

H. pylori Csd4 (HP1075), together with other peptidoglycan hydrolases, plays an important role in determining the helical cell shape. Its crystal structure has been determined in three different forms.
Keywords: csd4; csd5; HP1075; d,l-carboxypeptidase; Helicobacter pylori; peptidoglycan; meso-diaminopimelate; pgp1; cell shape
Abstract
Helicobacter pylori infection causes a variety of gastrointestinal diseases, including peptic ulcers and gastric cancer. Its colonization of the gastric mucosa of the human stomach is a prerequisite for survival in the stomach. Colonization depends on its motility, which is facilitated by the helical shape of the bacterium. In H. pylori, cross-linking relaxation or trimming of peptidoglycan muropeptides affects the helical cell shape. Csd4 has been identified as one of the cell shape-determining peptidoglycan hydrolases in H. pylori. It is a Zn2+-dependent d,l-carboxypeptidase that cleaves the bond between the γ-d-Glu and the mDAP of the non-cross-linked muramyltripeptide (muramyl-l-Ala-γ-d-Glu-mDAP) of the peptidoglycan to produce the muramyldipeptide (muramyl-l-Ala-γ-d-Glu) and mDAP. Here, the crystal structure of H. pylori Csd4 (HP1075 in strain 26695) is reported in three different states: the ligand-unbound form, the substrate-bound form and the product-bound form. H. pylori Csd4 consists of three domains: an N-terminal d,l-carboxypeptidase domain with a typical carboxypeptidase fold, a central β-barrel domain with a novel fold and a C-terminal immunoglobulin-like domain. The d,l-carboxypeptidase domain recognizes the substrate by interacting primarily with the terminal mDAP moiety of the muramyltripeptide. It undergoes a significant structural change upon binding either mDAP or the mDAP-containing muramyltripeptide. It it also shown that Csd5, another cell-shape determinant in H. pylori, is capable of interacting not only with H. pylori Csd4 but also with the dipeptide product of the reaction catalyzed by Csd4.
1. Introduction
Helicobacter pylori infects approximately half of the world’s population. Infection by H. pylori can cause a variety of upper gastrointestinal disorders such as chronic gastritis, gastric and duodenal ulcers, gastric adenocarcinoma and gastric mucosa-associated lymphoid tissue (MALT) lymphoma (Kusters et al., 2006 ▶). A triple therapy consisting of a proton-pump inhibitor such as omeprazole and the antibiotics clarithromycin and amoxicillin (or metronidazole) is effective in the eradication of H. pylori in most cases. However, increasing antibiotic resistance requires new therapies and the discovery of new antibiotics (Malfertheiner et al., 2012 ▶).
High motility of H. pylori is required for its colonization of the human stomach and allows the bacterium to survive in its preferred niche, the gastric mucosa (Ottemann & Lowenthal, 2002 ▶). The helical cell shape of H. pylori is thought to facilitate efficient colonization of the viscous epithelial mucus layer via a corkscrewing mechanism (Berg & Turner, 1979 ▶). Several cell-shape mutants that have lost the helical twist and/or curvature exhibit attenuated colonization (Wyckoff et al., 2012 ▶). The peptidoglycan layer of the bacterial cell wall not only withstands the turgor pressure as a stress-bearing structure but also maintains the cell shape (Vollmer & Bertsche, 2008 ▶; Sycuro et al., 2010 ▶). The essential component of peptidoglycan consists of a linear polysaccharide chain, which is a polymer of alternating N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM) disaccharides, with a pentapeptide linked to NAM (Vollmer, Blanot et al., 2008 ▶). In H. pylori the pentapeptide sequence is l-Ala1-γ-d-Glu2-mDAP3-d-Ala4-d-Ala5, where mDAP refers to meso-2,6-diaminopimelate. The Helicobacter peptidoglycan layer is cross-linked exclusively by 4→3 linkages (cross-links from the side chain of mDAP3 from one strand to the main-chain d-Ala4 of another; Costa et al., 1999 ▶) to form a mesh-like structure termed peptidoglycan sacculus (Meroueh et al., 2006 ▶).
