

Abstract

Multifunctional metal chelators that can modulate the amyloid β (Aβ) peptide aggregation and its interaction with metal ions such as copper and zinc hold considerable promise as therapeutic agents for Alzheimer’s disease (AD). However, specific rather than systemic metal chelation by these compounds is needed in order to limit any side effects. Reported herein are two novel small bifunctional chelators, 2-[2-hydroxy-4-(diethylamino)phenyl]benzothiazole (L1) and 2-(2-hydroxy-3-methoxyphenyl)benzothiazole (L2), in which the metal-binding donor atoms are integrated within a molecular framework derived from the amyloid-binding fluorescent dye thioflavin T (ThT). The metal-binding properties of L1 and L2 were probed by pH spectrophotometric titrations to determine their pKa values and the corresponding metal complex stability constants, and the isolated metal complexes were structurally characterized. The amyloid-fibril-binding properties of L1 and L2 were investigated by fluorescence titrations and ThT competition assays. Interestingly, L1 and L2 do not lead to the formation of neurotoxic Aβ42 oligomers in the presence or absence of metal ions, as observed by native gel electrophoresis, Western blotting, and transmission electron microscopy. In addition, L1 and L2 were able to reduce the cell toxicity of preformed Aβ42 oligomers and of the copper-stabilized Aβ42 oligomers. Given their ability to reduce the toxicity of soluble Aβ42 and Cu-Aβ42 species, L1 and L2 are promising lead compounds for the development of chemical agents that can control the neurotoxicity of soluble Aβ42 species in AD.
Short abstract
Two small bifunctional chelators, L1 and L2, were developed to specifically target the interactions of Cu2+ and Zn2+ ions with the Aβ42 peptide and to control the formation of neurotoxic Aβ42 aggregates. Unprecedentedly, these compounds promote the fibrillization of Cu2+-stabilized soluble Aβ42 oligomers, and most importantly they drastically reduce the neurotoxicity of soluble Aβ42 species, in both the presence or absence Cu2+ ions
Introduction
More than 5 million people in the United States and 24 million worldwide are affected by Alzheimer’s disease (AD), a disease characterized by memory loss and neurodegeneration.1−3 Insoluble aggregates of the 42 and 40 amino acid long amyloid β peptides (Aβ42 and Aβ40, respectively) are believed to be intimately involved in the onset of AD. However, while amyloid plaques, the ultimate product of Aβ aggregation, have been proposed to promote neurodegeneration and dementia,4 recent studies have shown that the soluble Aβ oligomers are more neurotoxic both in vitro and in vivo.5−8 In addition, while Aβ40 is present in larger amounts in amyloid plaques, the longer and more hydrophobic Aβ42 isoform has a higher tendency to generate neurotoxic oligomeric species.9,10
Interestingly, unusually high concentrations of copper, iron, and zinc have been found within the amyloid deposits in AD-affected brains.11 These metal ions have been shown to affect Aβ aggregation, as well as lead to the formation of reactive oxygen species (ROS).12−16 Although many studies have investigated the involvement of metal ions in Aβ aggregation and plaque formation, the molecular mechanisms of metal–Aβ species interactions, especially for the more neurotoxic Aβ42, are still not completely understood. Exley et al. have used fluorescence assays and transmission electron microscopy (TEM) to show that both sub- and superstoichiometric concentrations of Cu2+ prevent aggregation of Aβ42 into thioflavin T (ThT)-positive β-sheet-rich amyloid fibrils.17−19 We have also recently reported that Cu2+ ions slow down fibrillization of the Aβ42 peptide and stabilize the soluble Aβ42 species, as observed by native gel electrophoresis/Western blotting, TEM, cell viability studies,20,21 and pulsed hydrogen/deuterium exchange mass spectrometry studies.22 In this regard, metal chelators have been shown to reduce metal-mediated Aβ aggregation, ROS formation, and neurotoxicity in vitro.23−26 Recent efforts to control abnormal Aβ–metal interactions have focused on small molecules, bifunctional chelators (BFCs), that have binding affinity for both metal ions and Aβ aggregates.20,25,27−30 However, the majority of these BFCs have been studied for their effect on aggregation of the Aβ40 peptide,25,31 and fewer studies have focused on their interaction with the more neurotoxic Aβ42 peptide.20,21 Compounds that inhibit metal-mediated Aβ40 aggregation or promote disaggregation of amyloid fibrils were shown to lead to increased cell viability.25,31,32 However, this approach may not be optimal for the Aβ42 peptide, given the increased toxicity observed for soluble Aβ42 oligomers.20,21 Our current efforts are aimed at the development of BFCs that control the metal–Aβ42 neurotoxicity in vivo and take into consideration the formation of neurotoxic soluble Aβ42 oligomers and their proposed role in AD neuropathogenesis. Reported herein are two new BFCs, 2-[2-hydroxy-4-(diethylamino)phenyl]benzothiazole (L1) and 2-(2-hydroxy-3-methoxyhenyl)benzothiazole (L2), that were designed following a previously reported strategy of incorporating metal-binding atoms into the structural framework of an Aβ-interacting compound.23,33,34 L1 and L2 have a modified 2-(2-hydroxyphenyl)benzothiazole (HPB) molecular framework reminiscent of the amyloid-binding fluorescent dye ThT (Scheme 1), which was shown to be promising in the design of new Aβ-binding compounds.33 These BFCs fulfill the druglike criteria for further biological in vivo assays. The bifunctional character of the two compounds was confirmed by metal-chelating and Aβ-binding studies. The Cu2+ complexes of L1 and L2 were isolated and characterized spectroscopically and by X-ray structural determination. The amyloid-binding properties of L1 and L2 were explored by fluorescent titrations and by ThT competition assays. While L1 exhibits an acceptable binding affinity for Aβ fibrils, with the higher affinity binding site corresponding to a Ki of 260 nM, L2 binds very strongly to Aβ fibrils with a Ki of 15 nM. In addition, L1 and L2 moderately inhibit Aβ42 aggregation in the absence of metal ions, yet they promote aggregation in the presence of metal ions, as observed by native gel/Western blotting and TEM. In addition, L1 and L2 were able to reduce the cell toxicity of preformed Aβ42 oligomers as well as that of soluble copper-stabilized Aβ42 oligomers, which were shown recently to be neurotoxic.20,21 These promising properties of L1 and L2 make them suitable candidates for further in vivo investigation.
Scheme 1. Molecular Structures of ThT, Pittsburgh Compound B (PiB), Clioquinol (CQ), and the BFCs L1 and L2 Described in This Work.
Experimental Section
General Methods
All reagents were purchased from commercial sources and used as received unless stated otherwise. Solvents were purified prior to use by passing through a column of activated alumina using an MBraun Solvent Purification System. All solutions and buffers were prepared using metal-free Millipore water that was treated with Chelex overnight and filtered through a 0.22 μm nylon filter. 1H (300.121 MHz) and 13C (75 MHz) NMR spectra were recorded on a Varian Mercury-300 spectrometer. Chemical shifts are reported in ppm and referenced to residual solvent resonance peaks. UV–vis spectra were recorded on a Varian Cary 50 Bio spectrophotometer and are reported as λmax, nm (ε, M–1 cm–1). Electrospray ionization mass spectrometry (ESI-MS) experiments were performed using a Bruker M-axis QTOF mass spectrometer with an ESI source. ESI-MS was provided by the Washington University Mass Spectrometry NIH Resource (Grant P41RR0954), and elemental analyses were carried out at Intertek Chemical and Pharmaceuticals testing and analysis services. TEM analysis was performed at the Nano Research Facility at Washington University.
