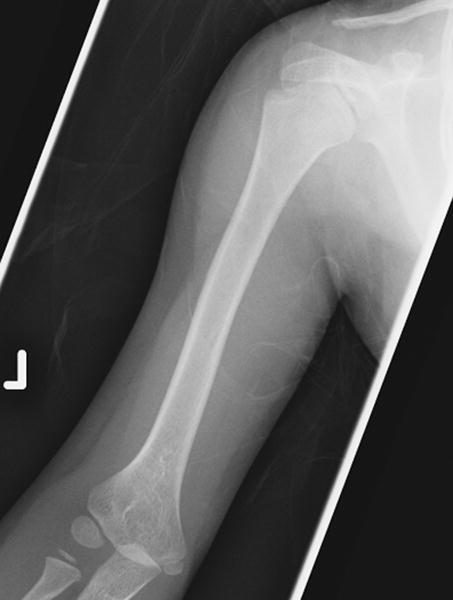
a: 17 year old boy with Progeria with enlarged proximal humerus: The proximal humerus is moderately enlarged bilaterally. (Also note narrow pulmonary apices, small clavicles with distal resorption, thin ribs with anterior resorption and cardiomegaly.)
b: 12 year old boy with Progeria with enlarged proximal and distal humerus: This patient shows mild enlargement of the proximal humerus with comparable enlargement of the distal humerus, particularly laterally. The diaphysis is mildly narrowed. (Also note pseudoarthrosis of the distal clavicle.)
c: 4 year old sister of patient in figure 4b who also has Progeria: This image, in comparison to figure 4c, reveals normally sized proximal and distal humerus without diaphyseal narrowing. (Also note mild resorption of the distal clavicle which is mildly narrowed.)
Table 2.
Age related observations
| Observation* | 2–5 years (%)** | 6–18 years (%) | p-value |
|---|---|---|---|
| Thin ribs | 4/18(22%) | 19/21(91%) | <0.0001 |
| Resorption of the anterior ribs | 2/18(11%) | 15/21(71%) | 0.0003 |
| Generalized osteopenia | 2/18(11%) | 11/19(58%) | 0.005 |
| Flexed fingers | 1/18(6%) | 13/21(62%) | 0.0003 |
| Ulnar minus variant | 2/18(11%) | 10/21(48%) | 0.02 |
| Sagittal suture diastasis*** | 6/11(55%) | 2/19(11%) | 0.03 |
| Enlarged femoral head | 3/18(17%) | 11/19(58%) | 0.02 |
| Pseudoarthrosis | 0/18(0%) | 9/21(43%) | 0.002 |
| Enlarged femoral greater trochanter | 1/18(6%) | 8/21(38%) | 0.02 |
Rates are indicated only for those observations with a statistical difference between the age groups.
Incidences are reported as percent (%) of patients within each age group with the presence of the observation.
This is not unexpected as the cranial sutures narrow and close with advancing age.
Of the eight newly recognized observations, a focal concave cortical defect at or near to the insertion of a major muscle group was the most common, seen in 14 of 39 (36%) patients at one or multiple sites. In the 9 patients with imaging of the forearm and lower leg, it was present in the proximal ulnar aspect of the radius (insertion of biceps) in 7 (fig 1a) and in the proximal tibial metadiaphysis (below the insertion of the patella tendon) in 5 (fig 1b,c). It was noted to affect the lateral aspect of the femoral greater trochanter (insertion of gluteus medius) in 4 children (fig 1d), the medial femoral neck (above the insertion of iliopsoas-MRI in one patient shows this to be at the attachment of the joint capsule) in 5 children (fig 1e) and the proximal humerus (insertion of deltoid) in 1 child (fig 1f). Of the 14 affected patients, 9 had the defect at 1 site, 3 children were affected at 2 sites, and 2 children were affected at 4 sites. When present, these defects were often, but not universally bilateral. In one patient, there was questionable internal partial ossification. Younger patients with minimal or no ossification of the femoral greater trochanter did not exhibit focal cortical concave defects at that site (Table 2).