In many bacteria, the peptidoglycan layer is remodelled by a number of cell-wall hydrolases as well as synthetases for peptidoglycan maturation, regulation of cell-wall growth, cell division, peptidoglycan turnover and recycling, cell lysis and the release of peptidoglycan fragments for host–pathogen interactions (Vollmer, Joris et al., 2008 ▶). In H. pylori, seven cell shape-determining genes have been identified to date: csd1, csd2, csd3/hdpA, ccmA, csd4, csd5 and csd6 (Sycuro et al., 2010 ▶, 2012 ▶, 2013 ▶; Bonis et al., 2010 ▶; Frirdich et al., 2012 ▶). Peptidoglycan cross-linking relaxation by these proteins is critical for the helical cell shape of H. pylori and for stomach colonization. H. pylori Csd1, Csd2 and Csd3/HdpA proteins belong to the M23B/LytM family of metallopeptidases and show d,d-endopeptidase (d,d-EPase) activity (Sycuro et al., 2010 ▶; Bonis et al., 2010 ▶). Csd3/HdpA has an additional d,d-carboxypeptidase (d,d-CPase) activity (Bonis et al., 2010 ▶). CcmA does not encode a putative enzyme and CcmA-like bactofilins form cytoplasmic filaments that bind a peptidoglycan-synthesis enzyme for localized activity (Koch et al., 2011 ▶; Kühn et al., 2010 ▶).
Csd4 is a Zn2+-dependent d,l-carboxypeptidase (d,l-CPase) which cleaves the γ-d-Glu2-mDAP3 bond of the non-cross-linked peptidoglycan muramyltripeptide (muramyl-l-Ala1-γ-d-Glu2-mDAP3) to produce the corresponding muramyldipeptide (muramyl-l-Ala-γ-d-Glu) and mDAP. On the other hand, Csd5 is a putative scaffolding protein that appears to be unique to H. pylori and closely related bacterial species (Sycuro et al., 2012 ▶). The deletion mutant of csd4 (Δcsd4) loses nearly all curvature, yielding a straight rod, despite having a normal growth rate (Sycuro et al., 2012 ▶). The curvature-inducing function of Csd4 does not depend on the M23B/LytM EPase activity of Csd1–Csd3 (Sycuro et al., 2012 ▶). The Δcsd4 mutant with the straight rod cell shape shows impaired stomach colonization (Sycuro et al., 2012 ▶). A Csd4 homologue in Campylobacter jejuni, Pgp1, exhibits a similar Zn2+-dependent d,l-CPase activity against the monomeric disaccharide tripeptide substrate and promotes the helical rod shape of this organism (Frirdich et al., 2012 ▶). It has 41% sequence identity and 60% similarity to H. pylori Csd4. These studies suggest that the d,l-CPase activity of Csd4 (or of its close homologues) is vital for generating the proper cell shape and colonization of helically shaped bacteria. In addition, Csd5 was identified as another cell-shape determinant (Sycuro et al., 2012 ▶) which does not contain any known enzymatic domain but does contain a bacterial SH3 domain that could allow interactions with other peptidoglycan peptidases or with peptidoglycan itself. It was suggested that Csd4 and Csd5 act at a similar stage of helical cell-shape specification (Sycuro et al., 2012 ▶). The csd4csd5 double mutant resembled the csd4 mutant both in global peptidoglycan changes and in having a straighter shape than the csd5 mutant (Sycuro et al., 2012 ▶), suggesting that Csd5 acts downstream of Csd4.
Despite the importance of these cell shape-regulating proteins of H. pylori in promoting stomach colonization for its survival, no structural studies on them have been reported. To provide the structural basis for understanding the molecular function of H. pylori Csd4, we have determined its crystal structure in three different states: (i) a ligand-free form (Csd4-unbound), (ii) a substrate-bound form (Csd–muramyltripeptide) complexed with the synthetic peptidoglycan fragment NAM-tripeptide (NAM-l-Ala-γ-d-Glu-mDAP) and (iii) a product-bound form (Csd4–mDAP) complexed with mDAP, one of the two products. H. pylori Csd4 consists of three domains: an N-terminal CPase domain (residues 22–267), a central β-barrel domain with a novel fold (residues 268–340) and a C-terminal immunoglublin-like domain (residues 341–438). Our structures reveal that the CPase domain of H. pylori Csd4 primarily recognizes the terminal mDAP moiety of the muramyltripeptide substrate and undergoes a significant structural change upon binding either mDAP or the mDAP-containing tripeptide. We also show that H. pylori Csd5 directly interacts with H. pylori Csd4 and with the muramyldipeptide generated by the enzymatic activity of Csd4. The structures disclosed herein could serve as a foundation for the discovery of novel inhibitors that would prove helpful in fighting infections by drug-resistant H. pylori.