X-ray Crystallography
Suitable crystals of appropriate dimensions were mounted in a Bruker Kappa Apex II CCD X-ray diffractometer equipped with an Oxford Cryostream LT device and a fine-focus Mo Kα radiation X-ray source (λ = 0.71073 Å). Preliminary unit cell constants were determined with a set of 36 narrow frame scans. Typical data sets consist of combinations of ϖ and ϕ scan frames with a typical scan width of 0.5° and a counting time of 15–30 s/frame at a crystal-to-detector distance of ∼4.0 cm. The collected frames were integrated using an orientation matrix determined from the narrow frame scans. Apex II and SAINT software packages35 were used for data collection and data integration. Final cell constants were determined by the global refinement of reflections from the complete data set. Data were corrected for systematic errors using SADABS.35 Structure solutions and refinement were carried out using the SHELXTL-PLUS software package.36 The structures were refined with full-matrix least-squares refinement by minimizing ∑w(Fo2 – Fc2)2. All non-hydrogen atoms were refined anisotropically to convergence. All hydrogen atoms were added in the calculated position and refined using appropriate riding models (AFIX m3). Additional crystallographic details can be found in the Supporting Information (SI).
Acidity and Stability Constant Determination
UV–vis pH titrations were employed for determination of the acidity constants of L1 and L2 and their stability constants with Cu2+ and Zn2+. For the acidity constants, solutions of BFCs (25 μM for L1 and 50 μM for L2, 0.1 M NaCl, pH 3) were titrated with small aliquots of 0.1 M NaOH at room temperature. At least 30 UV–vis spectra were collected in the pH 3–11 range. Because of the limited solubility of L1 and L2 in water, methanol (MeOH) stock solutions (10 mM) were used and titrations were performed in a MeOH–water mixture in which MeOH did not exceed 20% (v/v). Similarly, the stability constants for the L1–copper system were determined by titrating a solution of L1 (25 μM) and Cu(ClO4)2·6H2O (12.5 μM) with small aliquots of 0.1 M NaOH at room temperature. At least 30 UV–vis spectra were collected in the pH 3–11 range. The acidity and stability constants were calculated using the HypSpec computer program (Protonic Software GmbH, U.K.).37 Speciation plots of the compounds and their metal complexes were calculated using the program HySS2009 (Protonic Software GmbH, U.K.).38
Aβ Peptide Experiments
Aβ monomeric films were prepared by dissolving commercial Aβ42 (or Aβ40 for Aβ-fibril-binding studies) peptide (Keck Biotechnology Resource Laboratory, Yale University) in HFIP (1 mM) and incubating for 1 h at room temperature.39 This solution was then aliquoted out, and HFIP was allowed to evaporate overnight. The aliquots were vacuum-centrifuged, and the resulting monomeric films were stored at −80 °C. Aβ fibrils were generated by dissolving monomeric Aβ films in dimethyl sulfoxide (DMSO), diluting into the appropriate buffer, and incubating for 24 h at 37 °C with continuous agitation (the final DMSO concentration was <2%). For metal-containing fibrils, the corresponding metal ions were added before initiation of the fibrillization conditions. For inhibition studies, BFCs (50 μM, DMSO stock solutions) were added to Aβ solutions (25 μM) in the absence or presence of metal salts (CuCl2 or ZnCl2, 25 μM) and incubated for 24 h at 37 °C with constant agitation. For disaggregation studies, the preformed Aβ fibrils in the absence or presence of metal ions were treated with BFCs and further incubated for 24 h at 37 °C with constant agitation. For the preparation of soluble Aβ42 oligomers, a literature protocol was followed.7,39 A monomeric film of Aβ42 was dissolved in anhydrous DMSO, followed by the addition of DMEM-F12 media [1:1 (v/v), without phenol red; Invitrogen]. The solution (50–100 μM) was incubated at 4 °C for 24 h and then centrifuged at 10000g for 10 min. The supernatant was used as a solution of soluble Aβ42 oligomers.
Fluorescence Measurements
All fluorescence measurements were performed using a SpectraMax M2e plate reader (Molecular Devices). For ThT fluorescence studies, samples were diluted to a final concentration of 2.5 μM Aβ in phosphate-buffered saline (PBS) containing 10 μM ThT and the fluorescence was measured at 485 nm (λex = 435 nm). For Aβ-fibril-binding studies, a 5 μM Aβ fibril solution was titrated with small amounts of compound and their fluorescence intensity measured (λex/λem = 330/450 nm). For ThT competition assays, a 5 μM Aβ fibril solution with 2 μM ThT was titrated with small amounts of compound and the ThT fluorescence measured (λex/λem = 435/485 nm). For calculation of Ki values, a Kd value of 1.17 μM was used for the binding of ThT to Aβ40 fibrils.20,21
TEM
Glow-discharged grids (Formar/Carbon 300 mesh, Electron Microscopy Sciences) were treated with Aβ samples (25 μM, 5 μL) for 2–3 min at room temperature. Excess solution was removed using filter paper, and grids were rinsed twice with water (5 μL). Grids were stained with uranyl acetate (1% w/v, water, 5 μL) for 1 min, blotted with filter paper, and dried for 15 min at room temperature. Images were captured using a FEI G2 Spirit Twin microscope (60–80 kV, 6500–97000× magnification).
Native Gel Electrophoresis and Western Blotting
All gels, buffers, membranes, and other reagents were purchased from Invitrogen and used as directed except where otherwise noted. Samples were separated on 10–20% gradient Tris-tricine minigels. The gel was transferred to a nitrocellulose membrane in an ice bath, and the protocol was followed as suggested except that the membrane was blocked overnight at 4 °C. After blocking, the membrane was incubated in a solution (1:2000 dilutions) of 6E10 anti-Aβ primary antibody (Covance) for 3 h. Invitrogen’s Western Breeze Chemiluminescent kit was used to visualize the bands. An alkaline/phosphatase antimouse secondary antibody was used, and the protein bands were imaged using a Fujifilm LAS-1000CH luminescent image analyzer.
Cytotoxicity Studies (Alamar Blue Assay)
Mouse neuroblastoma Neuro2A (N2A) cell lines were purchased from the American Type Culture Collection (ATCC). Cells were grown in DMEM/10% FBS, which is the regular growth media for N2A cells. N2A cells were plated to each well of a 96-well plate (2.5 × 104/well) with DMEM/10% FBS. The media was changed to DMEM/N2 media 24 h later. After 1 h, the reagents (20 μM Aβ42 species, compounds, and metals) were added. Because of the poor solubility of the compounds in water or media, the final amount of DMSO used was 1% (v/v). After an additional incubation of 40 h, the Alamar Blue solution was added in each well and the cells were incubated for 90 min at 37 °C. The absorbance was measured at 570 nm (control optical density = 600 nm). For these toxicity studies, three types of Aβ42 species were tested: freshly made monomeric Aβ42 (MAβ42), Aβ42 oligomers (OAβ42), and Aβ42 fibrils (FAβ42). These Aβ42 species were prepared as described above.
