

Abstract
Vestibular schwannomas (VSs) represent Schwann cell (SC) tumors of the vestibular nerve, compromising 10% of all intracranial neoplasms. VSs occur in either sporadic or familial (neurofibromatosis type 2, NF2) forms, both associated with inactivating defects in the NF2 tumor suppressorgene. Treatment for VSs is generally surgical resection or radiosurgery, however the morbidity of such procedures has driven investigations into less invasive treatments. Historically, lack of access to fresh tissue specimens and the fact that schwannoma cells are not immortalized have significantly hampered the use of primary cultures for investigation of schwannoma tumorigenesis. To overcome the limited supply of primary cultures, the immortalized HEI193 VS cell line was generated by transduction with HPV E6 and E7 oncogenes. This oncogenic transduction introduced significant molecular and phenotypic alterations to the cells, which limit their use as a model for human schwannoma tumors. We therefore illustrate a simplified, reproducible protocol for culture of primary human VS cells. This easily mastered technique allows for molecular and cellular investigations that more accurately recapitulate the complexity of VS disease.
Keywords: Medicine, Issue 89, Primary Vestibular Schwannoma, Cranial Nerve Schwannoma, Primary Acoustic Neuroma, Cell Culture
Introduction
Vestibular schwannomas (VSs) represent Schwann cell (SC) tumors of the vestibular nerve, compromising 10% of all intracranial neoplasms1-3. VSs occur in either sporadic or familial (neurofibromatosis type 2, NF2) forms, both associated with inactivating defects in the NF2 tumor suppressorgene. Treatment for VSs is generally surgical resection or radiosurgery, however the morbidity of such procedures includes deafness, facial neuropathies, spinal fluid leak, imbalance, and tumor regrowth1. Additionally not all patients are acceptable surgical or radiation candidates. Such significant morbidity as well as a lack of alternative therapies has driven investigation into the unique molecular biology of VSs in hopes of developing novel treatments3,4.
Cell culture allows for rapid, facile, and in-depth analysis of molecular and cellular behavior and screening of potential therapeutic compounds. Historically, lack of access to fresh tissue specimens and the fact that schwannoma cells are not immortalized have significantly hampered the use of primary cultures for investigation of schwannoma tumorigenesis. To overcome the limited supply of primary cultures, the immortalized HEI193 VS cell line was generated by transduction with HPV E6 and E7 oncogenes5. This oncogenic transduction introduced significant molecular and phenotypic alterations to the cells, which limit their use as a model for human schwannoma tumors. SC cultures derived from transgenic mouse lines that lack a functional NF2 gene represent another alternative to investigate NF2-dependent SC tumorigenesis in vitro. These cultures however fail to recapitulate the heterogeneous nature of human VSs or account for human specific behavior. Most previous VS culture techniques required relatively long processing times and complicated selective culture techniques6-8. Here we present a simple, reproducible protocol for primary VS cell culture with complete processing in under 3 hr, with 95% tumor cell purity as determined by immunostaining.
Protocol
Ethics Statement: use of the human tumor specimens in this protocol was approved by the University of Iowa Institutional Review Board (IRB).
1. Setup the Day Before Tumor Harvest
Coat Cell Culture Plates: In a sterile culture hood, add enough poly-L-ornithine (0.01% solution) to cover the bottom of a polystyrene/plastic culture well/dish, replace the lids, and let sit at room temperature for at least 2 hr. Glass slides or coverslips can be substituted for polystyrene for experiments requiring imaging.
After 2 hr, remove the poly-L-ornithine solution with suction and allow the plates to dry completely in a sterile culture hoods. Add enough laminin solution (10 µg/ml in Ca2+/Mg2+-free HBSS [HBSS-/-]) to completely cover the culture plates, replace lids, then either 1) place in cool 4 °C refrigerator overnight, or 2) place in 37 °C incubator for at least 2 hr. If plates will be stored overnight or longer, wrap the plates in plastic wrap to prevent evaporation/desiccation.