2. Materials and methods
2.1. Protein expression and purification
The N-terminal 19 residues of Csd4 (HP1075) from H. pylori strain 26695 are predicted to form a signal peptide by the SignalP 4.1 server (Petersen et al., 2011 ▶). Therefore, we PCR-amplified the gene covering residues Met22–Val438 of Csd4 and cloned it into the pET-28a(+) vector (Novagen) using NdeI and XhoI restriction enzymes. The resulting recombinant Csd4 protein is fused with a His6-containing tag (MGSSHHHHHHSSGLVPRGSH) at its N-terminus. It was expressed in Escherichia coli Rosetta2(DE3) cells using Luria Broth culture medium. Protein expression was induced using 0.5 mM isopropyl β-d-1-thiogalactopyranoside and the cells were incubated for an additional 15 h at 30°C following growth to mid-log phase at 37°C. The cells were lysed by sonication in buffer A (50 mM Tris–HCl pH 7.9, 500 mM sodium chloride, 50 mM imidazole) containing 5%(v/v) glycerol and 1 mM phenylmethylsulfonyl fluoride. The crude lysate was centrifuged at 36 000g for 1 h. The supernatant was applied onto a HiTrap Chelating HP affinity-chromatography column (GE Healthcare) which had previously been equilibrated with buffer A. Upon eluting with an imidazole gradient in the same buffer, the Csd4 protein eluted at 250–300 mM imidazole concentration. The eluted protein was diluted fivefold with buffer B (20 mM sodium phosphate pH 6.0) and was applied onto a HiTrap SP HP column (GE Healthcare) which had previously been equilibrated with buffer B containing 100 mM sodium chloride. Upon eluting with a gradient of sodium chloride in buffer B, the protein was eluted at 450–500 mM sodium chloride concentration. The eluted protein was further purified using a HiLoad XK-16 Superdex 200 column (GE Healthcare) which had previously been equilibrated with 20 mM Tris–HCl pH 7.0, 200 mM sodium chloride. Selenomethionine (SeMet)-substituted protein was overexpressed and purified essentially as above except that M9 cell-culture medium was used.
The N-terminal 40-residue-truncated construct of Csd5 (HP1250) from H. pylori strain 26695 was also designed based on prediction of the transmembrane region (Pro15–Val37). The gene covering residues Ser41–Glu192 was amplified and cloned into the pET-28b(+) vector (Novagen) using NdeI and XhoI restriction enzymes, producing the recombinant Csd5 protein with a His6-containing tag (MGSSHHHHHHSSGLVPRGSHM) at its N-terminus. The Csd5 protein was overexpressed and purified essentially as above, except that the buffer used for gel filtration was 10 mM HEPES pH 7.5, 100 mM sodium chloride.
2.2. Crystallization and structure determination
Fractions containing the recombinant Csd4 were pooled and concentrated to 8.3 mg ml−1 for crystallization. All crystals of Csd4 (including SeMet-labelled Csd4) were grown by the sitting-drop vapour-diffusion method at 23°C by mixing 0.4 µl each of the protein solution and a reservoir solution consisting of 200 mM calcium chloride, 100 mM HEPES pH 7.5, 25%(w/v) polyethylene glycol 3350. We could only obtain crystals in the presence of calcium ions in the reservoir. Rod-shaped crystals grew to 0.2 × 0.1 × 0.1 mm in a week. Serendipitously, we obtained crystals of uncomplexed Csd4 (Csd4-unbound) and of Csd4 bound to mDAP (Csd4–mDAP) under the same conditions. It appeared that mDAP was present in E. coli at a sufficient concentration and bound to Csd4 in a minor fraction of the crystals even when we did not intentionally add mDAP during the purification and crystallization steps. For data collection of Csd4-unbound and Csd4–mDAP, crystals were dipped into reservoir solution containing 20%(v/v) glycerol for 1–2 min and were flash-cooled using a cold nitrogen-gas stream at 100 K. To obtain crystals of the Csd4–muramyltripeptide complex, crystals of native Csd4 were dipped for 90 s into 1.5 µl reservoir solution supplemented with 20%(v/v) glycerol and 50 mM NAM-tripeptide and were flash-cooled in a cold nitrogen-gas stream at 100 K for data collection. The NAM-tripeptide was synthesized as described previously (Lee et al., 2009 ▶).