Synthesis of L1
A mixture of 2-aminothiophenol (2.5 g, 20 mmol) and (diethylamino)salicylaldehyde (3.86 g, 20 mmol) in DMSO (20 mL) was heated with stirring at 150 °C for 6 h under dinitrogen. Then it was cooled to 0 °C for 8 h, and the orange solid crystallized out, was collected by filtration, and was washed with hexane (2.97 g, yield 50%). 1H NMR (CDCl3): δ 7.85 (d, 1H, ArH), 7.81 (d, 1H, ArH), 7.46 (d, 1H, ArH), 7.43 (d, 1H, ArH), 7.30 (d, 1H, phenol H), 6.30 (d, 1H, phenol H), 6.27 (s, 1H, phenol H), 3.41 (q, 2H, CH2CH3), 1.22 (t, 3H, CH2CH3). 13C NMR (CDCl3): δ 169.78, 159.98, 152.43, 151.52, 132.08, 130.04, 126.42, 124.35, 121.40, 121.18, 105.96, 104.34, 98.053, 44.72, 12.86. UV–vis [λmax, nm, (ε, M–1 cm–1)]: in MeCN, 262 (10300), 380 (43000); in PBS, 272 (9500), 375 (18000), 405 (19500). HRMS. Calcd for [M + H]+: m/z 299.1218. Found: m/z 299.1225.
Synthesis of L2
A mixture of o-vanilin (2.0 g, 0.0131 mol) and 2-aminothiophenol (1.64 g, 0.0131 mol) in ethanol (15 mL) was refluxed with stirring for 24 h. Then it was cooled to room temperature and poured in water. The sticky yellow solid was crystallized from MeOH to obtain yellow crystals (1.15 g, yield 37%). 1H NMR (CDCl3): δ 8.02 (d, 1H, ArH), 7.91 (d, 1H, ArH), 7.52 (t, 1H, ArH), 7.42 (t, 1H, ArH), 7.33 (d, 1H, ArH), 7.01 (d, 1H, ArH), 6.99 (t, 1H, ArH), 3.97 (s, 3H, OCH3). 13C NMR (CDCl3): δ 169.28, 151.50, 148.93, 148.19, 148.17, 132.59, 126.70, 125.56, 122.16, 121.45, 119.98, 119.14, 116.71, 114.06, 56.22. UV–vis [λmax, nm, (ε, M–1 cm–1)]: in MeCN, 307 (br, 10200), 340 (sh, 5500); in PBS, 305 (3500), 340 (sh, 1800), 390 (800). HRMS. Calcd for [M + H]+: m/z 258.0588. Found: m/z 258.0584.
Synthesis of [(L1)2CuII]·0.5H2O (1)
A solution of CuCl2·2H2O (0.011 g, 0.084 mmol) in MeCN (2 mL) was added to the stirring mixture of L1 (0.050 g, 0.167 mmol) and Et3N (0.036 g, 0.334 mmol) in MeCN (5 mL). The brown precipitate immediately obtained was filtered, washed with ether, and dried under vacuum (0.040 g, yield 54%). UV–vis [λmax, nm, (ε, M–1cm–1)]: in MeCN, 273 (sh, 18000), 391 (56800), 404 (sh, 50500), 540 (900). HRMS. Calcd for [M + H]+: m/z 658.1497. Found: m/z 658.1492. Anal. Calcd for C34H34N4O2S2Cu·0.5H2O: C, 61.52; H, 5.25; N, 8.44. Found: C, 61.63; H, 6.11; N, 8.44.
Synthesis of [(L1)ZnII(Cl2)](Et3NH) (2)
A solution of ZnCl2 (0.011 g, 0.084 mmol) in MeCN (2 mL) was added to the stirring mixture of L4 (0.050 g, 0.167 mmol) and Et3N (0.018 g, 0.167 mmol) in MeCN (5 mL). The red solution was kept for slow evaporation, and a few single crystals were obtained within 2 days (0.052 g, yield 58%). UV–vis [λmax, nm, (ε, M–1cm–1)]: in MeCN, 265 (19900), 391 (51000). ESI-MS. Calcd for [(L1)ZnCl + H]+: m/z 397.01. Found: m/z 397.01. Satisfactory elemental analysis data could not be obtained because of the presence of a small amount of free ligand.
Synthesis of [(L2)2CuII]·0.5H2O (3)
A solution of CuCl2·2H2O (0.013 g, 0.097 mmol) in MeCN (2 mL) was added to the stirring mixture of L2 (0.050 g, 0.194 mmol) and Et3N (0.078 g, 0.776 mmol) in MeOH (10 mL). The brown precipitate immediately obtained was filtered, washed with ether, and dried under vacuum (0.051 g, yield 91%). UV–vis [λmax, nm, (ε, M–1cm–1)]: in MeCN, 398 (26200), 515 (sh, 800), 700 (350). ESI-MS. Calcd for [M + Na]+: m/z 598.0. Found: m/z 598.1. Anal. Calcd for C28H20N2O4S2Cu·0.5H2O: C, 57.47; H, 3.62; N, 4.79. Found: C, 57.34; H, 3.62; N, 4.83.
Results and Discussion
Design, Synthesis, and Characterization of L1 and L2
Herein we employed the strategy of developing metal BFCs with amyloid-binding properties as potential therapeutic agents for AD. The fluorescent dye ThT is well-known for its amyloid-binding properties because of the 2-phenylbenzothiazole aromatic framework. Previously, an iodinated HPB and a few related compounds were shown to have potential as metal-chelating agents by Rodríguez-Rodríguez et al.33 However, in that report, only the solution coordination properties of those compounds were investigated in detail, while the Aβ-binding and antiaggregation properties were only qualitatively evaluated through epifluorecense microscopy and turbidity assays. In contrast, in addition to the solution coordination chemistry, we have also determined the solid-state structure of the copper and zinc complexes of our compounds, and we have also quantitatively determined their Aβ-binding affinities through fluorescence titrations. In addition, we have used both TEM and gel electrophoresis/Western blotting to provide a complete picture of the effect of our compounds on Aβ aggregation and formation of various sized Aβ aggregates, in both the absence and presence of metal ions. Most importantly, we have correlated the in vitro studies with cellular toxicity studies, thus making this work a comprehensive study of these compounds in the context of developing new compounds that can control the toxicity and aggregation of various Aβ species. We have also recently reported 2-phenylbenzothiazole/vanillin derivatives as BFCs that showed a strong affinity for Aβ fibrils.20 Herein we designed two new molecules, L1 and L2, that contain a 2-phenylbenzothiazole moiety for Aβ recognition and oxygen- and nitrogen-atom donors needed for metal chelation. Synthesis of L1 and L2 was accomplished upon oxidative cyclization of the corresponding arylaldehyde and 2-aminothiophenol (Scheme S1 in the SI).40 These compounds exhibit UV absorption in MeCN at 260 and 380 nm for L1 and at 305 and 345 nm for L2 (Figures S1 and S2 in the SI).40 While both L1 and L2 are 2-phenylbenzothiazole derivatives, L1 exhibits low-intensity emission at 450 nm and a shoulder at 525 nm, yet L2 shows a more intense emission at 525 nm in PBS (Figures S3 and S4 in the SI).40 The parent molecule HPB has emission properties similar to those of L2 (Figure S4 in the SI), which suggests that having a diethylamino group in L1 strongly affects its emission properties.
Another important aspect of the design of new drug molecules for central nervous system diseases is their ability to cross the blood brain barrier (BBB).13,26,41 Both L1 and L2 are small in size, and they also satisfy Lipinski’s rules for crossing the BBB (Table 1). The ability of a compound to cross the BBB can be evaluated by determining the blood–brain partitioning parameter log BB that is determined by taking into account the molecular weight of the molecule, charge, lipophilicity, and its hydrogen-bonding capability. Molecules having log BB values higher than −0.3 are generally expected to cross the BBB.33 The calculated log BB values for L1 and L2 are +0.33 and +0.087, respectively, suggesting that these BFCs are expected to cross the BBB.