2. Setup the Day of Tumor Harvest
Prepare Culture Media: Media is made fresh on day of tumor harvest and processing, stored at 4 °C, and warmed to 37 °C in incubator immediately prior to media additions/changes.
Schwannoma culture media is high-glucose (4.5 mg/ml) DMEM (with phenol red) with 0.1 mg/ml penicillin and 0.1 mg/ml streptomycin; N2 media supplement final dilution 1:100; Bovine Insulin 5 µg/ml; Fetal Bovine Serum to 10% total volume. Filter all media through 0.22 μm filter.
Prepare Specimen Transport Cooler: Send an ice filled cooler with 1-2 (depending on amount of tumor to be harvested) sterile 50 ml conical tubes with 25 ml HBSS with Ca2+/Mg2+ (HBSS+/+) to the operating room for tumor sample transfer and transport.
Prepare individual tumor dissociation tubes – fill sterile 2 ml round bottom tubes with 1 ml of HBSS+/+ and place on ice. (Tubes will be used for sharp dissection/dissociation of tumor samples).
For dissociation, prepare approximately one dissociation tube for every 0.5 cm3 of harvested tissue; for example, a tumor specimen measuring 1.0 x 1.0 x 1.0 cm (1 cm3) will require two HBSS+/+ dissociation tubes.
Ultraviolet light sterilization: Place tissue scissors, small forceps, scalpel handle, 1x normal Petri dish, P1000 pipette, and pipette tips in a sterile culture hood, and leave under ultraviolet light for ~15 min prior to arrival and processing of tumor tissue.
3. Specimen Isolation and Transport
Isolate tumor specimens by surgical resection.
Immediately place tumor specimens into the ice-cold HBSS+/+ as quickly as possible after removal from the patient. Once all tissue samples are collected, transport samples (on ice) from the operative theater to the lab for further processing.
4. Tissue Dissociation and Trituration
Following standard sterile technique in a laminar flow hood, bring conical tube with tumor specimen into the culture hood, and wash samples by inverting tube and tumor 50x. Allow fragments to fall to the bottom of the tube, and then remove HBSS+/+. Refill with 30 ml HBSS+/+, and agitate/invert 50x again. Repeat inversion/media removal for a total of 4x.
Re-suspend tumor fragments in 30 ml HBSS+/+ for the last time, then pour mixture into a sterile100 mm petri dish, and separate tumor into 0.5 cm3 groupings. If larger tumor fragments are encountered, use the tissue scissors or scalpel (#11 or #15 blade) to cut into smaller pieces.
Use the forceps to place the 0.5 cm3 tissue groups into the individual HBSS+/+ filled, 2 ml round bottom conical tubes, all on ice. Use the tissue scissors to mince tumor fragments into sub-1 mm fragments –the suspension should have a ‘snowglobe’ appearance (Figure 1). Mince the tumor only until the majority of the fragments are sub-1 mm; do not ‘over-mince’ the tumor samples. Place completely minced tissue tube back on ice, and repeat with all additional specimen tubes.
Transfer all fragmented tumor samples into a single 15 ml conical tube. A P1000 pipette with a cut/modified sterile tip works well for this step. Use extra HBSS+/+ to flush/rinse the 2 ml dissociation tubes to make sure all tumor sample has been collected in 15 ml tube. Spin down mixture in centrifuge at 23 °C, at 73 x g for 5 min.
Suction off HBSS+/+, leaving the cell pellet. Re-suspend fragments in 1:1 mixture of 0.25% trypsin:0.2% collagenase (dissolved in HBSS-/-) – add just enough to keep tumor fragments in tight suspension. Replace screw-top, and place in 37 °C incubator for 30 min. Agitate cell mixture at least once during incubation to keep fragments in suspension. Place the Schwannoma culture media into the incubator to warm it at this time as well.