Single-wavelength anomalous diffraction (SAD) data were collected to 1.60 Å resolution from a crystal of SeMet-substituted Csd4 (Table 1 ▶). X-ray data for Csd4-unbound, the Csd4–muramyltripeptide complex and the Csd4–mDAP complex were collected to 1.60, 1.55 and 1.41 Å resolution, respectively (Table 1 ▶). Raw X-ray diffraction data were processed and scaled using the HKL-2000 program suite (Otwinowski & Minor, 1997 ▶). SAD phases were calculated with AutoSol from the PHENIX software package (Adams et al., 2010 ▶) and were further improved by the automatic model-building program RESOLVE (Terwilliger, 2003 ▶) to build an initial model. The model of SeMet-substituted Csd4 was completed using iterative cycles of model building with Coot (Emsley et al., 2010 ▶) and refinement with REFMAC5 from the CCP4 suite (Murshudov et al., 2011 ▶; Winn et al., 2011 ▶). The structures of Csd4-unbound, Csd4–mDAP and Csd4–muramyltripeptide were determined by molecular replacement with MOLREP (Vagin & Teplyakov, 2010 ▶) using the structure of the SeMet-substituted Csd4. Stereochemistry of the refined models was evaluated using MolProbity (Chen et al., 2010 ▶). Data-collection and refinement statistics are given in Table 1 ▶.
Table 1. Statistics of data collection and refinement.
Values in parentheses are for the highest resolution shell.
| Data set | Csd4-unbound | Csd4muramyltripeptide | Csd4mDAP | SeMet (peak) |
|---|---|---|---|---|
| Data collection | ||||
| Beamline source† | PF BL-17A | PLS BL-7A | PLS BL-7A | PLS BL-7A |
| X-ray wavelength () | 1.0722 | 0.9800 | 1.2823 | 0.9795 |
| Space group | P212121 | P212121 | P212121 | P212121 |
| a, b, c () | 53.14, 66.55, 144.05 | 53.13, 66.38, 143.79 | 52.93, 66.67, 144.00 | 53.17, 66.57, 144.39 |
| Resolution range () | 30.01.60 (1.631.60) | 50.01.55 (1.581.55) | 50.01.41 (1.431.41) | 30.01.60 (1.631.60) |
| Total No. of reflections | 686326 (33531) | 375793 (19100) | 475587 (19787) | 1655884 (81493)‡ |
| No. of unique reflections | 68532 (3387) | 71540 (3537) | 94978 (4497) | 131002 (6572)‡ |
| Multiplicity | 10.0 (9.9) | 5.3 (5.4) | 5.0 (4.4) | 12.6 (12.4)‡ |
| Completeness (%) | 99.9 (100.0) | 95.7 (96.1) | 95.7 (91.9) | 100.0 (100.0)‡ |
| I/(I) | 47.0 (5.7) | 31.5 (4.1) | 36.6 (2.1) | 39.5 (4.6)‡ |
| Wilson B factor (2) | 27.0 | 24.3 | 25.1 | 24.9 |
| R merge § (%) | 7.0 (53.1) | 8.5 (58.9) | 7.7 (75.8) | 10.1 (68.7)‡ |
| R r.i.m. ¶ (%) | 7.4 (56.0) | 9.5 (65.2) | 8.5 (85.5) | 10.5 (71.6)‡ |
| R p.i.m. †† (%) | 2.3 (17.7) | 4.0 (27.3) | 3.7 (38.6) | 3.0 (20.2)‡ |
| SAD phasing | ||||
| Figure of merit (before/after density modification) | 0.39/0.64 | |||
| Model refinement | ||||
| PDB code | 4q6m | 4q6n | 4q6o | |
| No. of Csd4 monomers in asymmetric unit | 1 | 1 | 1 | |
| Resolution range () | 20.01.60 | 20.01.55 | 20.01.41 | |
| R work/R free ‡‡ (%) | 19.4/22.0 | 20.0/22.7 | 18.9/21.2 | |
| No. of non-H atoms | ||||
| Total | 3812 | 3860 | 4114 | |
| Protein | 3366 | 3366 | 3366 | |
| Water O | 437 | 446 | 708 | |
| Glycerol | 6 | 18 | 24 | |
| Calcium ion | 3 | 3 | 3 | |
| Muramyltripeptide | 27 | |||
| mDAP | 13 | |||
| Average B factor (2) | ||||
| Overall | 20.2 | 19.6 | 23.2 | |
| Protein | 18.9 | 18.1 | 19.9 | |
| Water O | 30.1 | 29.8 | 38.0 | |
| Glycerol | 39.5 | 39.9 | 48.5 | |
| Calcium ion | 16.3 | 17.5 | 2.3 | |
| Muramyltripeptide | 30.1 | |||
| mDAP | 19.9 | |||
| R.m.s. deviations from ideal geometry | ||||
| Bond lengths () | 0.009 | 0.008 | 0.007 | |
| Bond angles () | 1.35 | 1.32 | 1.31 | |
| R.m.s. Z-scores§§ | ||||
| Bond lengths | 0.435 | 0.404 | 0.339 | |
| Bond angles | 0.618 | 0.596 | 0.596 | |
| Ramachandran plot¶¶ (%) | ||||
| Favoured/outliers | 97.4/0.0 | 97.6/0.0 | 97.7/0.0 | |
| Poor rotamers¶¶ (%) | 0.3 | 0.8 | 1.0 | |
PF, Photon Factory, Japan; PLS, Pohang Light Source, Republic of Korea.