Dystrophic calcifications have rarely been reported in HGPS [18] but were present in 11 of the 39 (28%) patients (fig 2a). Several children had these abnormal calcifications in multiple sites. However, the most common site was distal to the tufts of the fingers (7/11). Particularly at that site, the calcifications were often “popcorn like” (fig 2b) and similar to that seen with dermatomyositis, scleroderma and thermal injury [19].
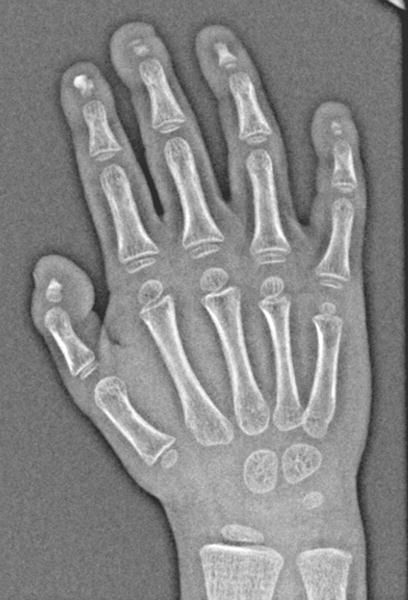
Fig 2.
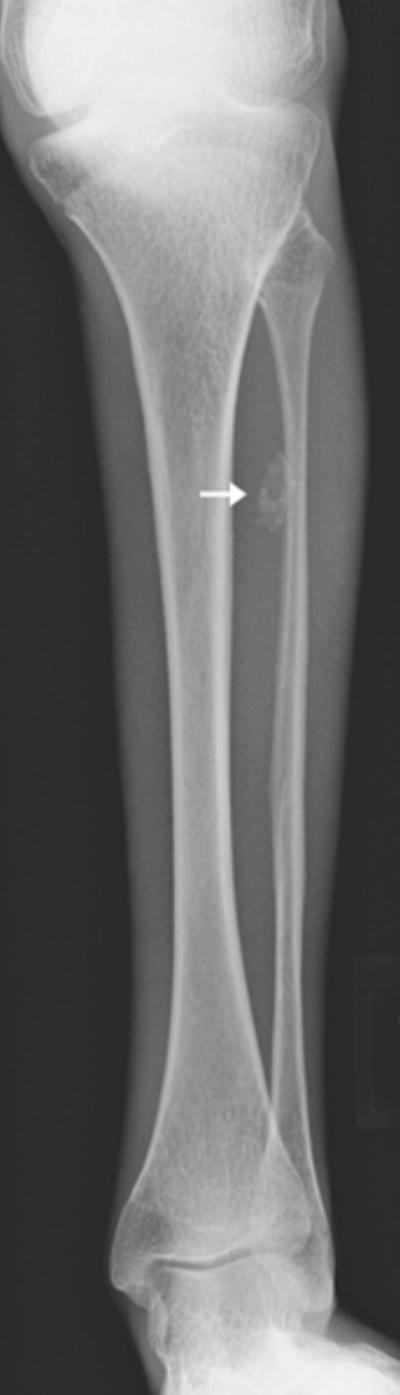
a: 17 year old boy with Progeria and dystrophic calcifications: Dystrophic soft tissue calcifications were seen over the chest, abdomen, and extremities. They were often linear, curvilinear or clumped (as in this child’s lower leg)(arrow).
b: 3 year old girl with Progeria and dystrophic calcifications: When calcifications were adjacent to the digital tufts, they frequently were “popcorn” like in their configuration as seen in the second digit. (Also note variable degrees of acroosteolysis of the tufts.)
