

Abstract
A combined DFT/electrostatic approach is employed to study the coupling of proton and electron transfer reactions in cytochrome c oxidase (CcO) and its proton pumping mechanism. The coupling of the chemical proton to the internal electron transfer between heme a and the binuclear center is examined for the O→E transition. The novel features of the His291 pumping model are proposed, which involve timely well-synchronized sequence of the proton-coupled electron transfer reactions. The obtained pKas and Ems of the key ionizable and redox-active groups at the different stages of the O→E transition are consistent with available experimental data. The PT step from E242 to H291 is examined in detail for various redox states of the hemes and various conformations of E242 side-chain. Redox potential calculations of the successive steps in the reaction cycle during the O→E transition are able to explain a cascade of equilibria between the different intermediate states and electron redistribution between the metal centers during the course of the catalytic activity. All four electrometric phases are discussed in the light of the obtained results, providing a robust support for the His291 model of proton pumping in CcO.
Keywords: Cytochrome c oxidase, Computational chemistry, Donor-acceptor systems, Gating mechanism, pKa and Em calculations, Proton-loading site, Proton pumping
1. Introduction
Cytochrome c oxidase (CcO) is one of the most fascinating biochemical systems where coupled electron (ET) and proton transfer (PT) reactions play a crucial role.[1, 2] The enzyme is redox-driven proton pump, where the reduction of oxygen to water is coupled to proton pumping across the membrane, generating in part the proton gradient utilized in the production of ATP by ATP-synthase. [2, 3]
CcO contains four redox-active metal centers: CuA, heme a (Fea), and the binuclear complex consisting of heme a3 (Fea3) and CuB, see Figure 1. Electrons supplied to CuA by reduced cytochrome c are sequentially transferred through Fea to the active site, where the reduction of oxygen takes place. [3–5] In the catalytic cycle, the transfer of each of the four electrons required for the reduction of an oxygen molecule is accompanied by the translocation of the two protons, of which one is consumed internally in the binuclear center for the chemical reduction of oxygen (“chemical or substrate proton”) and the other is pumped across the membrane (“vectorial or pumped proton”). The overall reaction can be expressed as follows:
| (1) |
where (in) and (out) indicate two sides of the membrane: the matrix (N-) and inter-membrane (P-) side, respectively. Three possible entrance proton transfer pathways (D-, K-, and H-pathways) have been identified in the X-ray structures of bovine and bacterial CcO at high resolution. [6–11] Both chemical and pumped protons are delivered along the D- and K-channels. [5, 12, 13] Presumably, 6–7 protons are delivered along the D-channel, whereas the D-channel provides all pumped protons. In contrast, ref. [7] and [14] suggest the primary role of H-pathway in translocation of the pumped protons from the N- to P-side of the membrane.
Figure 1.

Structure of the core catalytic subunits A and B from bovine cytochrome c oxidase embedded in the inner mitochondrial membrane. [7] Two entrance proton-conducting channels providing pumped and chemical protons are also shown.
The molecular mechanism of proton pumping of CcO still remains a subject of intense debate. Some recent reviews of the enzyme structure, function and kinetics, can be found e.g. in refs. [10, 15–23].
1.1. Recent theoretical studies. His291 model
In the work by this group, [24–27] on the basis of combined electrostatic and ab initio calculations a kinetic model of proton pumping by CcO has been proposed. The key elements of the model are H291 (a ligand to CuB center) and its the redox-dependent protonation state, and the two proton-conducting water chains connecting E242 both to the catalytic site (BNC) and to H291 (PLS). [25, 28] From the mechanistic point of view, the salt bridge PRDa3/R438 and a crystallographic water molecule W1 located between the propionates of heme a3 (Figure 2) are likely to be important for both the regulation of the transfer of the pumped proton to PLS, [24, 29], as well as for water exit from the hydrophobic catalytic cavity. [30] As establish experimentally, E242 plays a key role in conducting both the pumped and chemical protons. [31–33] The proposed “kinetic gating mechanism” suggests that E242 is connected to PLS and the active site (BNC) by two separate proton-conducting water chains with different proton conducting rates. The faster chain delivers the pumped protons to PLS, whereas the slower chain delivers the chemical protons to BNC. The difference in rates ensures that a proton is preloaded into the pump site PLS before the driving redox event, i.e. the protonation of the reduced oxygen intermediates at the active site by the chemical protons, occurs. [25] The structure and dynamics of proton-conducting water networks suggests that the rate of proton transfer from E242 to H291 is likely much faster than that between E242 and OH− in the catalytic center.
Figure 2.

The gating through conformational change of the E242 side-chain. The coordinates of red conformation (pointing downwards) are taken from the X-ray structure of bovine heart cytochrome c oxidase [7] and represent a proton loading conformation of E242 residue that is in position to closely interact with the D-channel residues and water molecule network. The blue conformation is obtained by the quantum-chemical optimization [34] and presumably represents the proton releasing conformation of residue E242. The internal water molecules of the hydrophobic cavity (not shown for clarity) may provide a proton-conducting network to the PLS (via PRDa3/R438 salt bridge and W1 water molecule), or to the BNC for the chemical reduction of oxygen intermediates. The proton-conducting rate of the former network is presumably faster than the rate of the latter. The protons on a H2O ligated in the BNC and Hδ1 proton on H291 residue, which may work as the PLS, are explicitly shown.
Recently, we have extended the proposed model by considering the rotational isomerization of E242 residue, a central proton intermediate, which can exist in two distinct conformations – downward and upward, which are assumed to be the proton input and output, respectively. [34] The two conformations seem to be of the great importance, as the Glu side-chain can form the H-bonded proton-conducting pathway only to one site at a time: either to the D-channel, or to PLS, or to BNC, see Figure 2. If Glu is connected to one of the pathways, the other two are temporary closed. The gating through conformational changes of the Glu side-chain [34] is similar to that in the E242 “valve model” proposed by the Wikström group. [35]
Several other computational groups have applied recently various methods to examine the pumping mechanism of CcO. Thus in Ref. [36] the transition state theory and energy diagrams have been used to analyze the proton pumping mechanism. The QM/MM has been employed to study the microscopic pKas of E242 in controlling the proton translocation, [37] while the generalized valence bond molecular dynamics, [38] Monte Carlo dynamic analysis, [39] quantum-chemical studies [40–46] and the continuum electrostatics [47–49] have been applied to study different aspects of this complex protein system.
In this paper, we report on the new calculations that combined first principles DFT and continuum electrostatics to evaluate the energetics of the proton transfer between E242 and H291 for different populations of the reduced/oxidized heme a and heme a3 in the course of the O→E transition studied recently in ref. [15]. The E242 conformational changes, thought to be of great importance for the unidirectionality and prevention of a backward proton leakage, [34, 36, 50–52] are taken into account and their importance for the kinetic gating mechanism proposed earlier [25] is discussed. The coupling of the chemical and pumped proton translocation to the internal electron transfer to the binuclear catalytic center (Fea3 to CuB) is examined. The midpoint potentials (Em) of the redox centers are evaluated at the different stages of the reaction cycle; all four electrometric phases observed in experiment [15] are discussed in the light of the obtained results. The new data provide a solid support for the novel features of the H291 model of CcO pumping.
The key question we examine in this study is whether the pumped proton translocation occurs already upon the reduction of heme a, as recently have been suggested on the basis of potentiometric data. [15, 53] For that purpose, we have monitored the PT reaction from E242 to H291 for different distributions of an electron on the two hemes during the corresponding ET event in bovine CcO.
1.2. Electrometric study: O→E transition
For convenience, here we briefly summarize the experimental data [15] on the O→E transition observed in potentiometric measurements. [12, 54–59] Upon single-electron injection, four kinetic phases are observed: a pure electronic kinetic phase, and three protonic phases, which overlapped with electron movement parallel to the membrane.
In ref. [15], phase 1 (10 μs) is associated with electron transfer from CuA to heme a, thereby moving a fraction of electron (β=0.70) to heme a. Further electron redistribution between CuA, Fea, and the Fea3/CuB binuclear center is coupled to three proton transfers, which generate three additional kinetic phases (Figure 3). The spectroscopic data indicate that by the end of the 150-μs phase, 60% of the electron is transferred to the Fea3/CuB center, while 40% still remains on heme a. This electron transfer is coupled to the translocation of a pumped proton from the N-side of the membrane to an unknown PLS above the BNC. E242 is apparently a proton donor for this proton transfer.
Figure 3.

Schematic representation of the electrometric results. The numbers correspond to the order of the kinetic phases observed in the experiment. [15] The BNC is OH− ligand of the binuclear catalytic center; the PLS is the proton-loading site of the pump, presumably H291. The donor of the pumped protons – E242 is shown in the two conformations, down and up, as discussed in the paper. For the O→E transition, the third phase apparently corresponds to the transfer of a chemical proton along the K-channel; in other transitions, the same proton transfer may take place along the D-channel.
The next 800-μs phase corresponds to the transfer of the remaining 40% of the electron from heme a to the BNC and an accompanying transfer of the chemical proton to the BNC. Finally, the last 2.6-ms phase is associated with the reprotonation of the donor site for chemical proton† and a displacement of the pumped proton from the PLS to the P-side of the membrane. Presumably, this happens due to repulsion between the chemical proton arrived to the BNC and the pumped proton preloaded to the PLS. [24, 60] Reprotonation of the proton donor site, before proton release from the PLS to the P-side of the membrane, further facilitates the proton pumping event. [34]
Although the experimental results are obtained for CcO from P. denitrificans, as it has been demonstrated recently, all species of the A-family show remarkable structural similarity. Moreover, their microscopic electrostatic and thermodynamic properties of the key amino-acid residues are almost identical, which strongly suggests similar mechanism in these species. [61]
2. Model and Methods
2.1. Studied model
Our His291 model [25] closely corresponds to the above interpretation of the electrometric study. The only inconsistency is related to the order of coupled electron and proton transfer steps associated with the ET from heme a to the BNC and the PT from E242 to PLS. Obviously, the reduction of heme a3 would make the translocation of a pumped proton more energetically feasible, because an electron closer to the PLS (presumed His291) would increase its pKa. In line with this, in ref. [52] the PT from Glu to PLS (PRAa3 was assumed for PLS) was found to be uphill in energy by 4.6 pK-units (275 meV). Nevertheless, the experimental data for the O→E transition can be interpreted to suggest a mechanism in which the translocation of the pumped proton occurs before the ET to the binuclear center, [15] contrary to our proposal. In contrast, ref. [58] supports the transfer of the pumped proton to PLS upon ET to the binuclear center. However, the latter experimental results are for the F→O transition, which may suggest that the oxidative and reductive halves of the catalytic cycle are not entirely identical in all mechanistic details, besides the obvious difference in chemistry and kinetic rates.