Friedel pairs were treated as separate observations.
R
merge = 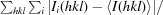
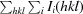 , where I(hkl) is the intensity of reflection hkl,
, where I(hkl) is the intensity of reflection hkl, 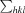 is the sum over all reflections and
is the sum over all reflections and 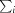 is the sum over i measurements of reflection hkl.
is the sum over i measurements of reflection hkl.
R
r.i.m. = 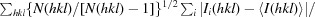 .
.
R
p.i.m. = 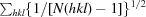
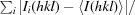 .
.
R
work = 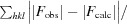
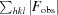 , where R
free is calculated for a randomly chosen 5% of reflections which were not used for structure refinement and R
work is calculated for the remaining reflections.
, where R
free is calculated for a randomly chosen 5% of reflections which were not used for structure refinement and R
work is calculated for the remaining reflections.
Values were obtained using REFMAC.
Values were obtained using MolProbity.
2.3. Identification of metal-binding sites by anomalous scattering
To test whether the metal-binding sites can be occupied by zinc ions, crystals of Csd4–mDAP were soaked for 80 s in 3 µl of a solution consisting of 200 mM sodium chloride, 100 mM HEPES pH 7.5, 25%(w/v) polyethylene glycol 3350, 2 mM EDTA, 20%(v/v) glycerol and then soaked for 80 s in 3 µl of a Zn2+-containing solution [100 mM zinc chloride, 100 mM sodium chloride, 100 mM HEPES pH 7.5, 25%(w/v) polyethylene glycol 3350, 20%(v/v) glycerol]. Two sets of SAD data (Zn1 and Zn2 in Supplementary Table S11) were collected from two different crystals at 100 K using an X-ray wavelength of 1.2823 Å on beamline 7A of Pohang Light Source. Raw data were processed and scaled using HKL-2000. Anomalous difference maps were calculated using FFT from the CCP4 suite (Read & Schierbeek, 1988 ▶; Winn et al., 2011 ▶). The two Zn-substituted Csd4 structures are essentially identical to the Csd4–mDAP structure in their overall structures. They contain different sets of Zn2+ and Ca2+ ions, possibly owing to different experimental conditions of EDTA treatment and Zn2+ substitution. The Zn1 model contains eight Zn2+ ions and one Ca2+ ion; the Ca2+ ion in the active site and the Ca2+ ion that stabilizes the β11–α7 loop are replaced by Zn2+. The Zn2 model has two Zn2+ and two Ca2+ ions; the Ca2+ ion bound to the C-terminal Ig-like domain is replaced by Zn2+. Other Zn2+ ions present in the Zn1 and Zn2 models are likely to be owing to nonspecific binding of Zn2+ ions by Csd4. Nonspecific binding of Zn2+ ions by proteins was exploited in solving the phase problem by Zn anomalous scattering (Cha et al., 2012 ▶).
2.4. Mass spectrometry
Mass spectra were acquired using a matrix-assisted laser desorption/ionization quadrupole ion-trap time-of-flight mass spectrometer (MALDI-QIT-TOF MS, AXIMA QIT; Shimadzu/Kratos, Manchester, England) equipped with a nitrogen laser (337 nm, 3 ns pulse width, maximum pulse rate 10 Hz). Mass spectra were obtained in the mass range (in Da) from 500 to 2000 m/z in positive-ion mode. Helium was used for trapping and cooling ions in the ion source. The pressure in the trap was held at 0.53 Pa. Each MS spectrum was constituted of an average of 200 profiles. All spectra were externally calibrated with bradykinin (m/z 757.3992), angiotensin II (m/z 1046.5418), angiotensin I (m/z 1296.6848), Glu-fibrinopeptide B (m/z 1570.6768) and N-acetyl renin substrate (m/z 1800.9432) in TOFMix (Shimadzu, Japan). Acquisition and data processing were controlled by the LaunchPad software. The analyte, NAM-tripeptide at 5 mM, was incubated with the recombinant Csd4 protein (5 µM) in the presence or absence of 2.5 mM metal ion (Zn2+, Ca2+ or Mn2+) dissolved in 10 mM Tris–HCl pH 7.9. A sample solution (1 µl) was mixed on the target with a fresh saturated matrix solution of 2,5-dihydroxybenzoic acid dissolved in 0.1%(v/v) trifluoroacetic acid and 50%(v/v) acetonitrile. For sample deposition, a 384-position stainless-steel sample plate was used.