Table 1. Lipinski Parameters for L1 and L2.
| property | L1 | L2 | Lipinski’s rulea |
|---|---|---|---|
| MW | 298.41 | 257.31 | ≤500 |
| c log P | 4.853 | 3.834 | ≤5 |
| HBD | 1 | 1 | ≤5 |
| HBA | 3 | 3 | ≤10 |
| PSA | 36.36 | 42.35 | ≤70 Å2 |
| log BB | 0.330 | 0.087 | >−0.3 |
MW = molecular weight; c log P = calculated octanol–water partition coefficient; HBD = hydrogen-bonding donor atoms; HBA = hydrogen-bonding acceptor atoms; PSA = polar surface area; log BB = −0.0148PSA + 0.152 c log P + 0.130 (Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J. Adv. Drug Delivery Rev.1997, 23, 3–25; Clark, D. E.; Pickett, S. D. Drug Discovery Today2000, 5, 49–58).
Acidity Constants of L1 and L2
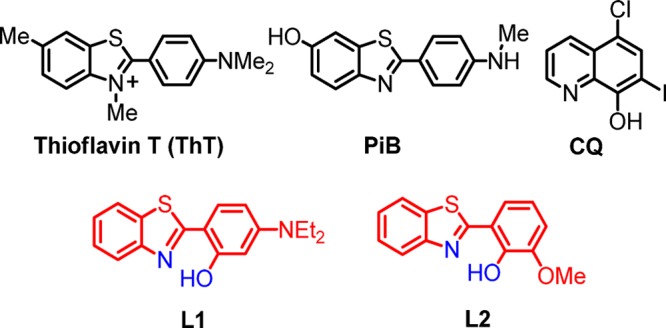
Because both L1 and L2 contain functional groups that can undergo protonation, the acidity constants (pKa) were determined by UV–vis spectrophotometric titrations. For both L1 and L2, UV–vis titrations from pH 3.0 to 11.0 reveal several changes in the spectra (Figures 1 and 2). For L1, the best fit to the data was obtained with pKa values of 3.556(2), 8.427(3), and 10.108(2) (Table 2). The first and second pKa values can be assigned to deprotonation of the nitrogen atoms of the benzothiazole and diethylamino groups, while the largest pKa value is likely due to phenol deprotonation.25,42 For L2, the best fit to the data was obtained with two pKa values of 3.154(2) and 9.360(1) (Table 2). These pKa values can be assigned to deprotonation of the nitrogen atoms of the benzothiazole and phenol groups, respectively.
Figure 1.
Variable-pH UV spectra of L1 (25 μM, 25 °C, I = 0.1 M NaCl) and the species distribution plot.
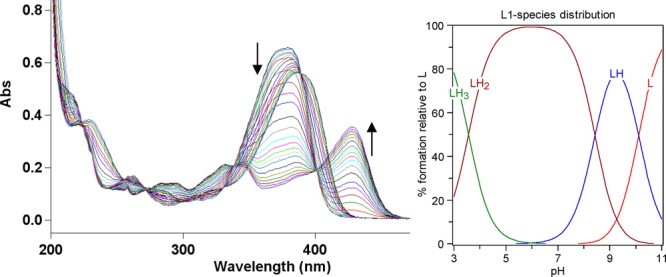
Figure 2.
Variable-pH UV spectra of L2 (50 μM, 25 °C, I = 0.1 M NaCl) and the species distribution plot.
Table 2. Acidity Constants (pKa Values) of L1 and L2 Determined by Spectrophotometric Titrations (Errors Are for the Last Digit)a.
| reaction | L1 | L2 |
|---|---|---|
| [H3L]2+ = [H2L]+ + H+ (pKa1) | 3.556(2) | |
| [H2L]+ = [HL] + H+ (pKa2) | 8.427(3) | 3.154(2) |
| [HL] = [L]− + H+ (pKa3) | 10.108(2) | 9.360(1) |
[HL] represents the neutral form of L1 and L2.
Characterization of Metal Complexes
The BFCs L1 and L2 were explored for their ability to chelate metal ions. The stoichiometry of L1–Cu2+ and L1–Zn2+ complexes in solution was determined by Job’s plot analysis.43 For L1–Cu2+, a break in the plot at 0.4 mole fraction of Cu2+ suggests the formation of both 1:1 and 2:1 L1–Cu2+ complexes in solution (Figure S4 in the SI).40 For L1–Zn2+, the break occurs at ∼0.3 mole fraction of Zn2+, suggesting the formation of a 2:1 L1–Zn2+ complex (Figure S5 in the SI).40 By contrast to L1, no Job’s plot analysis could be performed for L2–Cu2+ because of the limited solubility of this complex, while no significant spectral change was observed for the L2–Zn2+ system. The Cu2+ complexes of L1 or L2 were synthesized by reacting CuCl2·2H2O with the corresponding ligand in MeCN in the presence of an equivalent amount of Et3N. The 2:1 complex formation was confirmed by the ESI-MS signal at m/z 658.1 corresponding to [M + H]+, which was further supported by X-ray structural determination (see below). The UV–vis spectrum of the L1–Cu2+ complex 1 reveals a d–d transition at ∼700 nm and a more intense band at 540 nm, while a phenolate-to-Cu2+ charge-transfer band is observed at ∼400 nm (Figure S6 in the SI).40 Similarly, the UV–vis spectrum of the L2–Cu2+ complex 3 shows characteristic d–d transition bands at 700 and 515 nm as well as a phenolate-to-Cu charge-transfer band at ∼400 nm (Figure S7 in the SI).40 Although a single crystal suitable for X-ray crystallography could be obtained from the reaction mixture of L1 and Zn2+, the isolation of the zinc complexes of L1 or L2 did not lead to pure complexes to allow for their complete characterization.
Spectrophotometric titrations were also performed to determine the stability constants and solution speciation of Cu2+ with L1. The pKa values of the ligands were included in the calculations,42 and the best fit to the data shows that L1 binds Cu2+ with a stability constant of 11.99(2) for the M + L = ML equilibrium and a value of 20.99(2) for the ML + L = ML2 equilibrium (L represents the deprotonated form of L1). The speciation diagram for the Cu–L1 system is shown in Figure 3. In addition, the concentration of free Cu2+ (pCu = −log [Cufree]) can be determined at specific pH values and total ion concentrations, with pCu values of 6.7 and 8.2 being determined for the Cu–L1 system at pH 6.6 and 7.4, respectively. These pCu values are lower compared to those for the analogous HPB compound reported previously,33 likely because of the strong electron-withdrawing effect of the diethylamine group upon protonation. Unfortunately, the spectrophotometric titration of L2 in the presence of Cu2+ revealed few spectral changes at micromolar concentrations, while the use of higher concentrations led to precipitation of the L2–Cu2+ complex. However, it is expected that L2 exhibits a higher binding affinity to Cu2+ than L1 because of the electron-donating o-methoxy group present in L2. In addition, almost no changes were observed in the spectra of both L1 and L2 upon the addition of Zn2+; therefore, the corresponding stability constants could not be determined. Attempts were then made to determine the apparent stability constants for the Zn–L1 and Zn–L2 complexes by using competition assays with a zinc chelator, zincon. Zincon was used previously by Faller et al. in Zn–Aβ interaction studies and exhibits a Kd ≈ 10 μM for zinc.44 In our case, the addition of zincon to solutions of Zn–L1 and Zn–L2 complexes results in quantitative formation of the Zn–zincon complex. In addition, L1 and L2 do not compete significantly with zincon for zinc binding, even when present in large excess versus zincon, suggesting that they exhibit affinities for zinc that are at least 10 times lower than that of zincon.