The aforementioned discrepancy has motivated us to study the coupling between the PT and ET reactions in CcO in more details. Here we used a modified model shown in Figure 4. We particularly focus on the PT from Glu to His291 for different populations of the reduced/oxidized hemes (a and a3) during the coupled ET reaction.
Figure 4.
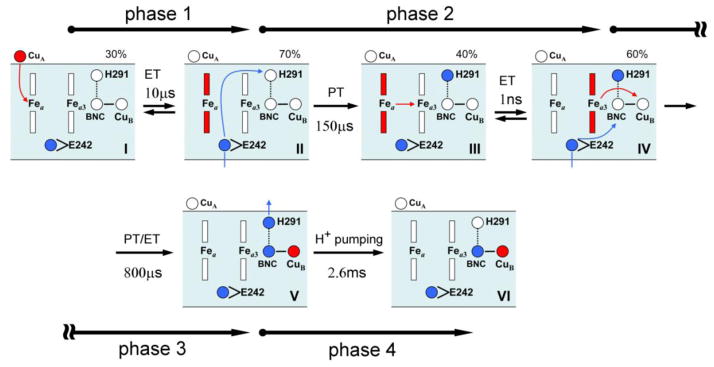
Schematics of the studied pumping model based on the kinetic measurements and interpretation of the experiments for the O→E transition. [15] The four redox-active centers (CuA, Fea, Fea3, CuB) and three key protonatable sites (E242, H291, OH− ligand in the BNC) of the proposed model are schematically shown. The reduced and oxidized metal centers are shown in red and white color, while the protonated and deprotonated sites are displayed in blue and white, respectively. The red arrows represent the electron transfer (ET) steps, as the blue arrows represent the proton translocations (PT).
2.2. DFT/electrostatic method
The calculations of the pKa values of H291 and E242 have been done using a method that combines DFT (Jaguar 5.5 program [62]) and continuum electrostatic calculations (MEAD suite [63] and Karlsberg program [64]), as described in our recent work. [26, 34] Briefly, in the calculations the whole system is divided into a quantum-mechanical (QM) part, and the surrounding medium, which consists of the rest of the protein, membrane, internal cavities and external aqueous phase. Density functional theory [65] is applied to a relatively small QM system, to optimize its geometry, evaluate the gas phase electronic energies (Gelec), calculate the reorganization energies (Gstrain), and obtain the atomic partial charges for the different redox and protonation states of the complex. Electrostatic calculations, on the other hand, are used to evaluate the reaction (Grf) and protein field (Gpf) solvation energies. Thus, the protonation energies of the two key residues (Glu and His) are calculated by DFT modified by the influence of the protein charges and dielectric inhomogeneities. The pKa’s then can be obtained relative to the appropriate model compound in a standard fashion.
The starting structure of the active site was taken from the X-ray crystal structure of bovine heart cytochrome c oxidase obtained by Yoshikawa, et al, at 2.3Å resolution (PDB ID: 2OCC). [7] The QM model used to calculate the pKa of H291 consists of the CuB center together with OH−/H2O ligand and three coordinated 4-methylimidazoles (representing H240, H290 and H291). Also, Y244 cross-linked to H240 is included as a part of the QM system by replacing it with phenol ring. When pKa of E242 is evaluated, only propionic acid representing Glu side-chain was treated by the DFT. Since, the system contains only two separate QM parts, the self-consistency in their pKas is straightforward to achieve.
The hybrid density functional B3LYP [66] and open shell electronic configurations (for oxidized CuB) are used with a restricted open shell variant of DFT. Geometry optimizations are done with the LACVP+* basis set, while the single point energies are calculated with the LACV3P+* basis set. [62, 67] Electronic reorganization of the solute and the corresponding set of the ESP fitted charges for the QM system are obtained by the DFT-SCRF method, [68] which surrounds the complex by a continuum dielectric. For a detailed set up of the calculations, see our recent publications, [26, 27, 69] while a review of the combined DFT/electrostatic method can be found elsewhere. [70–73]
2.3. Continuum electrostatic calculations
The electrostatic calculations have been performed by using program MEAD, [63, 74] as described in ref. [26]. To correctly describe the effects of the protein charges on the pKas of selected groups, the protonation state of all other titratable groups of the enzyme, for a given redox state of metal centers, is required. To this end, we performed the standard continuum electrostatic calculations as described in ref. [24]. In different redox states the equilibrium proton distribution is slightly different due to the proton uptake from aqueous phase, and much more different from the standard protonation state.
The electrostatic calculations were performed on chains A and B of bovine heart CcO (PDB ID: 2OCC). [7] The two chains were embedded into a membrane and solvated in aqueous phase. The membrane domain is modeled as a low dielectric slab of 40Å that covers the central part of the enzyme (Fig. 1). The two discrete rotational isomers of the Glu side-chain have been used in the present study, see Figure 2. They correspond to the proton loading and proton releasing conformers of residue E242 (see ref. [34] for details).
2.4. Redox midpoint potential calculations
Electrostatic energies are evaluated by solving the Poisson-Boltzmann equation:
| (2) |
with the charge distribution ρ(r⃗) and dielectric constants of εprot.= 4 and εsolv.= 80 to obtain the electrostatic potential ϕ(r⃗). The second term accounts for the ionic strength effects due to mobile ions in solution but it can be safely dropped, since we previously found that mobile ions do not make any significant contribution to the calculated potentials in the interior of CcO. [24]
Gibbs free energy of a specific protonation/redox state of the enzyme n⃗, is given as:
| (3) |
where n⃗ is the N-dimensional protonation/redox state vector (N= P + R; total number of protonatable sites plus redox-active groups) with elements corresponding to the deprotonated (reduced) and protonated (oxidized) states respectively, and referring to the neutral reference state. is the unitless formal charge of the deprotonated (reduced) form of the group μ, i.e. −1 for acids and 0 for bases and redox centers. Esol is the external redox potential and F is the Faraday constant. The first sum runs over all P protonatable sites, the second sum runs over all R redox groups, and the third term is associated with the charge-charge interactions between all N titratable sites in the system. The intrinsic quantities, and , are defined as Emor p Ka of the site in protein when all other sites are in their neutral reference states. [74]
The redox midpoint potential of hemes and Cu centers are calculated at pH 7 (Em,7), as thermodynamic average over the protonation and redox states of all titratable sites of the enzyme, as explained in detail elsewhere. [74–78] This method aims to reproduce the shift in Em (pKa) associated with transferring a model compound representing the redox (protonatable) group with known solution Em (pKa) value from the aqueous phase to the protein environment. The following model compounds in aqueous solution with the corresponding Em, sol values are used in the present study: 1) for the a-type heme, bis-histidine heme a model compound is used with the experimental value of −120 meV; 2) for histidine–H2O heme a3, a value of −20 meV is assigned; 3) for the redox potential of histidine–OH− heme a3, a value of 100 meV is applied. [79, 80] Redox midpoint potentials of the CuA and CuB centers are evaluated from the shift due to interactions with other charged sites in the protein (redox or protonatable). Their Ems can be expressed as the intrinsic midpoint reduction potential plus a shift in redox potential associated with site-site interactions. [81] Thus, the shift in their Ems arises from the coupling with other redox-centers and changes in the protonation state of the protein, primarily the neighboring cluster of residues, when the Cu center goes from the oxidized to reduced state.
2.5. Solvation energy calculations
Once the charges of the QM system have been calculated, its geometry optimized, and the equilibrium charge distribution of the protein has been determined, the solvation energy of the QM system in the protein can be calculated. For solvation calculations, the MEAD suite [63] has been used. The solvation energy in the protein consists of two main contributions – the Born solvation energy (reaction field), and the energy of interaction with the protein charges (protein field).
To find these contributions, the Poisson Equation (or PBE) is solved numerically. The dielectric constants of 1, 4 and 80, were used for the QM system, the protein-membrane, and the solvent region, respectively. [82–84] Also, we examined the dependence of the results on the various values of the dielectric constant associated with water filled cavities inside of the protein (εcavity = 15, 20, 40, and 80).
In these calculations, the RESP fitted charges are used for the QM-system, while partial charges of the protein atoms are taken from our own parameterization [76] consistent with CHARMM22 parameter set, [85] modified so as to reflect the equilibrium protonation state of the enzyme for a given redox state of the metal centers. [24] The protein charges [76] and atomic radii [85] that we use in this application have been used successfully before to calculate the electrostatic energies, protonation probabilities of titratable groups and redox potentials of cofactors in different proteins. [26, 64, 76, 81, 86]
2.6. Coupling of the heme redox states
We performed the calculations to evaluate pKas and energetics of the proton transfer between the key protonatable sites – E242 and H291, for different populations of the reduced and oxidized hemes during the course of the transition. The initial state consists of the oxidized CuA, reduced Fea, and oxidized binuclear center (as in OH state of the catalytic cycle). [59] In the final state, the electron resides on Fea3, after it was transferred from the reduced Fea to the binuclear center. Simultaneously, we have monitored the pKa changes of the key residues for the different populations of the reduced and oxidized hemes during the corresponding electron transfer event.
In order to drive an electron from Fea to Fea3, along the reaction coordinate, we introduced a coupling parameter η which is numerically equivalent to parameter γ [15, 87] used to describe a fraction of an electron on Fea3 during the transition. As a matter of fact, η is the reaction coordinate of the coupled electron and proton transfer reaction, as discussed in the next section. η can take values between 0 and 1, and it is a coupling parameter chosen in such a way that η =0 and η =1 correspond to the initial (Sinitial = a− [a3/CuB] ) and final (S final = a [a3/CuB]−) redox state of the enzyme during the ET. Any (intermediate S(η) state for the given distribution of an electron can be defined as combination of the two redox states of the enzyme:
| (4) |
Not only the charges of the oxidized and reduced state of heme moieties are combined according to Eq. 4, but also all other protein charges producing the protein field in the two states (Sinitial, Sfinal) have to be combined and weighted by parameter η in the same manner. So obtained, the weighted S(η) protein charges were used later in solvation energy calculations.