2.5. Surface plasmon resonance
Direct binding of Csd5 to Csd4 was assessed using a Reichert SR7500 surface plasmon resonance (SPR) dual-channel instrument (Reichert, Depew, New York, USA). Purified Csd4 in 20 mM sodium acetate pH 5.5 was immobilized using standard amino coupling at 10 µl min−1 on a carboxymethyl dextran hydrogel surface sensor chip (Reichert, Depew, New York, USA) until saturation was achieved. The running buffer C used in all SPR experiments was 10 mM HEPES pH 7.5, 100 mM sodium chloride. SPR experiments were performed at 25°C. Csd5 (0.3, 0.5, 1.0 and 2.0 µM) was injected over the Csd4 chip at 30 µl min−1 for 4 min for association analyses. Subsequently, the running buffer was flowed over the chip for an additional 4 min (30 µl min−1) for molecular-dissociation analyses. To remove metal ions that might be intrinsically bound to Csd4, the Csd4-immobilized chip was treated with running buffer C containing 20 mM EDTA, the running buffer C was flowed over the chip and Csd5 was injected over the Csd4 chip, showing no binding signal. Subsequently, running buffer C containing 5 mM calcium chloride was passed over the immobilized Csd4, which recovered the Csd4–Csd5 binding. Regeneration of the chip was carried out using 20 mM sodium hydroxide solution. Binding was detected as a change in the refractive index at the surface of the chip as measured in response units (RU). A reference flow cell was used to record the response by bovine serum albumin (BSA) as a positive control, and the response by BSA was subtracted from each sample. SPR data were fitted using the Scrubber2 software.
2.6. Fluorescence polarization binding assay
The FITC-labelled dipeptide (l-Ala-d-Glu), synthesized by Anygen Co. Ltd, Republic of Korea, was dissolved in and diluted with the binding buffer (10 mM HEPES pH 7.5, 100 mM sodium chloride) to 9.2 nM equally in each 100 µl reaction well. The recombinant Csd5 was also serially diluted in the binding buffer and mixed into each reaction well at concentrations ranging from 17.0 nM to 17.4 µM. To detect the change in light polarization of the FITC-labelled dipeptide, fluorescence measurements were made in a 96-well format plate reader (Molecular Devices) with excitation and emission wavelengths of 485 and 538 nm, respectively. A nonlinear graph of dipeptide concentration-dependent polarization was produced using Prism 6 (GraphPad).
2.7. Accession codes
The coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 4q6m, 4q6n and 4q6o for the Csd4-unbound, Csd4–muramyltripeptide and Csd4–mDAP structures, respectively, and 4q6p and 4q6q for the Zn-bound forms of Csd4.
3. Results and discussion
3.1. Structure determination of H. pylori Csd4
We have solved the crystal structure of H. pylori Csd4 using Se-SAD data (Table 1 ▶). Subsequently, we have refined three models of H. pylori Csd4: ligand-free (Csd4-unbound), substrate-bound (Csd4–muramyltripeptide) and product-bound (Csd4–mDAP) structures (Table 1 ▶). All of our Csd4 crystals contain one Csd4 monomer in the asymmetric unit. These models account for all of the amino-acid residues of the recombinant Csd4 (residues Met22–Val438) except for the N-terminal affinity tag (Fig. 1 ▶ a). The peptide portion of the bound NAM-tripeptide and mDAP as well as three metal ions bound to Csd4 are well defined by the electron density. Since the crystallization reservoir solution contained calcium chloride, we have assigned the three metal ions as Ca2+. Metallopeptidases utilize a catalytic Zn2+ bound to the active site. Therefore, our crystallized enzyme with a Ca2+ ion in the active site is a noncompetent variant. Further anomalous scattering experiments confirmed that these calcium ions could be substituted by zinc ions (Supplementary Fig. S1 and Supplementary Table S1).
Figure 1.