Growth disturbances of the distal radius and ulna were seen in 38% (15/39) of the children: mild instances of ulnar minus wrist deformity (fig 3a) were present in 12 of 39 (31%) children and Madelung like deformity was present in 3 of 39 (8%) (fig 3b). Enlargement of the proximal femur (coxa magna)(26%)(10/39)(fig 1d) and humerus (13%)(5/39)(fig 4a,b) has been reported previously [13]. However, in those patients in whom the distal femur and distal humerus were imaged (9/39), comparable enlargement of the distal long bones was consistently present when proximal enlargement was noted (fig 4b). An unusual elongation and overgrowth of the femoral greater trochanter was present in 9/39 children (23%)(fig 1d, 5).
Fig 3.
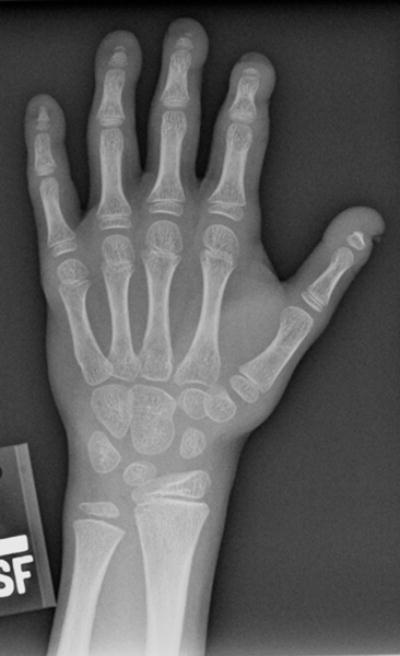
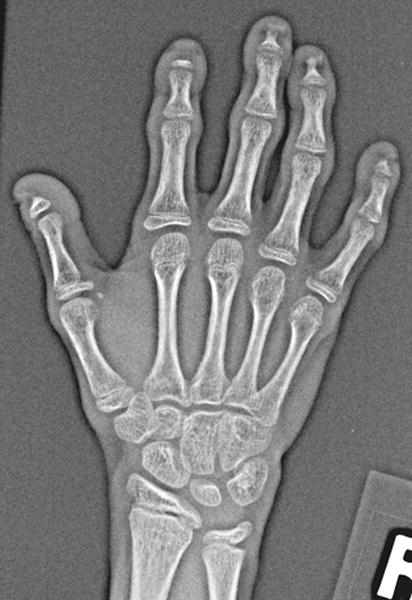
a: 8 year old boy with Progeria with ulnar minus variant: There is relative foreshortening of the ulna consistent with mild ulnar minus variant. (Also note variable degrees of acroosteolysis of the tufts.)
b: 10 year old girl with Progeria with Madelung like deformity: There is a mildly angular contour formed by the distal radius and ulna, with the median aspects of each sloping proximally, consistent with a mild Madelung’s deformity. Also note variable degrees of acroosteolysis of the tufts and dystrophic calcifications distal to several of the tufts.
Fig 5.
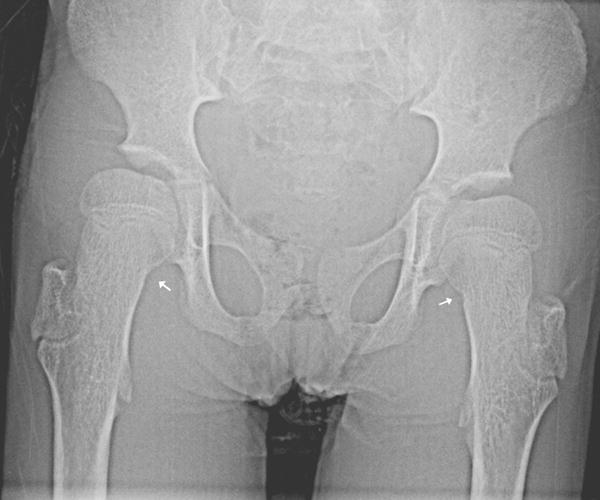
10 year old girl with Progeria with enlarged irregularly shaped greater trochanter: Once the femoral greater trochanter begins to calcify, it often becomes enlarged and elongated superiorly. (Also note small cortical notch defects in both medial femoral necks, left more pronounced than the right (arrows). Also there is mild hip dysplasia, with coxa valga and moderately enlarged proximal femurs (coxa magna).