Figure 3.

Variable-pH UV spectra of the L1–Cu2+ system ([L1] = 25 μM; [Cu2+] = 12.5 μM, 25 °C, I = 0.1 M NaCl) and the species distribution plot.
X-ray Structure of Metal Complexes
To further characterize the metal-binding properties of compounds L1 and L2, single-crystal X-ray structures were determined for complexes 1–3 (Figure 4 and Tables S1–S3 in the SI).40 Complex 1 affords single crystals by Et2O diffusion into a CH2Cl2 solution of 1 that crystallized out in the C2/c space group. An oxygen atom and a nitrogen atom from two ligand molecules coordinate to Cu2+. The Cu–O1 and Cu–N1 bond lengths are 1.980(3) and 1.876(2) Å, respectively, and the trans angles O1–Cu1–O1#1 and N1#1–Cu1–N1 are 153.3(2)° and 154.2(2)°, respectively, with these parameters suggesting a distorted square-planar geometry for Cu2+. For complex 2, the Zn2+ ion exhibits a distorted tetrahedral geometry and is coordinated to the benzothiazole nitrogen and phenol oxygen atoms of L1 and two chloride ions. The copper complex 3 afforded single crystals by Et2O diffusion into a CH2Cl2 solution and has structural properties similar to those of 1, yet it crystallized out in the P2/n space group. As shown for 1, in 3 an oxygen atom and a nitrogen atom from two ligand molecules coordinate to Cu2+. The Cu–O1 and Cu–N1 bond lengths are 1.873(4) and 1.965(4) Å, respectively, and the trans angles O1–Cu1–O1#1 and N1#1–Cu1–N1 are 145.1(2)° and 146.2(2)°, respectively, reflecting a distorted square-planar geometry for Cu2+.
Figure 4.

ORTEP representations (50% probability ellipsoids) of complexes 1–3. All hydrogen atoms, counterions, and solvent molecules are omitted for clarity. Selected bond distances: 1, Cu1–N1 1.980(3), Cu1–O1 1.876(2); 2, Zn–N1 2.005(1), Zn–O1 1.956(1), Zn–Cl(1) 2.233(1), Zn–Cl(2) 2.222(1); 3, Cu1–N1/N2 1.965(4), Cu1–O1/O3 1.873(4).
Interaction of L1 and L2 with Aβ Species
Both compounds L1 and L2 contain a 2-phenylbenzothiazole fragment reminiscent of the fluorescent dye ThT used to detect the β-sheet structure of fibrillar Aβ aggregates.45,46 In this context, the affinity of L1 and L2 toward Aβ fibrils was investigated by fluorescence assays. These studies were performed with Aβ40 fibrils, which form more homogeneous fibrillar structures without any nonfibrillar aggregates.45,47 Titration of L1 with Aβ fibrils was performed, and a saturation behavior is observed. The data were fitted with a one-site binding model to give a Kd of 4.70 ± 0.30 μM (Figure 5a), which suggests a moderate affinity of L1 for the Aβ fibrils. ThT fluorescence competition assays were also performed by the addition of L1 to a solution of Aβ fibrils in the presence of ThT. L1 shows a decrease in ThT fluorescence upon the addition of nanomolar amounts. Competitive-binding curve fitting yields a Ki value of 260 ± 40 nM (Figure 5b). It should be noted that the Ki value for L1 is smaller than the Kd value, which is most likely due to the fact that ThT binds with different affinities to more than one site to the Aβ fibrils.47 As such, we propose that L1 replaces ThT only from the higher affinity binding site. By constrast, L2 did not show any significant increase in the fluorescence intensity in the presence of Aβ fibrils yet replaces ThT from the Aβ fibrils and exhibits a Ki value of 15 ± 10 nM, indicating a very strong binding affinity to Aβ fibrils (Figure 5c).
Figure 5.

(a) Fluorescence titration assay of L1 with Aβ fibrils ([Aβ] = 5 μM; λex/λem = 330/450 nm). ThT fluorescence competition assays of Aβ fibrils with (b) L1 and (c) L2 ([Aβ] = 2 μM; [ThT] = 1 μM; λex/λem = 435/485 nm).
Effect of L1 and L2 on Aβ42 Aggregation
To investigate the effect of L1 and L2 on Aβ aggregation, we performed both inhibition and disaggregation studies (Scheme 3). Importantly, the Aβ42 peptide was employed in these studies because it was shown to form neurotoxic soluble Aβ oligomers.5,8,48 For inhibition experiments, freshly prepared Aβ42 fibrils were treated with metal ions, BFCs, or both, and the reactions was monitored by ThT fluorescence, native gel electrophoresis/Western blotting analysis, and TEM.
Scheme 3. Schematic Description of the Performed Inhibition and Disaggregation Experiments.

In inhibition experiments, a reduced ThT fluorescence was observed for the Cu2+- and Zn2+-containing fibrils, as reported previously (Figure 6).21,49,50 In the absence of metal ions, the presence of L1 leads to a low ThT fluorescence intensity, while L2 has a negligible effect on Aβ42 fibrillization, suggesting that L1 can inhibit fibrillization more efficiently than L2. In the presence of Cu2+ and either L1 or L2, a very low ThT fluorescence intensity is observed, which could be due to either limited Aβ42 fibril formation or fluorescence quenching by the paramagnetic Cu2+ ions. By comparison, the presence of Zn2+ and either L1 or L2 shows almost no effect on the ThT fluorescence intensity (Figure 6). Because the ThT fluorescence assays do not show a complete picture of the effect of BFCs on Aβ42 aggregation, native gel electrophoresis/Western blotting analysis and TEM were employed in order to characterize in more detail the various Aβ42 species formed during these aggregation studies. As such, the Aβ42 peptide forms well-defined Aβ42 fibrils, as confirmed by TEM (Figure 7, panel 1), while Aβ42 aggregation in the presence of Cu2+ shows the formation of a dramatically reduced number of Aβ42 fibrils (Figure 7, panel 2), thus supporting the low ThT fluorescence intensity observed above. By comparison, aggregation of Aβ42 in the presence of Zn2+ leads to the formation of large amorphous aggregates (Figure 7, panel 3). Native gel/Western blotting analysis shows that aggregation of Aβ42 in the absence of metal ions also forms high-molecular-weight oligomers, along with the insoluble aggregates that are observed at the top of the blot because they do not enter the gel (Figure 7, lane 1). As reported previously,20,21 the presence of Cu2+ ions inhibits the formation of large soluble and insoluble Aβ42 aggregates (Figure 7, lane 2).
Figure 6.

Normalized ThT fluorescence of the inhibition of Aβ fibrillization, measured upon incubaton at 37 °C for 24 h. Samples are as indicated on top of the lanes (PBS; [Aβ] = 25 μM; [M2+] = 25 μM; [compound] = 50 μM).
Figure 7.

Top: TEM images of the inhibition of Aβ42 aggregation by L1 and L2, in the presence or absence of metal ions ([Aβ42] = [M2+] = 25 μM; [compound] = 50 μM; PBS, 37 °C, 24 h, scale bar = 500 nm). Bottom: Native gel electrophoresis/Western blotting analysis. Panels and lanes are as follows: (1) Aβ42; (2) Aβ42 + Cu2+; (3) Aβ42 + Zn2+; (4) Aβ42 + L1; (5) Aβ42 + L1 + Cu2+; (6) Aβ42 + L1 + Zn2+; (7) Aβ42 + L2; (8) Aβ42 + L2 + Cu2+; (9) Aβ42 + L2 + Zn2+; (10) MW marker.