After incubation, add 100 µl FBS to each tube to inactivate the trypsin, and then centrifuge sample at 23 °C, 73 x g for 5 min. Using a pipette, carefully suction off as much supernatant as possible without disturbing cell pellet. Add the warmed culture media (N2/insulin/5% FBS) to the tumor fragments to a final ratio of 1:2 tumor fragments: culture media (for example, if there is 1 ml of tumor fragments, add in 2 ml of warmed media).
Prepare for tumor trituration by cutting the tip off of a P1000 pipette tip to a diameter of approximately 1-2 mm. Slowly triturate the cell suspension using progressively smaller and smaller diameter pipette tips, until you can easily pipette the solution through an unaltered P1000 pipette tip. Immediately move to a smaller tip once you can easily pipette the tumor solution through the tip – do not over triturate the sample. If a single larger piece of tissue blocks aspiration during trituration, tapping the pipette tip on the bottom of the conical tube often allows continued trituration.
5. Cell Plating
One 0.5 mm3 fragment of tissue is enough to seed 1 x 100 mm dish, or 4 x 8-well/4-well slides. Just prior to adding the media/cell mixture, remove the laminin solution from the culture plates but do not let the plates dry.
For each 100 mm dish, dissolve the tumor fragment in 10 ml warmed culture media.
For each well of a 4-well slide, use 750 μl warmed culture media (3 ml for the whole slide).
For each well of an 8-well slide, use 400 μl warmed culture media (3.2 ml for the whole slide).
Incubate the cultures in 37 °C, 100% humidity, 6% CO2. Leave the cultures alone for at least 36 hours, and then change the media if the solution is yellowed (acidic). Otherwise change the media at 72 hr, then every 2-3 days or when the media become acidic. If the media becomes acidic daily for several days in a row, this is generally an indication of potential bacterial or yeast contamination.
Representative Results
Correct tumor fragmentation is essential for optimal seeding and outgrowth in the tissue culture dishes (Figure 1). Identification of schwannoma cells during the first days after plating is often difficult, due to obscuring amounts of cellular debris and red blood cells. By the 4th or 5th day, cell extensions near adherent tumor fragments will create recognizable ‘lacing’ patterns among the debris. Subsequent media changes generally remove the majority of non-adherent cells by the second week. Using this method, 90% confluence can be expected between days 7-14, depending on the initial vitality of the tumor fragment prior to mincing and plating density (Figure 2). As described previously, schwannoma cultures appear to grow outward from small fragments of adherent tumor fragments9,10. Such outgrowth is detectable by the end of the first week. Figure 2 shows brightfield imaging of schwannoma cell outgrowth from a single tumor fragment, outlined in red in the lower right corner. Figure 3 illustrates the ‘bridging’ outgrowth that occurs between two different tumor fragments, in culture wells immunostained with polyclonal rabbit anti-S100 antibody. We routinely immunostain cultures with anti-S100 or anti-p75NTR antibodies to verify the purity of cultures and positively identify schwannoma cells for analysis (Figure 4). With these methods over >95% of the cells in these cultures represent schwannoma cells. Schwannoma cells also label with glial fibrillary acidic protein (GFAP) and ErbB2, among other markers11.