Alternatively, one can calculate the protonation state of the enzyme for the different fractions of an electron distributed on the two hemes by solving the PBE each time and later use the obtained equilibrium charge distributions to evaluate the corresponding protein field terms in the solvation electrostatic calculations. We have checked both methods and confirmed that the results are consistent between the two approaches.
3. Results
We have adopted the following abbreviations to simplify the notation: Glu “down” protonated/deprotonated – gH/g−; Glu “up” protonated/deprotonated – GH/G−; His protonated/deprotonated – PH/P− (P stands for the PLS). Also, the initial (a− [a3/CuB]) and final (a [a3/CuB]−) redox state of the enzyme are associated with the fully reduced state of heme a or heme a3, respectively.
3.1. Pumped proton vs. ET between hemes
The change of pKas of E242 and H291during the electron redistribution between heme a and heme a3 is shown in Figure 5 (See also Fig. S1). The pKas are plotted against the coupling parameter (η). The η value represents the population of the reduced heme a3 (or BNC), while 1 − η gives the population of the reduced heme a. CuA center is always oxidized, while CuB center is in the oxidized state with a hydroxyl ligand, as presumably is the case in the O→E transition.
Figure 5.

The pKas of E242 and H291 for the different fraction of an electron on heme a and heme a3 during the electron transfer. The order of the Glu pKa-curves from the top: a) Glu “up” protonated- GH, b) Glu “down” protonated- gH, c) Glu “down” deprotonated- g−, d) Glu “up” deprotonated- G−. The His p Ka-curves from the bottom: 1) His with GH, 2) His with gH, 3) His with g−, 4) His with G−. The pairs of curves: 1 and a, 2 and b, 3 and c, and, 4 and d, correspond to each other.
Since we consider the two conformational and two protonation states of E242, total four curves are obtained for each group. The pKa value is calculated for equilibrated state of the protein that corresponds to a given state of the group, e.g. gH, g−, GH, G−; therefore the pKa of GH and G− may not be exactly the same. The difference in pKa of the two E242 conformers is in the range of 1–3 pK-units depending on the protonation state of H291 and the redox state of the metal centers (for more details see [34]). The difference in conformational energy is around 3 kcal/mol in the protonated state, and 6 kcal/mol in the deprotonated state (cf. ref. [52]). In contrast to H291, E242 is roughly equidistant from the two hemes, which makes its pKa(η) dependence more moderate.
The calculated pKas correspond to a given distribution η of an electron between the metal centers. It is seen that when the electron is on heme a, the pKa of Glu is higher than that of H291, and when electron is on heme a3, the opposite is true. Moreover, for the electron on heme a, H291 is deprotonated for all protonation states of Glu. Thus upon electron transfer from heme a to heme a3, the proton can be transferred from E242 to H291. This is a typical situation for a coupled electron and proton transfer reactions. [88] Namely, without an electron, the proton transfer is unfavorable, likewise without the proton, electron transfer is unfavorable; however, transition of both electron and proton is favorable in energy. The transition in this case occurs in the course of thermal fluctuations. The calculated data reflect the statistics in the thermodynamic ensemble, and indicate that as a function of the reaction coordinate η, the coupled reaction has a transition state – pKa of His becomes higher than that of Glu - when η is roughly between 0.4 and 0.6. The fractional value of η underscores the statistical and coupled nature of the reaction.
Figure 6 is obtained by taking the difference in pKas between the corresponding Glu- and His-curves. This difference is the free energy of proton transfer from Glu to His in pK-units, , at different values of the reaction coordinate η. The ET energy cost for overall coupled reaction is not included here; hence the shown curves do not represent the total free energy change, but rather only part related to the corresponding proton transfer. The four curves shown correspond to pKa differences: a – 1, b – 2, c – 3, and d – 4 of the curves in Fig. 5, in the order from top to bottom. The difference between the curves is due to two possible conformation states of Glu, and different protein states equilibrated at the initial state of the reaction‡. This difference between the curves represents the uncertainty in the conditions under which proton transfer occurs; however, for all cases shown, the qualitative picture is clear: when electron is still on heme a, there is no driving force for the proton transfer, while there is a significant driving force when electron is on heme a3. The actual transition can occur as a coupled reaction, at the intermediate value of the reaction coordinate η.
Figure 6.

The free energy of proton translocation from E242 to H291 in pK-units. The ΔGPT curves are related to the corresponding pKa curves shown in Fig. 5. The curves from top to bottom correspond to the following initial conditions of the protein: GH, gH, g−, and G−. See Fig. 5 caption, and text for further details.
In all cases, E242 always possesses pKa > 9, and presumably gets rapidly reprotonated through the D-channel. [35] Hence, one can also say that H291 gets effectively protonated by a translocation of the pumped proton from the N-side of the membrane.
The curve depicting a transition: a−/[a3/CuB]/gH/P− to a/[a3/CuB]−/gH/PH (second curve from top) is the one most likely related to the experimental measurements and the schematics of the model presented in Figure 4. According to the experimental data, [15] by the end of the 150-μs phase, 60% of an electron is transferred to the Fea3/CuB site, while 40% still remains on Fea and this electron transfer is coupled to the translocation of the pumped proton from the N-side of the membrane to the PLS.
For the transition – a−/[a3/CuB]/gH/P− to a/[a3/CuB]−/gH/PH, a transfer of the pumped proton from E242 to H291 is isoenergetic at λ=0.63 (crossing point of the two pKa− curves, Fig. 5), when 63% of an electron is transferred to the Fea3/CuB center, while 37% still remains on Fea. However, if energy of the ET is taken into account, the energy of 14 meV (0.23 pK-units) separates the two states. The corresponding Glu and His pKas are both 9.66. Setting the proton affinity of the N-side of the membrane as the reference state is a natural choice, since on the N-side is pH=7. It means that a drop in energy for a direct translocation of the pumped proton from the N-side to H291 site is 2.66 pK-units. Also, based on pKa of E242, the repotonation g− →gH is downhill in energy by 2.66 pK-units at 63% of the reduced population of Fea3.
For the experimentally observed distribution of 40% to 60% for the electron residing on heme a vs. heme a3/CuB complex, the calculated pKas of Glu and His are 9.72 and 9.38, respectively. At physiological pH both sites are protonated and there is a stabilization of the state with the protonated His of 2.38 pK-units relative to the N-side of the membrane. Also, the repotonation g− →gH is favorable by 2.72 pK-units at this point, what set up the state with g− to +0.34 pK-units relative to the reference energy level. The energy of the ET itself is −10 meV (−0.17 pK-units, at room temperature), as expected from the experiment. [15] The obtained energy levels are in good agreement with results of the recent study from the Wikström group. [52]
We examined the dependence of the results on the various values of the dielectric constant associated with water filled cavities inside of the protein (εcavity = 15, 20, 40, and 80). The obtained pKas of both, E242 and H291, are somewhat higher for the lower values of εcavity, but this does not considerably affect their difference, i.e. ΔGPT. Since, lower values of εcavity have a slightly stronger effect on the pKa of H291, the obtained ΔGPT curves cross the x-axis at somewhat lower values of η. For εcavity = 15, the corresponding shift is about 0.10–0.15 depending on a transition which curve describes (data are not shown).
3.2. Chemical proton vs. the internal ET in the BNC
Based on the obtained results, the proton translocation from E242 to H291 (PLS) upon the reduction of heme a is an energy requiring step, which could be facilitated by an initially generated small population of the reduced heme a3. The proton transfer to the PLS considerably increases the redox potential of heme a3 (Em ~0.35V), thereby stabilizing the electron at the BNC at the significant level, which in turn further increases the driving force for proton transfer to the PLS. In other words, the increased population of the protonated His (PH) gives rise to the reduced population of heme a3, and vice versa. Therefore, one can say that the electron and the proton drive each other at this step to the more stable (intermediate) state of the enzyme where they occupy heme a3 (BNC) and H291 (PLS), respectively. By the end of 150-μs phase, an electron is equilibrated between the two hemes, and the transfer is incomplete primarily because of the small difference in the redox potential between the two hemes of only 10 mV. But also, the redox potential of the CuB complex is lower than the Em of heme a3 by 110 mV, what prevents an electron flow to the oxidized CuB and consequently a shift of the equilibrium heme a⇄ heme a3 towards the binuclear center, see Figure 7. After a chemical proton enters the binuclear center for the chemical reduction of OH− ligand (oxygen intermediate), the redox potential of the CuB complex compared with the Em of heme a3 becomes yet by 135 mV more positive and the electron ultimately joins the proton at the CuB complex (Fig. 7).
Figure 7.
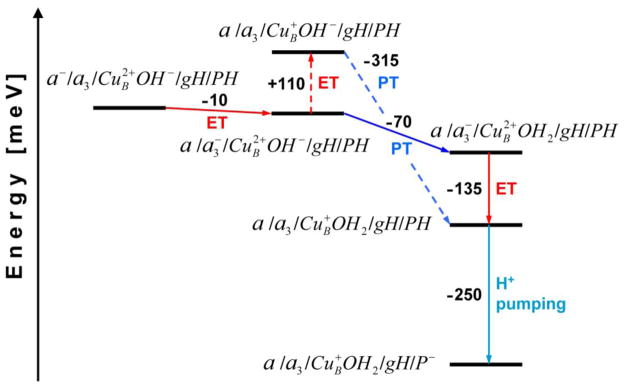
The energy diagram illustrating the transfer of a chemical proton and an electron transfer within the binuclear center at pH 7. The red, blue and turquoise lines refer to electron transfer (ET), proton transfer (PT), and proton pumping step, respectively. The dashed-line shows a thermodynamically less favorable path. The redox/protonation state changes of CcO are designated in the following order: heme a/heme a3/CuB–oxygen ligand/E242/H291.
The final step in the discussed pumping mechanism, after the reprotonation of the proton donor group, is a displacement of the preloaded proton from the H291 site to the P-side of the membrane due to electrostatic repulsion with the chemical proton in the BNC. The pumping step is exergonic by 250 meV.