The presence of either L1 or L2 in the absence of metal ions does not seem to significantly inhibit Aβ42 aggregation, although some morphological changes were observed for the Aβ42 fibrils (Figure 7, panels 4 and 7); the observed decrease in the ThT fluorescence intensity may thus be due to these morphological changes of the Aβ42 aggregates. Native gel/Western blotting analysis reveals a limited effect of L1 and L2 on Aβ42 aggregation (Figure 7, lanes 4 and 7). Interestingly, the presence of either L1 or L2 has a dramatic effect on the Cu2+-mediated oligomerization of Aβ42 and promotes the formation of larger Aβ42 aggregates, as observed by TEM (Figure 7, panels 5 and 8). Importantly, native gel/Western blotting analysis reveals bands at the top of the gel, suggesting the presence of insoluble Aβ42 aggregates (Figure 7, lanes 5 and 8), which were not observed in the presence of Cu2+ alone (Figure 7, lane 2). Thus, compounds L1 and L2 reduce the amount of soluble Aβ42 oligomers formed in the presence of Cu2+ by promoting the formation of insoluble Aβ42 aggregates. In addition, the presence of either L1 or L2 does not dramatically affect the formation of insoluble Aβ42 aggregates in the presence of Zn2+ (Figure 7, panels 6 and 9), which is supported by native gel/Western blotting analysis showing no formation of high-molecular-weight soluble Aβ42 oligomers, although insoluble aggregates are observed at the top of the gel (Figure 7, lanes 6 and 9). Overall, these studies suggest that the presence of either L1 or L2, in both the presence or absence of metal ions, does not promote the formation of soluble Aβ42 oligomers. Because the soluble Aβ42 oligomers are the most neurotoxic species, these compounds are expected to control the neurotoxicity of Aβ42 species in the presence of metal ions (vide infra).
Effect of L1 and L2 on Aβ40 Aggregation
Although it was shown in previous studies9,10,21 that the Aβ42 peptide is more prone to form neurotoxic soluble Aβ oligomers than the Aβ40 peptide, we have also investigated the effect of L1 and L2 on the metal-mediated aggregation of Aβ40. TEM analysis shows that Aβ40 forms well-defined fibrils either in the absence or presence of L1 or L2 (Figure S9 in the SI, panels 1 and 4). Importantly, the presence of Cu2+ and Zn2+ does not inhibit Aβ40 aggregation,21 yet they change the morphology of the insoluble aggregates that become more amorphous (Figure S9 in the SI, panels 2 and 3). Moreover, in the presence of Cu2+ and either L1 or L2, a small amount of Aβ40 fibrils are still formed (Figure S9 in the SI, panels 5 and 8), while the presence of Zn2+ and either L1 or L2 leads to the formation of a larger amount of fibrillar or amorphous aggregates, respectively (Figure S9 in the SI, panels 6 and 9). The TEM results are also supported by native gel electrophoresis/Western blotting analysis (Figure S10 in the SI), although this is less informative than in the case of the Aβ42 peptide because of the lack of formation of an appreciable amount of soluble Aβ oligomers under any experimental conditions. Therefore, because we are particularly interested in evaluating the role of metal ions and bifunctional compounds in controlling the formation of the neurotoxic soluble Aβ oligomers, the Aβ42 peptide was employed in the additional studies described below.
Disaggregation of Aβ42 Fibrils
The effect of L1 and L2 on preformed Aβ42 fibrils was also investigated in both the absence and presence of metal ions (Scheme 3). For disaggregation studies, Aβ42 fibrils were prepared by incubation for 24 h at 37 °C, in both the presence and absence of metal ions. Then, L1 or L2 was added and further incubated for an additional 24 h at 37 °C. TEM and native gel electrophoresis/Western blotting characterization was performed on all of these samples. First, either L1 or L2 in metal-free conditions does not disaggregate the preformed Aβ42 fibrils to an appreciable extent. Only some morphological changes were observed even after 24 h of incubation, and a significant amount of Aβ42 aggregates were still observed by TEM (Figure 8, panels 4 and 7). Native gel electrophoresis/Western blotting analysis reveals high-molecular-weight soluble Aβ42 species and insoluble aggregates, although to a lesser extent in the presence of L1 (Figure 8, lanes 4 and 7). In the presence of Cu2+, either L1 or L2 partially inhibits the Cu2+-promoted formation of small soluble aggregates and allows for enhanced Aβ42 aggregation (Figure 8, panels 5 and 8 vs panel 2). This is supported by native gel/Western blotting, which shows that while there is a slightly increased amount of low-molecular-weight (10–80 kDa) soluble Aβ42 species, more insoluble aggregates are also observed at the top of the gel (Figure 8, lanes 5 and 8). In the presence of Zn2+, either L1 or L2 seems to have a limited effect on the morphology of the insoluble Aβ42 aggregates, as observed by both TEM and Western blotting (Figure 8, panels/lanes 6 and 9). Overall, all of these in vitro studies suggest that compounds L1 and L2 should control the formation of Cu2+-stabilized soluble Aβ42 species and thus should control their neurotoxicity (vide infra).
Figure 8.

Top: TEM images of the disaggregation of Aβ42 aggregation by L1 and L2, in the presence or absence of metal ions ([Aβ42] = [M2+] = 25 μM; [compound] = 50 μM; PBS, 37 °C, 24 h for fibrilization and an additional 24 h for disaggregation, scale bar = 500 nm). Bottom: Native gel electrophoresis/Western blotting analysis. Panels and lanes are as follows: (1) Aβ42; (2) Aβ42 + Cu2+; (3) Aβ42 + Zn2+; (4) Aβ42 + L1; (5) Aβ42 + L1 + Cu2+; (6) Aβ42 + L1 + Zn2+; (7) Aβ42 + L2; (8) Aβ42 + L2 + Cu2+; (9) Aβ42 + L2 + Zn2+; (10) MW marker.
Effect of L1 and L2 on the Aβ42 Neurotoxicity
Because Cu2+–Aβ species have been shown to be neurotoxic,20,21,31,51,52 the development of metal-binding compounds that control the metal-dependent Aβ toxicity is desired. In this regard, the effect of L1 and L2 on the Aβ42 neurotoxicity in N2A cells was investigated using an Alamar Blue cell viability assay.53 First, we examined the toxicity of both L1 and L2 along with ethylenediaminetetraacetic acid (EDTA) and CQ at various concentrations ranging from 0.2 to 20 μM (Figure S11 in the SI).40 Both L1 and L2 show no appreciable cell toxicity (>80% cell survival) even up to 20 μM concentration. By comparison, the clinically tested compound CQ was quite toxic to cells even at 2 μM concentration, while EDTA does not show any toxicity up to 20 μM.25,31 For the preformed Aβ42 fibrils, no significant cell toxicity was observed in either the absence or presence of Cu2+ (Figure 9, lanes 1 and 2), supporting the previously reported diminished toxicity of Aβ42 fibrils.5,7,54−56 Moreover, compounds L1 and L2 do not affect the cell viability when added to the N2A cells in the presence of preformed Aβ42 fibrils (Figure 9, lanes 3 and 4). Even in the presence of preformed Aβ42 fibrils and Cu2+, the cell viability is not affected by either L1 or L2 (Figure 9, lanes 5 and 6). By contrast, CQ shows quite high cell toxicity to cells in the presence of Cu2+ and Aβ42 fibrils with a cell survival of only 26 ± 3% (Figure 9, lane 7). Thus, these cellular toxicity data suggest that L1 and L2 do not lead to any cell toxicity in the presence or absence of Aβ42 fibrils and Cu2+ ions, suggesting that these compounds do not disaggregate Aβ42 fibrils to generate neurotoxic soluble Aβ42 species.