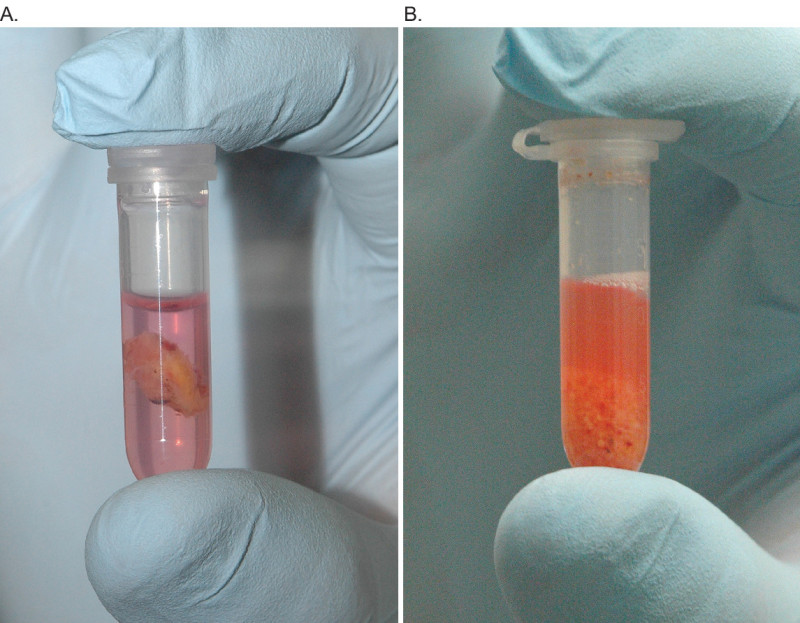 Figure 1. Comparison of the same tumor sample both before (A) and after (B) mincing. Sample prepared in 1.5 ml HBSS+/+ with Phenol Red in 2.0 ml round bottom conical tube.
Figure 1. Comparison of the same tumor sample both before (A) and after (B) mincing. Sample prepared in 1.5 ml HBSS+/+ with Phenol Red in 2.0 ml round bottom conical tube.
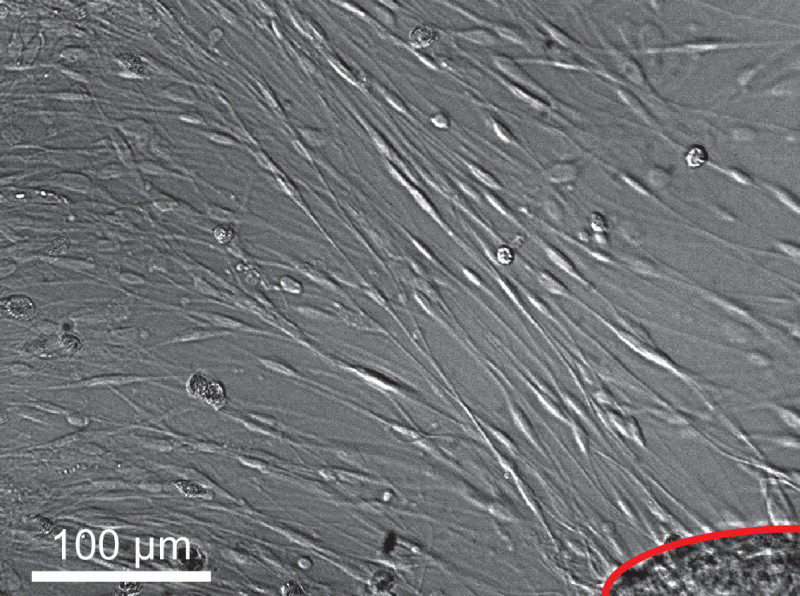 Figure 2. Typical appearance of cultures on brightfield microscopy. Spindled schwannoma cell outgrowth radiates from tumor fragment, image taken on culture day 10. Scale bar = 100 µm.
Figure 2. Typical appearance of cultures on brightfield microscopy. Spindled schwannoma cell outgrowth radiates from tumor fragment, image taken on culture day 10. Scale bar = 100 µm.
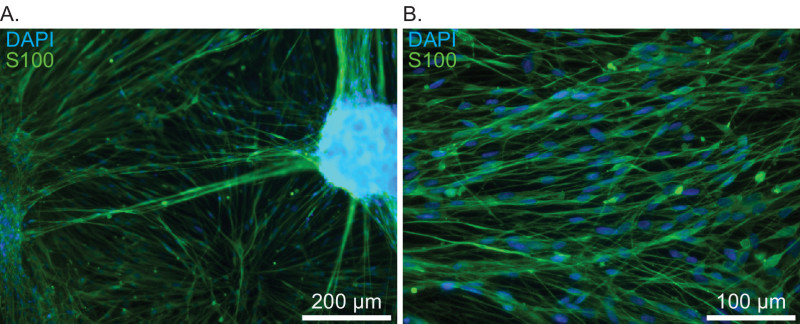 Figure 3. Immunostaining of primary vestibular schwannoma cultures with anti-S100 antibodies demonstrating typical culture and cell morphologies. A) Low power view illustrating outgrowth from plated tumor fragments, culture day 14. Scale bar = 200 µm. B) Higher power view of schwannoma cells with spindled morphology, culture day 14. Scale bar = 100 µm.