We did not explore here all different possibilities, such as for example, the existence of the two proton-loading sites, whereas the PLS1 gets a proton when electron is distributed between the hemes, but a proton moves further to the PLS2 after the electron reaches the CuB complex. We also did not study in all detail the coupling between the translocation of a chemical proton and the electron transfer within the binuclear center by considering the different populations of the reduced Fea3 and CuB, as we did for the hemes. The reason is that there is no experimental data which indicate such equilibration of an electron in the binuclear center.
3.3. Redox potential of the metal centers
In a separate set of calculations, the redox potentials of the metal centers have been examined, as described in the Method section. The obtained results on the redox potential calculations of CuA, heme a, heme a3, and CuB center are presented for different steps of the reaction cycle in the O to E transition. The data shown in Table 1 represent the midpoint potential values at pH 7 (Em,7). The evaluated Ems are obtained for the protonated state of E242 site in down conformation, gH, unless it is otherwise stated.
Table 1.
The calculated redox midpoint potentials (Em,7 at pH 7; in mV) of the redox-active cofactors for the different steps in the reaction cycle during the O to E transition in bovine CcO
| Calculation/Experiment | Transfer step | Redox–active centers
|
||||||
|---|---|---|---|---|---|---|---|---|
| CuA | Fea | Fea3 | CuB | |||||
| phase 1: | [ET] | 258a | ⥂ | 270 | 150 | 0 | ||
| 140b | 17[b] | |||||||
| phase 2: | [PTpump/ET] | 238 | 340 | ⥂ | 350 | 240 | ||
| phase 3: | [PTchem/ET] | 267 | 380 | 355 | → | 490 | ||
| phase 4: | [pumping] | 267 | 278 | 120 | 240 | |||
|
| ||||||||
| Exp.1:c | pH 7.0 | 212–258 | 254–281 | n.a.g | 227–272 | |||
| pH 7.0 | 224 | 267 | n.a. | 230 | ||||
| Exp.2:d | pH 7.0 | 230–265 | 267–359 | 300–360 | >400 | |||
| Exp.3:e | pH 7.0 | 270 | 342 | 330 | n.a. | |||
| Exp.4:f | pH 6.5 | 250 | 391 (238) | 383 (405) | 412 | |||
| pH 8.0 | 250 | 359 (222) | 350 (372) | 366 | ||||
The Em of the CuA center is adopted from ref. [89], see text for details.
The calculated Ems of heme a and heme a3 in phase 1, if residue E242 is in its deprotonated state.
Steady-state redox potential ranges of the metal centers in bovine CcO, taken from ref. [89], see also refs therein.
The range of Em values obtained from the equilibrium experiments performed in the anaerobic conditions or in the presence of an inhibitor (CO, CN−, NO, etc.) in the BNC of bovine CcO, summarized from refs. [90, 93, 95, 97].
The Ems of the metal centers in bovine CcO obtained by fitting the experimental data to theoretical model. [55]
Experimental Ems of the redox centers, at pH 6.5 and 8.0, in CcO from P. denitrificans. For heme a and heme a3 are listed their high-asymptotic redox potentials. The numbers in brackets refer to the low-potential and high-potential heme transitions, see ref. [98] for details.
Data are not available.
The intrinsic midpoint potential (Em,intr) of the CuA center is modeled based on the experimental measurements provided in ref. [89], whereas the value of +258 mV is fitted to the Em of CuA center for the equilibrium state when an electron is shared with heme a. The calculated strength of coupling between the two redox sites is 40 mV and it shows the anti-cooperative effect, i.e. the interaction between CuA and Fea causes the Em potential for one of these sites to be decreased by approximately 40 mV upon reduction of the other, in agreement with experiments. [90]
Upon reduction of the CuA center, there is an uptake of 0.68 H+ by the neighboring cluster of residues from the P-side of the membrane (cf. ref. [91]). Once the electron is completely transferred to heme a, the cluster around the CuA site releases 0.39 H+ back to the P-side, while the cluster around Fea accepts 0.33 H+. Consistent with the experimental finding, [92] there is no net proton uptake from solution upon the ET between CuA and heme a. After the PT from E242 to H291 occurs and the electron gets equilibrated between the two hemes, both protonatable clusters release the remaining fraction of proton back to the P-side. Taking the effects of protonation changes due to transient proton uptake from the P-side along with the electrostatic coupling between the CuA and Fea centers, the following potential ranges are obtained for CuA, 227–282 mV and heme a, 240–294 mV, both in good agreement with the experiments [89, 90, 93] and the Em of bovine cytochrome c in aqueous solution of 240–264 mV. [94–96] Specifically for the ROOO redox state of the enzyme the calculated Em of CuA and Fea are 282 and 240 mV, respectively. Provided that there is no the PT reaction from E242 to the PLS in the OROO state, the corresponding Ems are 227 and 294 mV. If the two redox centers share an electron as in the 10-μs phase, their equilibrium Ems are 258 and 270 mV, respectively. If both metal centers, CuA and Fea, are oxidized the corresponding Ems are 267 and 278 mV.
Previous electrostatic calculations using the ZINDO charges gained Em = 280 mV for heme a in the redox state of the enzyme corresponding to the fully oxidized BNC. [34] In the present calculations, using the DFT charge set for the metal centers, the Em of 270 mV is obtained for Fea that is in the equilibrium with the CuA center, what is in good agreement with the experiments, [89, 90] indicating that the system has been modeled properly in respect to the protein and cofactor charges used in both calculations.
Upon the full oxidation of the CuA center with 40% of the reduced population remaining on Fea, as in phase 2, the decreases from 258 to 238 mV. After the reoxidation of heme a, once the electron is completely transferred to the BNC as in phase 3, the Em of CuA raises to value of 267 mV. It is noteworthy to point out that the obtained Em variation of CuA and Fea at pH 7 is related in part to their experimental pH-dependence of −20 mV/pH unit for both redox sites. [90, 91]
According to data presented in Table 1, the electron is in phase 1 equilibrated between CuA and Fea, since the Em of Fea3 and the CuB center are too low. We also estimated the Em of the two hemes for the g− state of E242, concluding that the ET from CuA to Fea is not feasible unless the Glu residue is in the gH state.
The transfer of the pumped proton from E242 to His291 increases the Em of all three metal centers in subunit A by 70, 200 and 240 mV. At the same time the Em of CuA site slightly decreases. However, the electron is by the end of phase 2 equilibrated between the two hemes, which possess the highest potential in the enzyme at this stage. The calculated Em values at this point probably correspond to the high-asymptotic redox potentials observed in anaerobic redox titrations in equilibrium with protons. [90, 97, 98]
The entrance of the chemical proton in the BNC produces an additional increase in the redox potential of the metal centers buried in low dielectric of the membrane region. The CuB complex has by far the highest midpoint potential in phase 3 and consequently the electron flows therein to join the chemical proton. Interestingly, the Em of Fea also elevates by an additional 40 mV comparing to its Em value in previous phase, due to the coupling primarily with the oxidized state of Fea3. Namely, going from phase 2 to phase 3, the reduced population of Fea3 vanishes from 60% to 0% (fully oxidized). On the other hand, the Em of Fea3 increases by only 5 mV due to the coupling with oxidized Fea and reduced CuB center. The oxidized state of Fea also causes an increase in redox potential of the CuA site. The computationally obtained redox potentials might here also correspond to static equilibrium measurements with the heme Em values related to the high-potential heme transition. [98]
In the last phase, the expulsion of the pumped proton from H291 site (PLS) causes a significant drop in the redox potential of the CuB center, but also considerably affects the potential of both hemes, as well. Once again, the Em changes are caused by the deprotonation of His site and a mutual redox coupling of metal centers. Comparing the calculated Ems at this stage with the experimental results, one can note their resemblance to the steady-state redox potentials. [89]
4. Discussion of the model
4.1. Electronic and protonic phases
The sequence of the electronic and protonic phases and the corresponding transition steps examined are schematically shown in Figure 3. Here we summarize the obtained results, and comment on the relevant previous results.
Phase 1 is associated with the transfer of the injected electron via the CuA center to heme a. As established experimentally, only a fraction of electron β=0.70 moves to heme a in this phase. [15] Previous studies also indicate that the coefficient β is 0.70–0.80 in bacterial [59] and β ~ 0.50 in bovine oxidase. [55]
Previously, we explored the coupling between the protonation state of E242, and the redox state of heme a. Based on the classical electrostatics, we have found that upon the reduction of heme a, the pKa value of E242 changes from 10.4 to 13.2, [34] making it sensitive to the redox changes of the system. The pKas of 9.6 and 11.8 are obtained by the present DFT/electrostatic calculations. Furthermore, we find that before the reprotonation of E242, the electron transfer from CuA to heme a is not feasible. According to the calculations, the reduction of heme a is only possible if the E242 site is already in the protonated state. The strength of the coupling between the two groups is about 130 meV. This gives rise in the Em of heme a of 130 mV at neutral pH, what is in good agreement with the experiments [99] (see also refs. therein).
At this stage, the calculated redox potential of heme a of 270 mV vs. the experimentally measured midpoint potential of CuA (250 mV in P. denitrificans [98]) corresponds to an electron distribution of 0.68 vs. 0.32, respectively. The Em of CuA center in bovine CcO has been determined in experiments as 258 mV [89] and 270mV. [55] This gives the electron distribution ratio of 0.61/0.39 and 0.50/0.50 respectively, both in good agreement with the experimental value β ~ 0.50 in bovine CcO. [55]
Phase 2 is associated with the transfer of the pumped proton from the N-side of the membrane to an unknown PLS group (most likely H291 or PRAa3), via the D-channel and E242, and simultaneous redistribution of the injected electron between the redox centers of the enzyme. By the end of the phase 2, a fraction 0.6 of an electron is localized on the Fea3/CuB binuclear center and fraction 0.4 remains on heme a. It is assumed that first a proton gets translocated from Glu to the PLS in a concerted manner that would lower the energy barrier required for this reaction, [100] followed by the reprotonation of E242 site and a subsequent “equilibrated” electron transfer between the two hemes. Further support for the described sequence of events comes from the interpretation of the experimental results which indicate that the rate-limiting step in the reduction of the binuclear complex is a proton uptake with the time constant of 150 μs. [15] The electron transfer by itself is very fast (τ= 1 ns), [52, 101] due to the electron tunneling between the two hemes across a short distance.