Pseudoarthrosis of one or both distal clavicles has rarely been reported with HGPS [20] but was present in 9/39 patients (23%)(fig 6).
Fig 6.
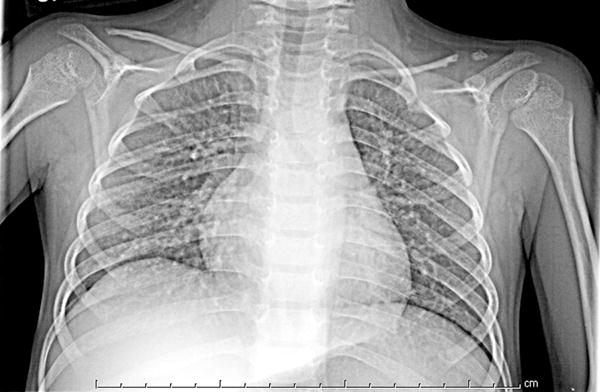
5 year old girl with Progeria with pseudoarthrosis: There is pseudoarthrosis of the distal left clavicle. Both clavicles are thin with distal resorption.
Three findings were observed in one patient and are recognized as normal variants and are not likely associated with HGPS. There were bifid ribs and congenitally fused ribs (seen in the same patient) [21], as well as ivory epiphyses [22].
Two observations previously reported as associated with Progeria were not observed in the present cohort. These were oval vertebrae [14, 15] and narrowed femoral diaphysis [14]. However, the femoral diaphyses were not specifically imaged in this study and therefore the observation of narrowing may have been inapparent.
Many of the “classic” manifestations of Progeria were confirmed [14]. These observations are typically reported in survey textbooks of syndromes and other dysmorphic conditions. For ease of reader access and comprehensive listing of observations, references to these “classic” manifestations will be to these textbooks.
Skull
Sagittal suture diastasis was present in 8/30 (27%). However with maturation of the children, the sagittal suture assumed a normal configuration. This is not unexpected as the cranial sutures narrow and close with advancing age. The oldest child with sagittal suture diastasis was 7.2 years. The earliest age at which the sagittal suture was closed was 2.5 years with mean age of closure of 8.8 years. Wormian bones were present in 4/35 patients (11%).
Chest
Narrow chest apices were present in 32/39 (82%). Small clavicles were found in 39/39 (100%), resorption of the distal clavicles in 32/39 (82%), thin ribs in 23/39 (59%) and resorption of the anterior ribs in 17/39 (44%). Rib fractures were present in 4/39 children (10%). Heart size was judged as enlarged in 12/39 patients (31%) and the pulmonary vasculature as prominent in 5/39 patients (13%).
Scoliosis
Kyphoscoliosis has been previously reported [14]. The extent of convexity of the spinal deformity (expressed in degrees of the subtended arc) in frontal plane deformity (scoliosis) and in the sagittal plane (kyphosis) was assessed using the Cobb method [23]. A scoliosis of the thoracolumbar spine (defined as the presence of an mild undulating curve (N = 3: major curve of 7, 12 and 13 degrees) or a Cobb angle of 15 degrees or greater)(N = 2: 16 and 18 degrees) and/or a kyphosis (defined as a Cobb angle of 45 degrees or greater)(N = 3: 57, 68, and 83 degrees) was present in 6/39 patients (15%). (Two of the patients with a scoliosis greater than 15 degrees also had a kyphosis of greater than 45 degrees.)
Hip
There were several observations pertaining to the hips that seem to be an integral component of the progeria phenotype (fig 1d, 1e, 5, 8). Coxa valga [14] was present in 37/39 (95%), hip dysplasia in 27/39 children (69%), an enlarged proximal femur (coxa magna) [13] in 10/39 (26%), an enlarged elongated femoral greater trochanter in 9/39 (23%) and avascular necrosis (AVN) [14] of the proximal femur in 7/39 (18%).