Figure 3. Immunostaining of primary vestibular schwannoma cultures with anti-S100 antibodies demonstrating typical culture and cell morphologies. A) Low power view illustrating outgrowth from plated tumor fragments, culture day 14. Scale bar = 200 µm. B) Higher power view of schwannoma cells with spindled morphology, culture day 14. Scale bar = 100 µm.
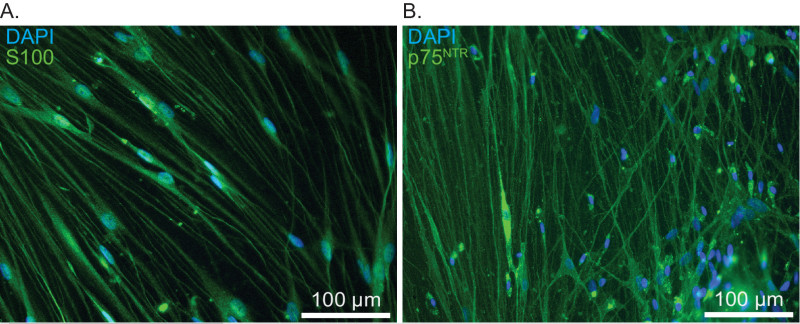 Figure 4. Immunostaining of primary vestibular schwannoma cultures to confirm culture purity and cell identity. A) Cultures immunostained with anti-S100 antibodies. Nuclei are labeled with DAPI. Scale bar = 100 µm. B) Cultures immunostained with anti-p75NTR antibodies. Nuclei are labeled with DAPI. Scale bar = 100 µm.
Figure 4. Immunostaining of primary vestibular schwannoma cultures to confirm culture purity and cell identity. A) Cultures immunostained with anti-S100 antibodies. Nuclei are labeled with DAPI. Scale bar = 100 µm. B) Cultures immunostained with anti-p75NTR antibodies. Nuclei are labeled with DAPI. Scale bar = 100 µm.
Discussion
History of in vitro Schwannoma Cultures
Efforts to establish in vitro schwannoma cultures began just a few decades after the initial acoustic neuroma open surgical resection occurred in 189412. The first recorded cultures were by Kredel in the 1920s, who unsuccessfully attempted to grow minced tumor by “hanging drop method”12. Later, Drs. Murray and Stout published their in vitro culture experience with neurilemomas grown on clotted human plasma. Interestingly, this technique revealed that Antoni A and Antoni B specific fragments grew differently, with unique cellular morphology and differential rates of agar liquefaction13. Cravioto and Lockwood published further observations of acoustic neuroma cultures in avian clots on coverglass slips and roller tubes. They were also the first to use Rose chambers for time-lapse imaging of tumor growth14. Publications in the 1970s investigated growth differences between typical monolayer culture and ‘en bloc’ growth of larger tumor fragments10. Baur combined the ‘en bloc’ and monolayer approaches in her publication, which also demonstrated the mitogenic effects of laminin-coated culture plates9. Most subsequent published protocols, as our own, utilize a similar protocol of seeding minced tumor fragments onto a culture surface treated with a charged amino acid (poly-L-ornithine, poly-L-lysine) and laminin preparation8. We have used this same method to culture schwannomas arising from the facial, trigeminal, vagus, and spinal nerves with similar results.
Benefits of Vestibular Schwannoma Primary Cultures
Studies of human schwannoma tumorigenesis often use homogenous immortalized cell lines and transgenic mice models. Such tools are beneficial but do not accurately reflect the genotypes, phenotypes, and complexity of human disease. By contrast, primary cultures likely provide a more realistic model. They more faithfully recapitulate the heterogeneity of genomic and molecular status associated with human tumors15.