The results of present study support this sequence of steps and predict an electron distribution ratio of 0.37 vs. 0.63 between Fea and Fea3, respectively. Obviously, the midpoint potential of heme a is initially much higher than the Em of heme a3 and accordingly in phase 1 the electron is distributed between CuA and Fea (Table 1). However, after the PT from E242 to the PLS occurs, the order in the redox potential changes due to a simultaneous increase in the Em of both hemes. To be more specific, the Em of heme a changes from 270 to 340 mV, while the Em of heme a3 increases from 150 to 350 mV. Eventually, their midpoint potentials get separated by only 10–14 mV. Thus, the proton uptake by a protonatable group, presumably the proton-loading site, close to the Fea3/CuB center increases their redox potentials particularly stabilizing the reduced population of heme a3, which in turn would further increase and stabilize the population of the protonated His (PH). The shift in the midpoint potentials of two hemes is consistent with the position of the PLS, which is H291 in our calculations. The evaluated Ems are obtained by assuming the prior reprotonation of E242 site.
According to the present calculations, even a small fraction of an electron on heme a3 would be sufficient to assist a proton transfer from E242 to H291 site. The elevated occupancy of the PH state causes an increase in occupancy of the reduced heme a3, and vice versa. Therefore, the electron and proton drive each other at this step to the more stable state where they tend to occupy Fea3 and H291, respectively. However, the ET is still not complete.
Phase 3 is the transfer of the chemical proton and the remaining fraction of the electron to the binuclear center (BNC). The transport of the chemical proton takes place by passing through the K-channel (in O→E[59] and E→R transitions[56]) or along the D-channel and E242 (in all other transitions) providing a proton to oxygen intermediates. This proton transfer is accompanied by the electron transfer joining each other at the CuB center. Figure 7 and discussion in “Chemical proton vs. the internal ET in the BNC” refer to the obtained results of calculations at this stage. We have demonstrated that in this phase of the O→E transition, the electron wants to go all the way down to the CuB center, whereas the electron ultimately joins the chemical proton. In Figure 7, two options are displayed:
The chemical proton enters the BNC increasing its redox potential and consequently driving the reduction of the CuB center. The calculated Ems of CuA, Fea, Fea3 and CuB center at this stage are 267, 380, 355 and 490 mV, respectively.
Energetically less favorable pathway, where first the CuB center gets reduced, followed by the entrance of a chemical proton in the BNC. Reduction of the CuB center is here uphill in energy by 110 meV and this is most likely the reason why in phase 2 the electron gets stacked (equilibrated) between the hemes, instead of proceeding further to the CuB center, see Table 1.
Among the other options, there is also possibility (similar to phase 2) that the increased population of the protonated OH− ligand (i.e. H2O) gives rise to the reduced population of the CuB center, and vice versa. Therefore, the electron and proton may drive each other at this step to the more stable intermediate state of the enzyme, where they form the reduced CuB and H2O in the BNC. Moreover, it could also mean that the PT and the ET reactions may proceed simultaneously at this phase.
Phase 4 consists of the reprotonation of the proton donor site K319 (in the K-channel) or E242 (through the D-channel) depending on the transition of the catalytic cycle, and a subsequent ejection of the pumped proton from the preloaded pumping site (in phase 2) due to electrostatic repulsion with the chemical proton in the BNC. [24] It should be mentioned that the electrometric measurements [15] cannot distinguish which one takes place first, a release of the pumped proton or the reprotonation of the proton donor group. Our calculations support a scenario, where the reprotonation of E242 (or K319) occurs before the ejection of the pumped proton from the PLS, additionally increasing the electrostatic repulsion with the preloaded pumping site. [34] As a result, the reprotonation of the proton donor site may control proton release from the proton loading site. The final pumping step is then exergonic by 250 meV based on the present calculations.
4.2. Identity of PLS
One of the key features of the considered model is the existence of the proton-loading site (PLS) located above the heme porphyrins. The identity of PLS have been recently examined in the theoretical kinetic study [87] showing that only a few sites may play the role – propionates of heme a3, H291, Trp126, Trp236, R438 and R439, all at roughly the same dielectric depth. The identity of the PLS is still not known, but the main candidates are H291, [24, 25] PRAa3 [36, 52] and PRDa3. [17, 51, 102, 103] One model proposed earlier suggests that the pumped proton might be kept distributed between the propionates of heme a3, H291 and water molecule W1 (Fig. 2), including a possible formation of the Zundel-cation or a larger cluster that involves a water W1 and some of the neighboring water molecules in the hydrophilic cavity above the Fea3/CuB complex. [24]
4.3. The nature of the “kinetic gating”
The key question that still remains unanswered is why the conducting rate is greater for the translocation of a proton from E242 to the PLS, rather then to the BNC. In other words, why does the transfer of the pumped proton occur before the translocation of the chemical proton?
It was initially assumed, [24, 25] but recently experimentally supported [15] (and computationally examined in MD studies [35, 104]) that the water network to the PLS has greater proton-conducting rate than the one to the BNC, despite the fact that OH− ligand in the BNC possesses greater proton affinity than the PLS itself. [24, 27] Therefore, here it is not the thermodynamic but rather kinetic criterion that decides in which direction a proton should go. As a result, this leads to the creation of a meta-stable intermediate state with a preloaded proton at PLS, from which the pumped proton is later ejected to the P-side of the membrane.[25, 48]
Kinetic gating is presumably based on the intrinsic properties of the hydrophobic cavity including the internal water molecules, rotational isomerization of E242 side-chain, the redox properties of the metal centers and the redox-dependent affinity switches. The redox-dependent affinity switches are related to the protonatable groups which may considerably change their pKas and protonation state upon the redox changes of the metal centers. Upon the ET to heme a, the rotational isomerization of the E242 side-chain is greatly facilitated, but also the mobile properties of the water molecules within the hydrophobic cavity get changed. This includes the water dipole reorientation, [105] the insertion of more water molecules in the given PT pathway, [51] but also the ability of the proton-conducting network to the PLS to be formed first and stay stable longer during the course of the transition than the competing network to the BNC. [30] Moreover, structurally and energetically it is easier to form and to maintain longer almost a linear H-bonded chain, as the one to the PLS, than a bent water molecules chain, as required for supplying a chemical proton to the BNC. It seems that the distribution and the mobility of water molecules, both within the D-channel and in the hydrophobic catalytic cavity, play a significant role in determining the proton-conducting rates along the different pathways. [10, 102]
An additional but related question refers to the number of water molecules in the hydrophobic catalytic cavity. It seems that the cavity can accommodate up to 7 water molecules, but most likely in the steady-state of the working enzyme, there are only 4–5 water molecules. [28, 35, 37, 51, 106, 107] From MD studies, [51, 105–107] it appears that only two water molecules may be sufficient to provide the PT pathway to the PLS, while three of them are probably required to form a pathway to the BNC. Due to their increased mobility, some of water molecules can interexchange or jump between the two pathways, forming one or other water chain, i.e. to open or close pathways separately. Finally, the newly formed water molecule released from the BNC may also interfere with the water molecule networks in the hydrophobic cavity. This raises a question of the mechanistic role of the R438/PRDa3 salt bridge which opens to release the excessive water molecules [30] and closes to prevent a proton leakage, [34, 36, 48, 50–52] and its function in the redirection of pumped protons. [24, 29]
Thus, there are several possible mechanisms, which all likely to work together, that can justify the kinetic gating mechanism. [25] However, a robust explanation why one proton-conducting network is more efficient than the other, despite a fact that formation of a water molecule in the BNC is thermodynamically more favorable§ than protonation of the PLS, [24, 47, 49] is still missing.
The solution is most likely related to the internal water molecules inside of the D-channel and hydrophobic cavity. Any mutation [10, 32, 108] disturbing the water networks existing in wild-type CcO may lead to changes of the rates along the proton-conducting pathways below and above the E242 site. It appears that the fast water network to the PLS is more sensitive to these changes and requires a more precise timing than the slower one to the BNC.
We have discussed here only the kinetic gating situation, which is the most relevant for the present study. Furthermore, the proton paths, transition states and the corresponding energy barriers have not been studied. However, several models have been proposed lately, discussing other aspects, alternative mechanisms, different gating situations and kinetic energy barriers, e.g. refs [36, 51, 52, 109]. Also, a recent computational study [110] has cast some doubts on the E242 “valve model” and the water-wire gating via water reorientation.
5. Conclusion
According to new calculations presented here, the proton translocation from E242 to H291 upon the reduction of heme a is an endergonic process, which requires to be either directly coupled to an exergonic process (such as an ET) or to a series of the consecutive energetically favorable steps, assuming at the same time that the corresponding activation barrier of the transition state is not too high. The calculations show that the endergonicity of the pumped proton transport can be lessen by an initially generated small population of the reduced heme a3. Thus, even a small fraction of an electron on heme a3 would be sufficient in facilitating the PT and stabilizing the protonated state of the PLS. The increased occupancy of the His protonated state (PH) would in turn elevate the occupancy of the reduced heme a3, and vice versa. Therefore, the electron and proton might drive each other at this stage, as in a typical coupled electron and proton transfer reaction, [88] to the more stable intermediate state where they occupy Fea3 and H291, respectively. However, by the end of the 150-μs phase, the PH state exhibits a 100% occupancy, but the ET is not complete since 40% of the electron still remains on heme a. At this point, the transfer GH/P−→G−/PH is favorable by 2.6 pK-units (155 meV), what is sufficient to achieve a complete PT between the two protonatable sites. The difference in the midpoint potential between Fea and Fea3 of only 10 mV along with the low Em of the CuB center cause the equilibration of the injected electron between two hemes.
The transfer of the remaining 40% of electron from heme a to the binuclear center is accompanied by the chemical proton transfer to the BNC. During the redistribution of the injected electron in the O→E transition, the fully reduced state of heme a3 is perhaps just a short-living transient state formed only upon the entrance of a chemical proton in the BNC. However after the chemical proton transfer occurs, the electrostatic and thermodynamic properties of the active site significantly change, thereby affecting additionally the pKas of the key protonatable sites. Under these circumstances the redox potential of the CuB center becomes the highest in the system and the electron directly goes to the CuB center joining the chemical proton. Consequently, the pKa of H291 decreases due to electrostatic repulsion with the chemical proton, thereby giving rise to the pumping event.