Humerus
A similar alteration in long bone morphology was also recognized in the proximal humerus. The humeral head was enlarged [7] in 5/39 (13%) and the diaphysis was narrowed [14] in 5/39 (13%)(fig 4b). Only enlargement of the proximal humerus and femur have been previously reported [13]. However, in the current study, in those instances were both ends of the bone were imaged, if the proximal end of the long bone was enlarged, the distal metaphysis and epiphysis were comparably enlarged (fig 4b). In every patient in whom enlargement of both the ends of the humerus was noted, there was comparable enlargement of both the ends of the femur. There were 5 patients with mild enlargement of the ends of the femur in whom the ends of the humerus were not notably enlarged.
Hand
Acroosteolysis of phalangeal tufts was present in 36/39 (92%)(fig 3b). Flexion of the fingers associated with the generalized flexion contractures that occur in Progeria [17] were present on bone age images of 14/39 (36%). Bone age estimates were performed using the standards of Greulich and Pyle [24]: 18/39 (46%) were within 1 standard deviation (SD) of the norm, 11/39 (28%) were within 2 SD of the norm, 6/39 (15%) were within 3 SD above the norm, 1/39 (3%) was within 3 SD below the norm, 3/39 (8%) were 3 or more SD above the norm and none were 3 or more SD below the norm.
Because HGPS is a progressive disease which can exhibit age-associated bone abnormalities [13], we compared abnormalities between younger and older age groups. Consequently the children were divided into two groups of approximately equal size based on age at initial imaging; 18 patients aged 2–5 years (39% male) and 21 aged 6–18 years (38% male). Table 2 lists 8 skeletal findings that were significantly more common in the older patient group, and one that was more common in the younger patient group.
Osteopenia
Generalized osteopenia was judged as present in 15/39 patients (38%) and accentuated osteopenia as present at the proximal humerus and or femur in 4/39 patients (10%).
Discussion
The objective of this study is to provide a comprehensive survey of the skeletal dysmorphisms observed in patients with HGPS evaluated using conventional radiography. This study presents the only prospective radiographic examination of approximately 15–20% of the world’s HGPS population, encompassing significant gender, age, and ethnic diversity. The only other prospective study of children with HGPS was relatively limited and identified only acro-osteolysis, clavicular resorption, and coxa valga in all 15 of its subjects [17]. This current study extends the previous retrospective radiographic study of HGPS [13] by identifying new HGPS-related skeletal dysmorphisms and findings that are apparently age-related. These observations may allow us to examine more closely the root causes of the skeletal abnormalities associated with HGPS. In addition, as children of all ages with HGPS undergo new treatment trials initiated at all ages, we can closely follow the progression, regression or delayed onset of skeletal abnormalities in response to the therapies. Only a sufficiently large and uniformly studied baseline cohort can yield these types of treatment analyses.
Of the eight newly recognized observations, a focal concave cortical defect at or near to the insertion of a major muscle group was the most common, seen in 14/39 patients (36%). Although many of the defects are at the insertion of muscles, some are merely relatively close to an insertion site. Whether the defects relate to abnormal tension, are a component of a skeletal dysplasia or an as yet unrecognized abnormality of mesodermal proliferation is unknown.
Dystrophic calcifications, previously rarely recognized as a component of HGPS, were present in 11/39 (28%) patients. The precise etiology of these calcifications is unclear but may relate to microvascular compromise as suggested in dermatomyositis, scleroderma and thermal injury [19, 25–27].
Ulnar minus variant [28] and Madelung’s deformity [29], both abnormalities of the distal radius and/or ulna, have been reported with various syndromes [30, 31] and in isolation.
Enlargement of the distal ends of the femur and humerus and its association with an enlarged proximal femur and proximal humerus has not been previously noted. It is unclear whether this represents a skeletal dysplasia affecting the metaphysis or whether the intervening diaphysis is disproportionately narrowed. An unusual elongation and overgrowth of the greater trochanter, present in 23% (9/39) of our cohort, also has not previously been described and may be associated with the coxa valga and coxa magna frequently observed at the proximal femur of our patient cohort.