Figure 9.

Cell viability (% DMSO control) of N2A cells determined by the Alamar Blue assay, upon incubation with (1) Aβ42 fibrils, (2) Aβ42 fibrils + Cu2+, (3) Aβ42 fibrils + L1, (4) Aβ42 fibrils + L2, (5) Aβ42 fibrils + Cu2+ + L1, (6) Aβ42 fibrils + Cu2+ + L2, (7) Aβ42 fibrils + Cu2+ + CQ, (8) Aβ42 oligomers, (9) Aβ42 oligomers + L1, (10) Aβ42 oligomers + L2, (11) Aβ42 oligomers + L1 + Cu2+, and (12) Aβ42 oligomers + L2 + Cu2+. Conditions: [compound] = 20 μM; [Cu2+] = 20 μM; [Aβ42] = 20 μM. The t-test analysis reveals values of p < 0.001 for the pairs of data sets marked with asterisks.
As shown previously,20,21 the preformed soluble Aβ42 oligomers show a limited cell survival of 45 ± 6% (Figure 9, lane 8). Also, we have shown previously that while Cu2+ by itself is not toxic to N2A cells, the presence of Cu2+ and either monomeric Aβ42 or soluble Aβ42 oligomers exhibits enhanced cell toxicity.17,18 However, the addition of either L1 or L2 to the N2A cells along with the preformed Aβ42 oligomers drastically reduces the neurotoxicity of soluble Aβ42 species to 100 ± 15% and 103 ± 15% cell viability, respectively (Figure 9, lanes 9 and 10). Even in the presence of Cu2+, which has been shown to stabilize the soluble Aβ42 oligomers,22 L1 and L2 were able to eliminate the neurotoxicity of soluble Aβ42 oligomers (Figure 9, lanes 9 and 10). These results are particularly exciting because, to the best of our knowledge, these are the first BFCs that reduce the toxicity of soluble Aβ42 oligomers, in both the presence or absence of Cu2+ ions. These BFCs thus represent promising lead compounds for the development of potential therapeutic agents that can control the metal-mediated neurotoxicity of soluble Aβ42 species in AD and are currently the focus of our ongoing in vivo studies.
Conclusion
In summary, two small BFCs, L1 and L2, were developed to specifically target the interactions of Cu2+ and Zn2+ ions with the Aβ42 peptide and to control the formation of neurotoxic soluble Aβ42 oligomers. The bifunctionality of L1 and L2 (i.e., metal chelation and Aβ interaction) was established through various spectroscopic studies such as spectrophotometric titrations, structural characterization, and fluorescent assays. We have previously shown that BFCs, which have high affinity for both metal ions and Aβ fibrils, can cause significant oligomerization via disaggregation of Aβ42 fibrils and lead to enhanced neurotoxicity.20 However, the BFCs described herein bind to metal ions and Aβ fibrils only with moderate affinity and are having the desired opposite effect of promoting fibrillization of Cu2+-stabilized soluble Aβ42 oligomers, and most importantly they drastically reduce the neurotoxicity of soluble Aβ42 species, in both the presence or absence Cu2+ ions. Because, to the best of our knowledge, these BFCs are the first to control the metal-mediated neurotoxicity of Aβ42 oligomers, L1 and L2 are promising lead compounds for the development of chemical agents that can control the neurotoxicity of soluble Aβ42 species in AD.
Acknowledgments
We thank the Oak Ridge Associated Universities for a Ralph E. Powe Junior Faculty Award (to L.M.M.), the Washington University Knight Alzheimer’s Disease Research Center (Pilot Research NIH Grant P50-AG05681 to L.M.M.), the Alzheimer’s Association for New Investigator Research Grant NIRP-12-259199 (to L.M.M.), and the NIH Grant P30NS69329 (to J.K.) for financial support. L.M.M. is a Sloan Fellow.
Supporting Information Available
X-ray crystallographic data in CIF format, synthetic schemes for L1 and L2, UV–vis and fluororescence spectra of L1 and L2, Job’s plots for L1 in the presence of metal ions, UV–vis spectra of metal complexes, toxicity data of compounds, and X-ray data for 1–3. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Alzheimer’s Disease Facts and Figures: Annual Report, 2013, from www.alz.org.
- Ferri C. P.; Prince M.; Brayne C.; Brodaty H.; Fratiglioni L.; Ganguli M.; Hall K.; Hasegawa K.; Hendrie H.; Huang Y.; Jorm A.; Mathers C.; Menezes P. R.; Rimmer E.; Scazufca M. Lancet 2005, 366, 2112–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob-Roetne R.; Jacobsen H. Angew. Chem., Int. Ed. 2009, 48, 3030–3059. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. Science 2002, 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Selkoe D. J. J. Neurochem. 2007, 101, 1172–1184. [DOI] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [DOI] [PubMed] [Google Scholar]
- Lambert M. P.; Barlow A. K.; Chromy B. A.; Edwards C.; Freed R.; Liosatos M.; Morgan T. E.; Rozovsky I.; Trommer B.; Viola K. L.; Wals P.; Zhang C.; Finch C. E.; Krafft G. A.; Klein W. L. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y. S.; Chang L.; Viola K. L.; Lacor P. N.; Lambert M. P.; Finch C. E.; Krafft G. A.; Klein W. L. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E.; Pickford F.; Kim J.; Onstead L.; Eriksen J.; Yu C.; Skipper L.; Murphy M. P.; Beard J.; Das P.; Jansen K.; DeLucia M.; Lin W. L.; Dolios G.; Wang R.; Eckman C. B.; Dickson D. W.; Hutton M.; Hardy J.; Golde T. Neuron 2005, 47, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperstein I.; Broersen K.; Benilova I.; Rozenski J.; Jonckheere W.; Debulpaep M.; Vandersteen A.; Segers-Nolten I.; Van Der Werf K.; Subramaniam V.; Braeken D.; Callewaert G.; Bartic C.; D’Hooge R.; Martins I. C.; Rousseau F.; Schymkowitz J.; De Strooper B. EMBO J. 2010, 29, 3408–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell M. A.; Robertson J. D.; Teesdale W. J.; Campbell J. L.; Markesbery W. R. J. Neurol. Sci. 1998, 158, 47–52. [DOI] [PubMed] [Google Scholar]
- Bush A. I.; Pettingell W. H.; Multhaup G.; Paradis M. D.; Vonsattel J. P.; Gusella J. F.; Beyreuther K.; Masters C. L.; Tanzi R. E. Science 1994, 265, 1464–1467. [DOI] [PubMed] [Google Scholar]
- Atwood C. S.; Moir R. D.; Huang X. D.; Scarpa R. C.; Bacarra N. M. E.; Romano D. M.; Hartshorn M. K.; Tanzi R. E.; Bush A. I. J. Biol. Chem. 1998, 273, 12817–12826. [DOI] [PubMed] [Google Scholar]