Supplementary Material
Acknowledgments
We gratefully acknowledge helpful discussions with Dr. Jason Quenneville on electronic structure calculations. We are also grateful to Prof. Donald Bashford and Dr. Björn Rabenstein for their help and advices on electrostatic calculations. D.P. wishes to thank Prof. Marilyn Gunner with whom he worked in the past few years. This work has been supported in part by the NSF grant PHY 0646273, and NIH GM054052.
Abbreviations
- CcO
cytochrome c oxidase
- DFT
density functional theory
- QM
quantum-mechanical
- PBE
Poisson-Boltzmann Equation
- SCRF
self-consistent reaction field
- Em
midpoint potential
- ATP
adenosine-5′-triphosphate
- BNC
binuclear center
- PLS
proton-loading site
- PRAa3 and PRDa3
propionates A and D of heme a3
- ET
electron transfer
- PT
proton transfer
- N-side
negatively charged inner side of the membrane
- P-side
positively charged outer side of the membrane
- ROOO and OROO
the redox state of CuA, Fea, Fea3 and CuB center with R meaning reduced and O meaning oxidized. Bovine notation is used throughout the paper, unless it is otherwise stated
Footnotes
In the different transitions of the catalytic cycle, K319 (in K-channel) or E242 (in D-channel) play the role of a donor for the chemical (substrate) proton.
The effect of further possible variations in the initial and the final conditions of the protein is shown in Figure S2 in SM.
Although for the final conclusion a more elaborated study such as a large scale QM calculations, which would include all relevant residues around the binuclear center, is required.
Supplementary Material available:
SM contains details on the calculated protonation free energy of E242 and H291 for different reduction levels of the hemes. This material is available online free of charge at http://www.elsevier.com/bbabio/.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wikström M. Proton pump coupled to cytochrome c oxidase in mitochondria. Nature. 1977;266:271–273. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- 2.Babcock GT, Wikström M. Oxygen activation and the conservation of energy in cell respiration. Nature. 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 3.Ferguson-Miller S, Babcock GT. Heme/Copper Terminal Oxidases. Chem Rev. 1996;7:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 4.Ostermeier C, Iwata S, Michel H. Cytochrome c oxidase. Curr Op Struct Biol. 1996;6:460–466. doi: 10.1016/s0959-440x(96)80110-2. [DOI] [PubMed] [Google Scholar]
- 5.Zaslavsky D, Gennis RB. Proton pumping by cytochrome oxidase: progress, problems and postulates. Biochim Biophys Acta. 2000;1458:164–179. doi: 10.1016/s0005-2728(00)00066-9. [DOI] [PubMed] [Google Scholar]
- 6.Ostermeier C, Harrenga A, Ermler U, Michel H. Structure at 2.7 Å resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody Fv fragment. Proc Natl Acad Sci USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Redox-coupled structural changes in bovine heart cytochrome c oxidase. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 8.Svensson-Ek M, Abramson J, Larsson G, Tornroth S, Brzezinski P, Iwata S. The x-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter Sphaeroides. J Mol Biol. 2002;321:329–335. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 9.Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, Aoyama H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Yao M, Ishimura Y, Yoshikawa S. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc Natl Acad Sci USA. 2003;100:15304–15309. doi: 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dürr KL, Koepke J, Hellwig P, Müller H, Angerer H, Peng G, Olkhova E, Richter OMH, Ludwig B, Michel H. A D-Pathway Mutation Decouples the Paracoccus denitrificans Cytochrome c Oxidase by Altering the Side-Chain Orientation of a Distant Conserved Glutamate. Journal of Molecular Biology. 2008;384:865–877. doi: 10.1016/j.jmb.2008.09.074. [DOI] [PubMed] [Google Scholar]
- 11.Qin L, Liu J, Mills DA, Proshlyakov DA, Hiser C, Ferguson-Miller S. Redox-dependent conformational changes in cytochrome c oxidase suggest a gating mechanism for proton uptake. Biochemistry. 2009;48:5121–5130. doi: 10.1021/bi9001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konstantinov AA, Siletsky S, Mitchell D, Kaulen A, Gennis RB. The role of the two proton input channels in cytochrome c oxidase from Rhodobacter Sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc Natl Acad Sci USA. 1997;94:9085–9090. doi: 10.1073/pnas.94.17.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wikström M, Jasaitis A, Backgren C, Puustinen A, Verkhovsky MI. The role of the D- and K-pathways of proton transfer in the function of the heam-copper oxidases. Biochim Biophys Acta. 2000;1459:514–520. doi: 10.1016/s0005-2728(00)00191-2. [DOI] [PubMed] [Google Scholar]
- 14.Shimokata K, Katayama Y, Murayama H, Suematsu M, Tsukihara T, Muramoto K, Aoyama H, Yoshikawa S, Shimada H. The proton pumping pathway of bovine heart cytochrome c oxidase. Proc Natl Acad Sci USA. 2007;104:4200–4205. doi: 10.1073/pnas.0611627104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belevich I, Bloch DA, Belevich N, Wikström M, Verkhovsky MI. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc Natl Acad Sci USA. 2007;104:2685–2690. doi: 10.1073/pnas.0608794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han S, Takahashi S, Rousseau DL. Time dependence of the catalytic intermediates in cytochrome c oxidase. J Biol Chem. 2000;275:1910–1919. doi: 10.1074/jbc.275.3.1910. [DOI] [PubMed] [Google Scholar]
- 17.Wikström M. Cytochrome c oxidase: 25 years of the elusive proton pump. Biochim Biophys Acta. 2004;1655:241–247. doi: 10.1016/j.bbabio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 18.Gennis RB. Coupled proton and electron transfer reactions in cytochrome oxidase. Front Biosci. 2004;9:581–591. doi: 10.2741/1237. [DOI] [PubMed] [Google Scholar]
- 19.Brzezinski P. Redox-driven membrane-bound proton pumps. Trends Biochem Sci. 2004;29:380–387. doi: 10.1016/j.tibs.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 20.Belevich I, Verkhovsky MI, Wikström M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature. 2006;440:829–832. doi: 10.1038/nature04619. [DOI] [PubMed] [Google Scholar]
- 21.Hosler JP, Ferguson-Miller S, Mills DA. Energy Transduction: Proton Transfer Through the Respiratory Complexes. Annu Rev Biochem. 2006;75:165–187. doi: 10.1146/annurev.biochem.75.062003.101730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brzezinski P, Gennis RB. Cytochrome c oxidase: Exciting progress and remaining mysteries. J Bioenerg Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharpe MA, Ferguson-Miller S. A chemically explicit model for the mechanism of proton pumping in heme – copper oxidases. J Bioenerg Biomembr. 2008;40:541–549. doi: 10.1007/s10863-008-9182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Popovic DM, Stuchebrukhov AA. Electrostatic study of proton pumping mechanism of bovine heart cytochrome c oxidase. J Am Chem Soc. 2004;126:1858–1871. doi: 10.1021/ja038267w. [DOI] [PubMed] [Google Scholar]
- 25.Popovic DM, Stuchebrukhov AA. Proton pumping mechanism and catalytic cycle of cytochrome c oxidase: Coulomb pump model with kinetic gating. FEBS Letters. 2004;566:126–130. doi: 10.1016/j.febslet.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 26.Popovic DM, Quenneville J, Stuchebrukhov AA. DFT/electrostatic calculations of pKa values in cytochrome c oxidase. J Phys Chem B. 2005;109:3616–3626. doi: 10.1021/jp046535m. [DOI] [PubMed] [Google Scholar]
- 27.Quenneville J, Popovic DM, Stuchebrukhov AA. Combined DFT and electrostatics study of the proton pumping mechanism in cytochrome c oxidase. Biochim Biophys Acta. 2006;1757:1035–1046. doi: 10.1016/j.bbabio.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Zheng X, Medvedev DM, Swanson J, Stuchebrukhov AA. Computer simulation of water in cytochrome c oxidase. Biochim Biophys Acta. 2003;1557:99–107. doi: 10.1016/s0005-2728(03)00002-1. [DOI] [PubMed] [Google Scholar]
- 29.Lee HJ, Ojemyr L, Vakkasoglu A, Brzezinski P, Gennis RB. Properties of Arg481 Mutants of the aa(3)-Type Cytochrome c Oxidase from Rhodobacter sphaeroides Suggest That neither R481 nor the Nearby D-Propionate of Heme a(3) Is Likely To Be the Proton Loading Site of the Proton Pump. Biochemistry. 2009;48:7123–7131. doi: 10.1021/bi901015d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wikström M, Ribacka C, Molin M, Laakkonen L, Verkhovsky M, Puustinen A. Gating of proton and water transfer in the respiratory enzyme cytochrome c oxidase. Proc Natl Acad Sci USA. 2005;102:10478–10481. doi: 10.1073/pnas.0502873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hellwig P, Behr J, Ostermeier C, Richter OMH, Pfitzner U, Odenwald A, Ludwig B, Michel H, Mantele W. Involvement of glutamic acid 278 in the redox reaction of the cytochrome c oxidase from Paracoccus denitrificans investigated by FT-IR spectroscopy. Biochemistry. 1998;37:7390–7399. doi: 10.1021/bi9725576. [DOI] [PubMed] [Google Scholar]