Non-traumatic (congenital) pseudoarthrosis, present in 23% (9/39) of our cohort, rarely has been reported in association with HGPS [20], although not infrequently seen in other multiple syndromes including fibrous dysplasia, neurofibromatosis type 1 and as an isolated phenomenon [32].
We acknowledge several study limitations. Some observations, although present, may have been affected by imaging variables. Heart size (12/39 patients: 31%) [14] and pulmonary vascular prominence (5/39 patients: 13%) were judged to be increased. However the degree of prominence was only moderate and frequently associated with low lung volumes which may accentuate these subjective assessments. More precise assessment of cardiovascular status has been presented in two prior natural history studies [17,33]. Decreased skeletal mineralization, assessed as generalized osteopenia [14] was judged as present in 15/39 children (38%). This subjective assessment was considered as mild to moderate in 14/39 and severe in degree in only one patient. Focally accentuated osteopenia at the proximal epiphyses of the humerus and femur [13] was judged as possibly present in 4/39 children (10%). It must be emphasized however, that osteopenia is a subjective observation based on conventional radiographic images. More objective assessment of skeletal mineralization in a subset of 26 of these patients, based on DEXA and pQCT evaluations, revealed that the bone mineral density was moderately low to low when adjusted for the equivalent height age or actual cross-sectional bone geometry but that the structural geometry of the bone was significantly abnormal. The decreased size and thickness of the bone structure contributed to the apparent appearance of low bone mass [16].
HGPS exhibits abnormalities in cells and tissues of mesodermal origin such as the vasculature, cartilage, extracellular matrices and bone, where cells not only express the disease-causing protein progerin, but also exhibit a wide array of in vitro and in vivo dysfunction. Although many of the precise mechanisms culminating in pathophysiology remain poorly defined, the commonality of several of our observations raises possibilities. The clustering of abnormalities at the ends of long bones (including avascular necrosis) suggests that impairment in development at secondary centers of growth (apophyses and epiphyses) may relate to vascular compromise as is known to occur in both the large and small vessels in patients with HGPS [14]. The presence of dystrophic soft tissue calcification could also be secondary to microvascular compromise leading to postinfarction calcification. The cortical defects seen at several sites may relate to abnormalities of mesodermal proliferation.
This comprehensive radiographic review of 39 children with HGPS constitutes the largest aggregate of patients studied collectively and approximately 15 – 20% of the world’s population of patients with HGPS. As such it provides insight into the spectrum of skeletal dysmorphisms associated with this disease. In addition to confirming several commonly recognized manifestations of the syndrome, it provides an appreciation of their frequency and an estimate of the incidence of radiographic characteristics not previously available. Eight previously unreported observations have been recognized, several of which occurred frequently in this population. Two previously rarely recognized observations were frequently present.
Several of the observations are more common at older ages suggesting that at least a part of the skeletal phenotype may relate to an on-going metabolic insult and/or buildup of abnormal tissues. This radiographic review allows us to better understand the spectrum of skeletal dysmorphisms associated with this disease and to monitor the progression and/or regression of the skeletal manifestations of the disease in response to treatment.
Conclusion
This report confirms commonly recognized manifestations of Progeria and provides an appreciation of their frequency. For several of the observations, the incidence increased with age. Additionally, eight previously unreported observations are recognized.
Acknowledgments
We are grateful to the children with progeria and their families for their participation in this study. We thank the Family Inn (Cambridge, MA) and Devon Nicole House (Boston, MA) for housing families; Susan Campbell, MS, Nancy Wolf-Jenssen, and Nancy Grossman for medical records coordination; Kyra Johnson, Kelly Littlefield, Kiera McKendrick, Angela Kraybill, and William Fletcher for coordinator services.