Purity of Schwannoma Cultures
A challenge of primary tissue culture is maintaining target cell purity. Schwannomas overwhelmingly consist of neoplastic Schwann cells6, however cultures are not immune from fibroblast contamination. Historically, methods for optimizing schwannoma cell purity fall into three categories: 1) selection by chemically defined medium and specific growth factors; 2) immunopurification (e.g., with magnetic bead cell sorting7), 3) a combination of both the preceding methods. In our experience fibroblasts or other non-schwannoma cells generally comprise less that 5% of cells in the culture, regardless of patient characteristics (age, sex), sporadic or NF2-associated tumors, primary or secondary tumor resection, or cystic vs solid tumor types. If fibroblasts appear to be taking over the culture, removal of the serum for 1-2 media changes will significantly reduce this population without adversely affecting schwannoma cell survival. Pre-treating cell culture surfaces with laminin also facilitates schwannoma cell growth9. Minimal passaging of the cultures also helps to decrease unwanted cellular contamination – cryostorage is best completed within the first two culture passages, and culture experimentation is not recommended beyond passage 4-5. We find optimal results when culture experimentation is completed by passage 1-2 with non-cryostored cells.
We use both immunostaining (anti-S100, DAPI) and microscopic appearance of cellular and nuclear morphology as quality control measures in our schwannoma cultures. Occasionally it is helpful to immunostain a subset of cultures for monocyte (CD68) or fibroblast specific markers to further verify culture purity9. During experimental analyses of cell behavior (e.g., proliferation or cell death), we always immunostain with anti-S100 or anti-p75NTR antibodies to verify that the findings are schwannoma cell specific.
Culture morphology and growth patterns
Schwannoma tumor cultures typically display unique growth patterns reminiscent of Antoni A (high cell density, tightly organized, indications of Verocay body formations) and rarely of Antoni B (comparatively sparse nuclei, abundant cellular processes, less organization) patterns seen in histopathology of resected schwannoma tumors16. Nearly all schwannoma cells display the classic long, spindled, bipolar cell shape associated with both Schwann and schwannoma cells6,17 however other cell morphologies, as described by Cravioto (Amoeboid Microglia-like Cells, Spindle-Shaped Cells, Racket-Shaped Cells, and Kite-Shaped Cells) sometimes occur14. Overall, cell growth and proliferation are very slow in our base medium (DMEM/10%FBS/Insulin/N2 supplement). Addition of known mitogenic factors such as forskolin and β1-neuregulin significantly increases cell proliferation18.
Critical Protocol Steps
Our schwannoma tissue culture protocol is simple and fairly forgiving; however there are several key steps that ensure success. First, correct tumor handling is critical. Expect best results when tumor is placed directly into ice-cold HBSS+/+ as soon as possible once removed from the patient. This is contrary to the usual surgical practice of collecting resected tumor in sterile saline then sending the aggregate tissue for processing in pathology at the end of the case. Second, keep tissue samples as cool as possible during processing. It is vital that the resected tumor is kept ice cold, and that processing occurs as expediently as possible following removal. Do as much of the tissue processing as possible on ice as well. Third, aggressively wash the tumor samples. This helps reduce the risk of bacterial or yeast contamination. Fourth, do not over-mince tumor specimens. Tumor trituration depends on a semi-subjective assessment of when to continue on with a smaller diameter P1000 pipette tip.
Disclosures
No conflicts of interest to disclose.