- 32.Pawate AS, Morgan J, Namslauer A, Mills D, Brzezinski P, Ferguson-Miller S, Gennis RB. A mutation in subunit I of cytochrome oxidase from Rhodobacter sphaeroides results in an increase in steady-state activity but completely eliminates proton pumping. Biochemistry. 2002;41:13417–13423. doi: 10.1021/bi026582+. [DOI] [PubMed] [Google Scholar]
- 33.Mills DA, Tan Z, Ferguson-Miller S, Hosler J. A role for subunit III in proton uptake into the D pathway and a possible proton exit pathway in Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry. 2003;42:7410–7417. doi: 10.1021/bi0341307. [DOI] [PubMed] [Google Scholar]
- 34.Popovic DM, Stuchebrukhov AA. Two conformational states of Glu242 and pKas in bovine cytochrome c oxidase. Photochem Photobiol Sci. 2006;5:611–620. doi: 10.1039/b600096g. [DOI] [PubMed] [Google Scholar]
- 35.Kaila VRI, Verkhovsky MI, Hummer G, Wikström M. Glutamic acid 242 is a valve in the proton pump of cytochrome c oxidase. Proc Natl Acad Sci USA. 2008;105:6255–6259. doi: 10.1073/pnas.0800770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siegbahn PEM, Blomberg MR. Energy diagrams and mechanism for proton pumping in cytochrome c oxidase. Biochim Biophys Acta. 2007;1767:1143–1156. doi: 10.1016/j.bbabio.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh N, Prat-Resina X, Gunner MR, Cui Q. Microscopic pK(a) Analysis of Glu286 in Cytochrome c Oxidase (Rhodobacter sphaeroides): Toward a Calibrated Molecular Model. Biochemistry. 2009;48:2468–2485. doi: 10.1021/bi8021284. [DOI] [PubMed] [Google Scholar]
- 38.Xu J, Voth G. Redox-coupled proton pumping in cytochrome c oxidase: Further insights from computer simulation. Biochim Biophys Acta. 2008;1777:196–201. doi: 10.1016/j.bbabio.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olsson MHM, Warshel A. Monte Carlo simulations of proton pumps: On the working principles of the biological valve that controls proton pumping in cytochrome c oxidase. Proc Natl Acad Sci USA. 2006;103:6500–6505. doi: 10.1073/pnas.0510860103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore DB, Martinez TJ. Ab Initio Study of Coupled Electron Transfer/Proton Transfer in Cytochrome c Oxidase. J Phys Chem A. 2000;104:2367–2374. [Google Scholar]
- 41.Blomberg MRA, Siegbahn PEM, Babcock GT, Wikström M. Modeling cytochrome oxidase: A quantum chemical study of the O-O bond cleavage mechanism. J Am Chem Soc. 2000;122:12848–12858. [Google Scholar]
- 42.Siegbahn PEM, Blomberg MRA, Blomberg ML. Theoretical Study of the Energetics of Proton Pumping and Oxygen Reduction in Cytochrome Oxidase. J Phys Chem B. 2003;107:10946–10955. [Google Scholar]
- 43.Makhov DV, Popovic DM, Stuchebrukhov AA. Improved Density Functional Theory/Electrostatic Calculation of the His291 Protonation State in Cytochrome c Oxidase: Self-Consistent Charges for Solvation Energy Calculation. J Phys Chem B. 2006;110:12162–12166. doi: 10.1021/jp0608630. [DOI] [PubMed] [Google Scholar]
- 44.Fee JA, Case DA, Noodleman L. Toward a Chemical Mechanism of Proton Pumping by the B-Type Cytochrome c Oxidases: Application of Density Functional Theory to Cytochrome ba(3) of Thermus thermophilus. Journal of the American Chemical Society. 2008;130:15002–15021. doi: 10.1021/ja803112w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blomberg MR, Siegbahn PEM. A quantum chemical study of the mechanism for proton-coupled electron transfer leading to proton pumping in cytochrome c oxidase. Molecular Physics. 2010;108:2733–2743. [Google Scholar]
- 46.Ali-Torres J, Rodriguez-Santiago L, Sodupe M. Computational calculations of pKa values of imidazole in Cu(II) complexes of biological relevance. Phys Chem Chem Phys. 2011;13:7852–7861. doi: 10.1039/c0cp02319a. [DOI] [PubMed] [Google Scholar]
- 47.Kannt A, Lancaster RD, Michel H. The coupling of electron transfer and proton translocation: electrostatic calculations on Parracocus denitrificants cytochrome c oxidase. Biophys J. 1998;74:708–721. doi: 10.1016/S0006-3495(98)73996-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Popovic DM, Stuchebrukhov AA. Proton exit channels in bovine cytochrome c oxidase. J Phys Chem B. 2005;109:1999–2006. doi: 10.1021/jp0464371. [DOI] [PubMed] [Google Scholar]
- 49.Song Y, Michonova-Alexova E, Gunner MR. Calculated proton uptake on anaerobic reduction of cytochrome c oxidase: Is the reaction electroneutral? Biochemistry. 2006;45:7959–7975. doi: 10.1021/bi052183d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaila VRI, Verkhovsky MI, Hummer G, Wikström M, Gutman M. Prevention of leak in the proton pump of cytochrome c oxidase. Biochim Biophys Acta. 2008;1777:890–892. doi: 10.1016/j.bbabio.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 51.Pisliakov AV, Sharma PK, Chu ZT, Haranczyk M, Warshel A. Electrostatic basis for the unidirectionality of the primary proton transfer in cytochrome c oxidase. Proc Natl Acad Sci USA. 2008;105:7726–7731. doi: 10.1073/pnas.0800580105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaila VRI, Verkhovsky MI, Hummer G, Wikström M. Mechanism and energetics by which glutamic acid 242 prevents leaks in cytochrome c oxidase. Biochim Biophys Acta. 2009;1787:1205–1214. doi: 10.1016/j.bbabio.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 53.Brändén G, Brändén M, Schmidt B, Mills DA, Ferguson-Miller S, Brzezinski P. The protonation state of a heme propionate controls electron transfer in cytochrome c oxidase. Biochemistry. 2005;44:10466–10474. doi: 10.1021/bi0502745. [DOI] [PubMed] [Google Scholar]
- 54.Zaslavsky D, Kaulen AD, Smirnova IA, Vygodina T, Konstantinov AA. Flash-induced membrane potential generation by cytochrome c oxidase. FEBS Lett. 1993;336:389–393. doi: 10.1016/0014-5793(93)80843-j. [DOI] [PubMed] [Google Scholar]
- 55.Jasaitis A, Verkhovsky MI, Morgan JE, Verkhovskaya ML, Wikström M. Assignment and charge translocation stoichiometries of the electrogenic phases in the reaction of cytochrome c with dioxygen. Biochemistry. 1999;38:2697–2706. doi: 10.1021/bi982275l. [DOI] [PubMed] [Google Scholar]
- 56.Ruitenberg M, Kannt A, Bamberg E, Ludwig B, Michel H, Fendler K. Single-electron reduction of the oxidized state is coupled to proton uptake via the K-pathway in paracoccus denitrificans cytochrome c oxidase. Proc Natl Acad Sci USA. 2000;97:4632–4636. doi: 10.1073/pnas.080079097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruitenberg M, Kannt A, Bamberg E, Fendler K, Michel H. Reduction of cytochrome c oxidase by a second electron leads to proton translocation. Nature. 2002;417:99–102. doi: 10.1038/417099a. [DOI] [PubMed] [Google Scholar]
- 58.Siletsky SA, Pawate AS, Weiss K, Gennis RB, Konstantinov AA. Transmembrane charge separation during the ferryl-oxo -> oxidized transition in a nonpumping mutant of cytochrome c oxidase. J Biol Chem. 2004;279:52558–52565. doi: 10.1074/jbc.M407549200. [DOI] [PubMed] [Google Scholar]
- 59.Bloch D, Belevich I, Jasaitis A, Ribacka C, Puustinen A, Verkhovsky MI, Wikström M. The catalytic cycle of cytochrome c oxidase is not the sum of its two halves. Proc Natl Acad Sci USA. 2004;101:529–533. doi: 10.1073/pnas.0306036101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rich PR. Towards an understanding of the chemistry of oxygen reduction and proton translocation in the iron – copper respiratory oxidases. Aust J Plant Physiol. 1995;22:479–486. [Google Scholar]
- 61.Popovic DM, Leontyev IV, Beech DG, Stuchebrukhov AA. Similarity of cytochrome c oxidases in different organisms. Proteins: Structure, Function and Bioinformatics. 2010;78:2691–2698. doi: 10.1002/prot.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jaguar 5.5. Schrödinger, L. L. C; Portland, OR: 1991–2003. [Google Scholar]
- 63.Bashford D. An object-oriented programming suite for electrostatic effects in biological molecules. In: Ishikawa Y, Oldehoeft RR, Reynders JVW, Tholburn M, editors. Scientific Computing in Object-Oriented Parallel Environments. Vol. 1343. Springer; Berlin: 1997. pp. 233–240. [Google Scholar]
- 64.Rabenstein B, Knapp EW. Calculated pH-dependent population of CO-myoglobin coformers. Biophys J. 2001;80:1141–1150. doi: 10.1016/S0006-3495(01)76091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kohn K, Sham LJ. Self-consistent equations including exchange and correlation effects. Physical Review. 1965;140:A1133–1136. [Google Scholar]
- 66.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. Journal of Chemical Physics. 1993;98:5648–5652. [Google Scholar]
- 67.Hay PJ, Wadt WR. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J Chem Phys. 1985;82:299–310. [Google Scholar]
- 68.Tannor DJ, Marten B, Marphy R, Friesner RA, Sitkoff D, Nicholls A, Ringnalda M, Goddard WA, Honig B. Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J Am Chem Soc. 1994;116:11875–11880. [Google Scholar]
- 69.Quenneville J, Popovic DM, Stuchebrukhov AA. Redox-dependent pKa of CuB histidine ligand in cytochrome c oxidase. J Phys Chem B. 2004;108:18383–18389. [Google Scholar]
- 70.Chen JL, Noodleman L, Case DA, Bashford D. Incorporating solvation effects into density functional electronic structure calculations. J Phys Chem. 1994;98:11059–11068. [Google Scholar]
- 71.Mouesca JM, Chen JL, Noodleman L, Bashford D, Case DA. Density functional/Poisson-Boltzmann calculations of redox potentials for iron-sulfur clusters. J Am Chem Soc. 1994;116:11898–11914. [Google Scholar]