Funding: This project was funded by The Progeria Research Foundation (PRFCLIN2007-01 and Grant #PRFCLINTRIAL003-080109), the National Heart, Lung and Blood Institute (1RC2HL101631-01), the Dana-Farber Cancer Institute Stop&Shop Pediatric Brain Tumor Program, by a National Center for Research Resources to the Children’s Hospital Boston General Clinical Research Center (MO1-RR02172), and a grant from the National Center for Research Resources, National Institutes of Health, to the Harvard Catalyst Clinical & Translational Science Center (Harvard Catalyst) (UL1 RR025758-01).
Footnotes
Disclosures: LBG is the parent of a child with HGPS who participated in this study.
References
- 1.De Sandre-Giovannoli A, Bernard R, Cau P, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 2.Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldman RD, Goldman AE, Shumaker DK. Nuclear lamins: building blocks of nuclear structure and function. Novartis Found Symp. 2005;264:3–16. discussion 16–21, 227–230. [PubMed] [Google Scholar]
- 4.Bridger JM, Kill IR. Aging of Hutchinson-Gilford progeria syndrome fibroblasts is characterised by hyperproliferation and increased apoptosis. Exp Gerontol. 2004;39:717–724. doi: 10.1016/j.exger.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Goldman RD, Shumaker DK, Erdos MR, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McClintock D, Gordon LB, Djabali K. Hutchinson-Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody. Proc Natl Acad Sci U S A. 2006;103:2154–2159. doi: 10.1073/pnas.0511133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olive M, Harten I, Mitchell R, et al. Cardiovascular Pathology in Hutchinson-Gilford Progeria: Correlation With the Vascular Pathology of Aging. Arterioscler Thromb Vasc Biol. 2010 doi: 10.1161/ATVBAHA.110.209460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amati F, Biancolella M, D’Apice MR, et al. Gene expression profiling of fibroblasts from a human progeroid disease (mandibuloacral dysplasia, MAD #248370) through cDNA microarrays. Gene Expr. 2004;12:39–47. doi: 10.3727/000000004783992189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Csoka AB, English SB, Simkevich CP, et al. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell. 2004;3:235–243. doi: 10.1111/j.1474-9728.2004.00105.x. [DOI] [PubMed] [Google Scholar]
- 10.Park WY, Hwang CI, Kang MJ, et al. Gene profile of replicative senescence is different from progeria or elderly donor. Biochem Biophys Res Commun. 2001;282:934–939. doi: 10.1006/bbrc.2001.4632. [DOI] [PubMed] [Google Scholar]
- 11.Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140:2603–2624. doi: 10.1002/ajmg.a.31346. [DOI] [PubMed] [Google Scholar]
- 12.Kieran MW, Gordon L, Kleinman M. New approaches to progeria. Pediatrics. 2007;120:834–841. doi: 10.1542/peds.2007-1356. [DOI] [PubMed] [Google Scholar]
- 13.Gordon LB, McCarten KM, Giobbie-Hurder A, et al. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics. 2007;120:824–833. doi: 10.1542/peds.2007-1357. [DOI] [PubMed] [Google Scholar]
- 14.Taybi H, Lachman R, editors. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasia. 4. St. Louis, Missouri: Mosby Year-Book, Inc; 1996. Progeria; pp. 401–403. [Google Scholar]
- 15.Jones K, editor. Smith’s Recognizable Patterns of Human Malformation. 6. Philadelphia, Pennsylvania: Elsevier Saunders; 2006. Progeria Syndrome (Hutchinson-Gilford Syndrome) pp. 146–149. [Google Scholar]