- Hureau C.; Faller P. Biochimie 2009, 91, 1212. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Su B.; Wang X.; Smith M.; Perry G. Cell. Mol. Life Sci. 2007, 64, 2202–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crichton R. R.; Dexter D. T.; Ward R. J. Coord. Chem. Rev. 2008, 252, 1189–1199. [Google Scholar]
- House E.; Collingwood J.; Khan A.; Korchazkina O.; Berthon G.; Exley C. J. Alzheimer’s Dis. 2004, 6, 291–301. [DOI] [PubMed] [Google Scholar]
- Exley C. J. Alzheimer’s Dis. 2006, 10, 173–177. [DOI] [PubMed] [Google Scholar]
- Exley C. Coord. Chem. Rev. 2012, 256, 3114–3114. [Google Scholar]
- Sharma A. K.; Pavlova S. T.; Kim J.; Finkelstein D.; Hawco N. J.; Rath N. P.; Kim J.; Mirica L. M. J. Am. Chem. Soc. 2012, 134, 6625–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A. K.; Pavlova S. T.; Kim J.; Kim J.; Mirica L. M. Metallomics 2013, 5, 1529–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Rempel D. L.; Zhang J.; Sharma A. K.; Mirica L. M.; Gross M. L. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 14604–14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Rodríguez C.; Telpoukhovskaia M.; Orvig C. Coord. Chem. Rev. 2012, 256, 2308–2332. [Google Scholar]
- Braymer J. J.; DeToma A. S.; Choi J.-S.; Ko K. S.; Lim M. H. Int. J. Alzheimer’s Dis. 2011, 2011, ID 623051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.-S.; Braymer J. J.; Nanga R. P. R.; Ramamoorthy A.; Lim M. H. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 21990–21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott L. E.; Orvig C. Chem. Rev. 2009, 109, 4885–4910. [DOI] [PubMed] [Google Scholar]
- Dedeoglu A.; Cormier K.; Payton S.; Tseitlin K. A.; Kremsky J. N.; Lai L.; Li X. H.; Moir R. D.; Tanzi R. E.; Bush A. I.; Kowall N. W.; Rogers J. T.; Huang X. D. Exp. Gerontol. 2004, 39, 1641–1649. [DOI] [PubMed] [Google Scholar]
- Liu Y. Z.; Kochi A.; Pithadia A. S.; Lee S.; Nam Y.; Beck M. W.; He X. M.; Lee D.; Lim M. H. Inorg. Chem. 2013, 52, 8121–8130. [DOI] [PubMed] [Google Scholar]
- Pithadia A. S.; Lim M. H. Curr. Opin. Chem. Biol. 2012, 16, 67–73. [DOI] [PubMed] [Google Scholar]
- Pithadia A. S.; Kochi A.; Soper M. T.; Beck M. W.; Liu Y. Z.; Lee S.; DeToma A. S.; Ruotolo B. T.; Lim M. H. Inorg. Chem. 2012, 51, 12959–12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindo S. S.; Mancino A. M.; Braymer J. J.; Liu Y. H.; Vivekanandan S.; Ramamoorthy A.; Lim M. H. J. Am. Chem. Soc. 2009, 131, 16663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott L. E.; Telpoukhovskaia M.; Rodriguez-Rodriguez C.; Merkel M.; Bowen M. L.; Page B. D. G.; Green D. E.; Storr T.; Thomas F.; Allen D. D.; Lockman P. R.; Patrick B. O.; Adam M. J.; Orvig C. Chem. Sci. 2011, 2, 642. [Google Scholar]
- Rodriguez-Rodriguez C.; de Groot N. S.; Rimola A.; Alvarez-Larena A.; Lloveras V.; Vidal-Gancedo J.; Ventura S.; Vendrell J.; Sodupe M.; Gonzalez-Duarte P. J. Am. Chem. Soc. 2009, 131, 1436–1451. [DOI] [PubMed] [Google Scholar]
- Braymer J. J.; DeToma A. S.; Choi J.-S.; Ko K. S.; Lim M. H. Int. J. Alzheimer’s Dis. 2011, 2011, 623051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruker Analytical X-ray: Madison, WI, 2008.
- Sheldrick G. M. Acta Crystallogr. 2008, A64, 112–122. [DOI] [PubMed] [Google Scholar]
- Gans P.; Sabatini A.; Vacca A. Ann. Chim. 1999, 45. [Google Scholar]
- Alderighi L. Coord. Chem. Rev. 1999, 184, 311. [Google Scholar]
- Klein W. L. Neurochem. Int. 2002, 41, 345–352. [DOI] [PubMed] [Google Scholar]
- See the SI.
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Adv. Drug Delivery Rev. 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Storr T.; Merkel M.; Song-Zhao G. X.; Scott L. E.; Green D. E.; Bowen M. L.; Thompson K. H.; Patrick B. O.; Schugar H. J.; Orvig C. J. Am. Chem. Soc. 2007, 129, 7453–7463. [DOI] [PubMed] [Google Scholar]
- Huang C. Y. Methods Enzymol. 1982, 87, 509. [DOI] [PubMed] [Google Scholar]
- Talmard C.; Bouzan A.; Faller P. Biochemistry 2007, 46, 13658–13666. [DOI] [PubMed] [Google Scholar]
- Klunk W. E.; Wang Y. M.; Huang G. F.; Debnath M. L.; Holt D. P.; Mathis C. A. Life Sci. 2001, 69, 1471–1484. [DOI] [PubMed] [Google Scholar]
- LeVine H. 3rd. Methods Enzymol. 1999, 309, 274–284. [DOI] [PubMed] [Google Scholar]
- Lockhart A.; Ye L.; Judd D. B.; Merritt A. T.; Lowe P. N.; Morgenstern J. L.; Hong G. Z.; Gee A. D.; Brown J. J. Biol. Chem. 2005, 280, 7677–7684. [DOI] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [DOI] [PubMed] [Google Scholar]
- Mold M.; Ouro-Gnao L.; Wieckowski B. M.; Exley C.. Sci. Rep. 2013, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House E.; Mold M.; Collingwood J.; Baldwin A.; Goodwin S.; Exley C. J. Alzheimer’s Dis. 2009, 18, 811–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush A. I. Neurobiol. Aging 2002, 23, 1031–1038. [DOI] [PubMed] [Google Scholar]
- Huang X. D.; Cuajungco M. P.; Atwood C. S.; Hartshorn M. A.; Tyndall J. D. A.; Hanson G. R.; Stokes K. C.; Leopold M.; Multhaup G.; Goldstein L. E.; Scarpa R. C.; Saunders A. J.; Lim J.; Moir R. D.; Glabe C.; Bowden E. F.; Masters C. L.; Fairlie D. P.; Tanzi R. E.; Bush A. I. J. Biol. Chem. 1999, 274, 37111–37116. [DOI] [PubMed] [Google Scholar]
- Dahlgren K. N.; Manelli A. M.; Stine W. B.; Baker L. K.; Krafft G. A.; LaDu M. J. J. Biol. Chem. 2002, 277, 32046–32053. [DOI] [PubMed] [Google Scholar]
- Hsia A. Y.; Masliah E.; McConlogue L.; Yu G.-Q.; Tatsuno G.; Hu K.; Kholodenko D.; Malenka R. C.; Nicoll R. A.; Mucke L. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S.; Koh M. T.; Kotilinek L.; Kayed R.; Glabe C. G.; Yang A.; Gallagher M.; Ashe K. H. Nature 2006, 440, 352–357. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Davis J.; Aucoin D.; Sato T.; Ahuja S.; Aimoto S.; Elliott J. I.; Van Nostrand W. E.; Smith S. O. Nat. Struct. Mol. Biol. 2011, 17, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.