- 72.Li J, Nelson MR, Peng CY, Bashford D, Noodleman L. Incorporating protein environments in density functional theory: A self-consistent reaction field calculation of redox potentials of [2Fe2S] clusters in ferredoxin and phthalate dioxygenase reductase. J Phys Chem A. 1998;102:6311–6324. [Google Scholar]
- 73.Li J, Fisher CL, Konecny R, Bashford D, Noodleman L. Density functional and electrostatic calculations of manganese superoxide dismutase active site complexes in protein environments. Inorg Chem. 1999;38:929–939. doi: 10.1021/ic980731o. [DOI] [PubMed] [Google Scholar]
- 74.Bashford D, Gerwert K. Electrostatic calculations of the pKa values of ionizable groups in bacteriorhodopsin. J Mol Biol. 1992;224:473–486. doi: 10.1016/0022-2836(92)91009-e. [DOI] [PubMed] [Google Scholar]
- 75.Antosiewicz J, Briggs JM, Elcock AH, Gilson MK, McCammon JA. Computing ionization states of proteins with a detailed charge model. J Comput Chem. 1996;17:1633–1644. [Google Scholar]
- 76.Popovic DM, Zaric SD, Rabenstein B, Knapp EW. Artificial cytochrome b: Computer modeling and evaluation of redox potentials. J Am Chem Soc. 2001;123:6040–6053. doi: 10.1021/ja003878z. [DOI] [PubMed] [Google Scholar]
- 77.Gunner MR, Alexov E. A pragmatic approach to structure based calculation of coupled proton and electron transfer in proteins. Biochim Biophys Acta. 2000;1458:63–87. doi: 10.1016/s0005-2728(00)00060-8. [DOI] [PubMed] [Google Scholar]
- 78.Song Y, Mao J, Gunner MR. Electrostatic Environment of Hemes in Proteins: pKas of Hydroxyl Ligands. Biochemistry. 2006;45:7949–7958. doi: 10.1021/bi052182l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gibney BR, Isogai Y, Rabanal F, Reddy KS, Grosset AM, Moser CC, Dutton PL. Self-assembly of heme a and heme b in a designed four-helix bundle: Implications for a cytochrome c oxidase maquette. Biochemistry. 2000;39:11041–11049. doi: 10.1021/bi000925r. [DOI] [PubMed] [Google Scholar]
- 80.Reedy CJ, Gibney BR. Heme protein assemblies. Chem Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 81.Popovic DM, Zmiric A, Zaric SD, Knapp EW. Energetics of radical transfer in DNA photolyase. J Am Chem Soc. 2002;124:3775–3782. doi: 10.1021/ja016249d. [DOI] [PubMed] [Google Scholar]
- 82.Sharp K, Honig B. Electrostatic interactions in macromolecules: Theory and applications. Ann Rev Biophys Biophys Chem. 1990;19:301–332. doi: 10.1146/annurev.bb.19.060190.001505. [DOI] [PubMed] [Google Scholar]
- 83.Simonson T, Brooks CL. Charge separation and the dielectric constant of proteins: Insights from molecular dynamics. J Am Chem Soc. 1996;118:8452–8458. [Google Scholar]
- 84.Shurki A, Warshel A. Structure/function correlations of proteins using MM, QM/MM, and related approaches: Methods, concepts, pitfalls, and current progress. Adv Protein Chem. 2003;66:249–259. doi: 10.1016/s0065-3233(03)66007-9. [DOI] [PubMed] [Google Scholar]
- 85.MacKerell AD, Jr, Bashford D, Bellot M, Dunbrack J, RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodholm B, Reiher I, Roux WEB, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 86.Vagedes P, Rabenstein B, Åqvist J, Marelius J, Knapp EW. The deacylation step of acetylholinesterase: Computer simulation studies. J Am Chem Soc. 2000;122:12254–12262. [Google Scholar]
- 87.Sugitani R, Medvedev ES, Stuchebrukhov AA. Theoretical and computational analysis of the membrane potential generated by cytochrome c oxidase upon single electron injection into the enzyme. Biochim Biophys Acta. 2008;1777:1129–1139. doi: 10.1016/j.bbabio.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hammes-Schiffer S, Stuchebrukhov AA. Theory of coupled electron and proton transfer reactions. Chem Rev. 2010;110:6939–6960. doi: 10.1021/cr1001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mason MG, Nicholls P, Cooper CE. The steady-state mechanism of cytochrome c oxidase: redox interactions between metal centres. Biochem J. 2009;422:237–246. doi: 10.1042/BJ20082220. [DOI] [PubMed] [Google Scholar]
- 90.Blair DF, Ellis J, WR, Wang H, Gray HB, Chan SI. Spectroelectrochemical study of cytochrome c oxidase: pH and temperature dependences of the cytochrome potentials. Characterization of site-site interactions. J Biol Chem. 1986;261:11524–11537. [PubMed] [Google Scholar]
- 91.Capitanio N, Capitanio G, Minuto M, Nitto ED, Palese LL, Nicholls P, Papa S. Coupling of Electron Transfer with Proton Transfer at Heme a and CuA (Redox Bohr Effects) in Cytochrome c Oxidase. Studies with the Carbon Monoxide Inhibited Enzyme. Biochemistry. 2000;39:6373–6379. doi: 10.1021/bi0003137. [DOI] [PubMed] [Google Scholar]
- 92.Verkhovsky MI, Jasaitis A, Verkhovskaya ML, Morgan JE, Wikström M. Proton translocation by cytochrome c oxidase. Nature. 1999;400:480–483. doi: 10.1038/22813. [DOI] [PubMed] [Google Scholar]
- 93.Rich PR, West IC, Mitchell P. The location of CuA in mammalian cytochrome c oxidase. FEBS Lett. 1988;233:25–30. doi: 10.1016/0014-5793(88)81349-8. [DOI] [PubMed] [Google Scholar]
- 94.Flatmark T. Multiple Molecular Forms of Bovine Heart Cytochrome c: A Comparative Study of Their Physicochemical Pproperties and Their Reactions in Biological Systems. J Biol Chem. 1967;242:2454–2459. [PubMed] [Google Scholar]
- 95.Wilson DF, Erecinska M, Dutton PL. Thermodynamic relationships in mitochondrial oxidative phosphorylation. Annu Rev Biophys Bioeng. 1974;3:203–230. doi: 10.1146/annurev.bb.03.060174.001223. [DOI] [PubMed] [Google Scholar]
- 96.Battistuzzi G, Borsari M, Cowan JA, Ranieri A, Sola M. Control of Cytochrome c Redox Potential: Axial Ligation and Protein Environment Effects. J Am Chem Soc. 2002;124:5315–5324. doi: 10.1021/ja017479v. [DOI] [PubMed] [Google Scholar]
- 97.Moody AJ, Rich PR. The effect of pH on redox titrations of haem a in cyanide-liganded cytochrome-c oxidase: experimental and modelling studies. Biochim Biophys Acta. 1990;1015:205–215. doi: 10.1016/0005-2728(90)90022-v. [DOI] [PubMed] [Google Scholar]
- 98.Gorbikova EA, Vuorilehto K, Wikström M, Verkhovsky MI. Redox Titration of All Electron Carriers of Cytochrome c Oxidase by Fourier Transform Infrared Spectroscopy. Biochemistry. 2006;45:5641–5649. doi: 10.1021/bi060257v. [DOI] [PubMed] [Google Scholar]
- 99.Verkhovsky MI, Morgan JE, Wikström M. Control of electron delivery to the oxygen reduction site of cytochrome c oxidase: A role for protons. Biochemistry. 1995;34:7483–7491. doi: 10.1021/bi00022a023. [DOI] [PubMed] [Google Scholar]
- 100.Olsson MHM, Sharma PK, Warshel A. Simulating redox coupled proton transfer in cytochrome c oxidase: Looking for the proton bottleneck. FEBS Letters. 2005;579:2026–2034. doi: 10.1016/j.febslet.2005.02.051. [DOI] [PubMed] [Google Scholar]
- 101.Pilet E, Jasaitis A, Liebl U, Vos MH. Electron transfer between hemes in mammalian cytochrome c oxidase. Proc Natl Acad Sci USA. 2004;101:16198–16203. doi: 10.1073/pnas.0405032101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wikström M, Verkhovsky MI. Mechanism and energetics of proton translocation by the respiratory heme-copper oxidases. Biochim Biophys Acta. 2007;1767:1200–1214. doi: 10.1016/j.bbabio.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 103.Fadda E, Yu CH, Pomès R. Electrostatic control of proton pumping in cytochrome c oxidase. Biochim Biophys Acta. 2008;1777:277–284. doi: 10.1016/j.bbabio.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 104.Olkhova E, Hutter MC, Lill MA, Helms V, Michel H. Dynamic Water Networks in Cytochrome c Oxidase from Paracoccus denitrificans Investigated by Molecular Dynamics Simulations. Biophys J. 2004;86:1873–1889. doi: 10.1016/S0006-3495(04)74254-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wikström M, Verkhovsky MI, Hummer G. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim Biophys Acta. 2003;1604:61–65. doi: 10.1016/s0005-2728(03)00041-0. [DOI] [PubMed] [Google Scholar]
- 106.Tashiro M, Stuchebrukhov AA. Thermodynamic properties of internal water molecules in the hydrophobic cavity around the catalytic center of cytochrome c oxidase. J Phys Chem B. 2005;109:1015–1022. doi: 10.1021/jp0462456. [DOI] [PubMed] [Google Scholar]
- 107.Sugitani R, Stuchebrukhov AA. Molecular dynamics simulation of water in cytochrome c oxidase reveals two water exit pathways and the mechanism of its transport. Biochim Biophys Acta. 2009;1787:1140–1150. doi: 10.1016/j.bbabio.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vakkasoglu AS, Morgan JE, Han D, Pawate AS, Gennis RB. Mutations which decouple the proton pump of the cytochrome c oxidase from Rhodobacter sphaeroides perturb the environment of glutamate 286. FEBS Lett. 2006;580:4613–4617. doi: 10.1016/j.febslet.2006.07.036. [DOI] [PubMed] [Google Scholar]
- 109.Brändén G, Pawate AS, Gennis RB, Brzezinski P. Controlled uncoupling and recoupling of proton pumping in cytochrome c oxidase. Proc Natl Acad Sci USA. 2006;103:317–322. doi: 10.1073/pnas.0507734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yang S, Cui Q. Glu-286 Rotation and Water Wire Reorientation Are Unlikely the Gating Elements for Proton Pumping in Cytochrome c Oxidase. Biophys J. 2011;101:61–69. doi: 10.1016/j.bpj.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.