Overall structure of H. pylori Csd4. (a) Ribbon diagram of the Csd4-unbound structure. β-Strands, α-helices, 310-helices and loops are shown in cyan, red, yellow and grey, respectively. Three calcium ions are shown as purple spheres. The metal-binding residues in the N-terminal CPase domain are shown as stick models. The blue and black boxes indicate the previously unknown Ca2+-binding sites of the CPase domain and the Ca2+-binding motif of the C-terminal Ig-like domain. The secondary-structure elements were defined by DSSP (Kabsch & Sander, 1983 ▶). (b) Electrostatic surface diagram of Csd4-unbound. The signal peptide is modelled as a black ribbon diagram. The black and cyan dotted boxes indicate the active-site cleft of the CPase domain and the Ca2+-binding channel of the Ig-like domain. The grey box indicates the highly positively charged surface of the central β-barrel domain. Electrostatic potential at the molecular surface was calculated using APBS (Baker et al., 2001 ▶). (c) Ribbon diagram of the Ca2+-binding motif (left) and the surface representation of the Ca2+-bound channel (right) in the Ig-like domain. The OMIT mF o − DF c map (contoured at 10σ) for the calcium ion is coloured green. The Ca2+-binding residues are shown as stick models. The calcium ion and the bound waters are shown as purple and red spheres, respectively. All figures representing the protein structure were drawn using PyMOL (v.1.3r1; Schrödinger).
The three models are highly similar to each other, with r.m.s. deviations of 0.17–0.38 Å for pairwise comparisons over 417 Cα-atom pairs. Interestingly, large Cα deviations occur in the β9–α5 loop, with values of 4.2 and 3.7 Å for Asp200 and Glu202, respectively (Supplementary Fig. S2). This region is well defined by the electron density in all three models and the observed large structural difference is induced by ligand binding, as discussed in more detail below. Csd4 exists as monomers in the crystals, as the largest surface areas buried at the interface between two neighbouring monomers within the crystals are 502, 467 and 481 Å2 per monomer (2.7, 2.5 and 2.6% of the monomer surface area) for the Csd4-unbound, Csd4–muramyltripeptide and Csd4–mDAP structures, respectively. This conclusion from the crystallographic data is also supported by the results of size-exclusion chromatography of the protein in solution.
3.2. Three-domain structure of H. pylori Csd4
H. pylori Csd4 consists of three domains: an N-terminal CPase domain (Met22–Asn267), a central β-barrel domain (Asp268–Glu340) with a novel fold and a C-terminal immunoglobulin (Ig)-like domain (Phe341–Val438) (Fig. 1 ▶ a). The three domains are arranged in a serial order to form a curved overall structure (Fig. 1 ▶ b, left panel). When we performed DALI searches (Holm & Rosenström, 2010 ▶) with individual domains, only the N-terminal CPase domain showed significant structural similarity to other known proteins, as described below. A DALI search with the whole chain of Csd4 gave essentially the same result as with the N-terminal CPase domain only.
The N-terminal CPase domain with a CPase fold has a central seven-stranded β-sheet (β1↑–β4↓–β2↓–β5↓–β10↓–β8↑–β9↑), which is highly twisted and spans the entire CPase domain from top to bottom (Fig. 1 ▶ a). This gives rise to a concave face, which accommodates helices α3 and α5 and a short β-sheet (β6↑–β7↑) as well as the active-site cleft containing a metal ion (Figs. 1 ▶ a and 2 ▶ a). The convex side of the sheet harbours four helices (α1, α4, α6 and α7) and a short β-sheet (β3↑-β11↑) which contributes to another metal-binding site, thereby stabilizing the β11–α7 loop (indicated by a blue box in Fig. 1 ▶ a and Supplementary Fig. S1b). The N-terminal domain of Csd4 has a canonical metallo-CPase-like architecture. It shows structural similarity to members of the CPase B protein family such as the activated porcine pancreatic carboxypeptidase B (CPase B) as a surrogate of human TAFIa (thrombin-activatable fibrinolysis inhibitor; Bunnage et al., 2007 ▶; PDB entry 2jew; Z-score of 18.6, r.m.s. deviation of 2.2 Å and sequence identity of 15% for 185 equivalent Cα positions) and human CPase A4 (Pallarès et al., 2005 ▶; PDB entry 2bo9; Z-score of 17.3, r.m.s. deviation of 2.5 Å and sequence identity of 14% for 187 equivalent Cα positions).
Figure 2.

Metal-dependency of H. pylori Csd4 as a carboxypeptidase. (a) Ribbon diagram of the active site in the Csd4-unbound structure coloured as in Fig. 1 ▶(a). The OMIT mF o − DF c map (contoured at 5σ) for the calcium ion is coloured green. (b–e) Mass spectra of muramylpeptides after the reaction catalyzed by Csd4 in the presence of EDTA (b) or in the presence of Zn2+ (c), Ca2+ (d) or Mn2+ (e). Green and blue arrows indicate the peaks corresponding to the muramyldipeptide and muramyltripeptide (Tri), respectively.