Acknowledgments
Support: NIDCD R01DC009801, P30DC010362, 5T32DC000040-17
References
- Arthurs BJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011;34(3):265–277. doi: 10.1007/s10143-011-0307-8. [DOI] [PubMed] [Google Scholar]
- Evans GR, Lloyd SK, Ramsden RT. Neurofibromatosis type 2. Adv Otorhinolaryngol. 2001;70:91–98. doi: 10.1159/000322482. [DOI] [PubMed] [Google Scholar]
- Fong B, et al. The molecular biology and novel treatments of vestibular schwannomas. J Neurosurg. 2011;115(5):906–914. doi: 10.3171/2011.6.JNS11131. [DOI] [PubMed] [Google Scholar]
- Jacob A, et al. Preclinical validation of AR42, a novel histone deacetylase inhibitor, as treatment for vestibular schwannomas. Laryngoscope. 2012;122(1):174–189. doi: 10.1002/lary.22392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DG. Neurofibromatosis 2 [Bilateral acoustic neurofibromatosis, central neurofibromatosis. NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599–610. doi: 10.1097/GIM.0b013e3181ac9a27. [DOI] [PubMed] [Google Scholar]
- Anniko M, Noren G. The Human Acoustic Neurinoma in Organ-Culture .1. Methodological Aspects. Acta Oto-Laryngologica. 1981;91(1-2):47–53. doi: 10.3109/00016488109138503. [DOI] [PubMed] [Google Scholar]
- Manent J, et al. Magnetic cell sorting for enriching Schwann cells from adult mouse peripheral nerves. J Neurosci Methods. 2003;123(2):167–173. doi: 10.1016/s0165-0270(02)00349-7. [DOI] [PubMed] [Google Scholar]
- Nair S, et al. Primary cultures of human vestibular schwannoma: selective growth of schwannoma cells. Otol Neurotol. 2007;28(2):258–263. doi: 10.1097/01.mao.0000247811.93453.6a. [DOI] [PubMed] [Google Scholar]
- Baur AM, et al. Laminin promotes differentiation, adhesion and proliferation of cell cultures derived from human acoustic nerve schwannoma. Acta Otolaryngol. 1995;115(4):517–521. doi: 10.3109/00016489509139359. [DOI] [PubMed] [Google Scholar]
- Cravioto H, et al. Experimental neurinoma in tissue culture. Acta Neuropathol. 1972;21(2):154–164. doi: 10.1007/BF00687569. [DOI] [PubMed] [Google Scholar]
- Kaasinen S, et al. Culturing of acoustic neuroma--methodological aspects. Acta Otolaryngol Suppl. 1995;520 Pt 1:25–26. [PubMed] [Google Scholar]
- Kredel FE. Tissue Culture of Intracranial Tumors with a Note on the Meningiomas. Am J Pathol. 1928;4(4):337–340. [PMC free article] [PubMed] [Google Scholar]
- Murray MR, Stout AP, Bradley CF. Schwann cell versus fibroblast as the origin of the specific nerve sheath tumor: Observations upon normal nerve sheaths and neurilemomas in vitro. Am J Pathol. 1940;16(1):41–60. [PMC free article] [PubMed] [Google Scholar]
- Cravioto H, Lockwood R. The behavior of acoustic neuroma in tissue culture. Acta Neuropathol. 1969;12(2):141–157. doi: 10.1007/BF00692502. [DOI] [PubMed] [Google Scholar]
- Lee JD, et al. Genetic and epigenetic alterations of the NF2 gene in sporadic vestibular schwannomas. PLoS One. 2012;7(1):30418. doi: 10.1371/journal.pone.0030418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wippold FJ, 2nd , et al. Neuropathology for the neuroradiologist: Antoni A and Antoni B tissue patterns. AJNR Am J Neuroradiol. 2007;28(9):1633–1638. doi: 10.3174/ajnr.A0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennel HD. Short-term tissue culture observations of experimental primary and transplanted nervous system tumors. J Neuropathol Exp Neurol. 1980;39(6):639–660. doi: 10.1097/00005072-198011000-00003. [DOI] [PubMed] [Google Scholar]
- Rosenbaum C, et al. Isolation and characterization of Schwann cells from neurofibromatosis type 2 patients. Neurobiol Dis. 1998;5(1):55–64. doi: 10.1006/nbdi.1998.0179. [DOI] [PubMed] [Google Scholar]