- 16.Gordon CM, Gordon LB, Snyder BD, et al. Hutchinson-gilford progeria is a skeletal dysplasia. J Bone Miner Res. 2011;26:1670–1679. doi: 10.1002/jbmr.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merideth MA, Gordon LB, Clauss S, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358:592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenthal IM, Bronstein IP, Dallenback FD, Pruzansky S, Rosewald AK. Progeria : report of a case with cepalometric roentgenograms and abnormally high concentrations of lipoproteins in serum. Pediatrics. 1956;18:565–577. [PubMed] [Google Scholar]
- 19.Poznanski A, editor. The Hand in Radiologic Diagnosis. Piladelphia, Pennsylvania: W.B. Saunders Company; 1984. Chapter 21: Metabolic Bone Disease and Other Abnormalities of the Hand; pp. 839–894. [Google Scholar]
- 20.Margolin FR, Steinbach HL. Progeria Hutchinson-Gilford Syndrome. AJR Am J Roentgenol. 1968;103:173–178. [PubMed] [Google Scholar]
- 21.Resnick D. Chapter 83: Additional Congenital or Heritable Anomalies and Syndromes. In: D R, editor. Diagnosis of Bone and Joint Disorders. 4. Philadelphia, Pennsylvania: W.B. Saunders Comp; 2002. pp. 4561–4631. [Google Scholar]
- 22.Taybi H, Lachman R, editors. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasia. St. Louis, Missouri: Mosby Year-Book, Inc; 1996. Epiphysis: Dense, Sclerotic, Ivory; p. 1029. [Google Scholar]
- 23.Ozonoff M. Chapter 82: Spinal Anomalies and Curvatures. In: Resnick D, editor. Diagnosis of Bone and Joint Disorders. 4. Philadelphia, Pennsylvania: W.B. Saunders Comp; 2002. pp. 4534–4559. [Google Scholar]
- 24.Greulich W, Pyle S, editors. Radiographic Atlas of Skeletal Development of the Hand and Wrist. Stanford, California: Stanford University Press; 1959. [Google Scholar]
- 25.Louis D, Greene T, Poznanski A. Burns, Frostbite, Foreign Bodies and Other Traumatic Lesions of the Hand. In: Poznanski A, editor. The Hand in Radiologic Diagnosis. Piladelphia, Pennsylvania: W.B. Saunders Company; 1984. pp. 708–733. [Google Scholar]
- 26.Staple T. Joint Disorders, Chapter 20. In: AK P, editor. The Hand in Radiologic Diagnosis. Philadelphia, Pennsylvania: W.B. Saunders Comp; 1984. pp. 791–838. [Google Scholar]
- 27.Taybi H. Scleroderma. In: Taybi H, Lachman R, editors. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasia. 4. St. Louis, Missouri: Mosby Year-Book, Inc; 1996. pp. 448–450. [Google Scholar]
- 28.Resnick D. Chapter 65: Internal Derangements of Joints. In: Resnick D, editor. Diagnosis of Bone and Joint Disorders. 4. Philadelphia, Pennsylvania: W.B. Saunders Comp; 2002. pp. 3019–3375. [Google Scholar]
- 29.Poznanski A, editor. The Hand in Radiologic Diagnosis. Piladelphia, Pennsylvania: W.B. Saunders Company; 1984. Chapter 8: Normal Variation and Congenital Anomalies of the Wrist; pp. 181–208. [Google Scholar]
- 30.Taybi H, Lachman R, editors. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasia. 4. St. Louis, Missouri: Mosby Year-Book, Inc; 1996. Ulna/Ulnar Ray: Aplasia, Hypoplasia; p. 1044. [Google Scholar]
- 31.Taybi H, Lachman R, editors. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasia. 4. St. Louis, Missouri: Mosby Year-Book, Inc; 1996. Madelung Deformity; p. 1036. [Google Scholar]
- 32.Taybi H, Lachman R, editors. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasia. 4. St. Louis, Missouri: Mosby Year-Book, Inc; 1996. Pseudoarthosis; pp. 1039–1040. [Google Scholar]
- 33.Gerhard-Herman M, Smoot LB, Wake N, et al. Mechanisms of Premature Vascular Aging in Children With Hutchinson-Gilford Progeria Syndrome. Hypertension Jan. 2012;59(1):92–7. doi: 10.1161/HYPERTENSIONAHA.111.180919. Epub 2011 Nov 14. [DOI] [PMC free article] [PubMed] [Google Scholar]