The central β-barrel domain appears to have a novel fold, because the highest Z-score was only 3.3 with RAS-related C3 botulinum toxin substrate 1 (Grizot et al., 2001 ▶; PDB entry 1hh4, chain D). It comprises two antiparallel β-sheets: a major twisted sheet (β16↓–β17↑–β18↓–β14↓) and a minor sheet (β15↑–β12↓–β13↓). Interestingly, the central β-barrel domain has a highly positively charged surface (shown by a grey box in Fig. 1 ▶ b), contributing to a high overall isoelectric point (pI) of the protein (theoretically 9.0). The theoretical pI of the central β-barrel domain is 10.0, while those of the N- and C-terminal domains are 6.7 and 8.1, respectively.
The overall fold of the C-terminal Ig-like domain containing two β-sheets (β20↑–β19↓–β22↑–β24↓ and β21↓–β26↓–β25↑-β23↓) resembles an Ig fold, even though the structural similarity is rather low. The highest similarity was detected in the Ig-like Y_Y_Y domain of the Bacteroides thetaiotaomicron histidine kinase (Lowe et al., 2012 ▶; PDB entry 4a2l; Z-score of 5.2, r.m.s. deviation of 2.9 Å for 78 equivalent Cα positions and sequence identity of 13%). However, the key residues (three Tyr residues) of the Y_Y_Y domain are not well conserved in the C-terminal domain of Csd4; the equivalent residues are Arg380, Asn382 and Tyr419. The C-terminal domain of Csd4 might be a member of a new subfamily of the Ig-like fold superfamily (Pfam CL0159). The Ig-like domain of Csd4 is decorated with several winding loops, including the β2–β25 loop, and two additional 310-helices. Unexpectedly, these structural elements form a Ca2+-binding channel (Fig. 1 ▶ c, right panel). The OMIT electron-density map of Csd4-unbound clearly indicates that a calcium ion is bound to the C-terminal Ig-like domain and is coordinated by six ligand atoms, i.e. the side-chain atoms of the highly conserved Asn382 (located on β23) and Glu396 (located on the β23–β24 loop) and the main-chain carbonyl O atoms of Val383 and Phe386 (Fig. 1 ▶ c, left panel; Supplementary Fig. S1c). The surface of the calcium ion-bound channel in the C-terminal Ig-like domain (Fig. 1 ▶ c, right panel) is lined with the β23–β24 and β24–β25 loops and strands β23 and β25. It is further contributed to by the long side chains of residues Asn291, His292, Phe337 and Ile339 from strands β14 and β18 of the central β-barrel domain (Fig. 1 ▶ c, right panel).
3.3. Unique features of the Csd4 d,l-carboxypeptidase domain
The N-terminal CPase domain of Csd4 has a highly twisted β-sheet, forming an active-site cleft on a concave face (indicated by a black dotted box in Fig. 1 ▶ b). The access to the active-site cleft is reminiscent of a funnel, the rim of which is formed by four surface loops (α2–α3, β2–α1, β7–β8 and β9–α5). In the Csd4-unbound structure, which represents the substrate-free state, a metal ion is bound to the active-site cleft with a broad entrance (Fig. 2 ▶ a). It was reported that Csd4 requires divalent metal ions to cleave the peptide bond (d-Glu2-mDAP3) of a disaccharide-tripeptide (NAG-NAM-l-Ala1-γ-d-Glu2-mDAP3; Sycuro et al., 2012 ▶). We tested the CPase activity of Csd4 with the synthetic NAM-tripeptide as the substrate in the presence of Zn2+, Ca2+ and Mn2+. Based on the mass analysis, we found that Csd4 trims the NAM-tripeptide to the NAM-dipeptide (NAM-l-Ala1-γ-d-Glu2) in the presence of any one of these metal ions, releasing mDAP (Figs. 2 ▶ c–2 ▶ e). The highest activity was observed with Zn2+. No such reaction was observed in the presence of EDTA (Fig. 2 ▶ b). Clear electron density is present at the metal-binding site of the CPase domain in our Csd4-unbound (Fig. 2 ▶ a), Csd4–muramyltripeptide and Csd4–mDAP structures. We interpreted it as Ca2+ because the crystallization condition contained a high concentration of Ca2+. We have confirmed this interpretation by collecting anomalous data at the Zn absorption edge (1.2823 Å). The anomalous difference map clearly showed that this metal ion is not Zn2+ in the Csd4-unbound crystal. Further anomalous scattering experiments on the crystal that was soaked in Zn2+ after EDTA treatment indicated that this metal ion can be replaced by Zn2+ (Zn1 data set in Table S1; Supplementary Fig. S1a). These results support that Csd4 employs a catalytic zinc ion in the active site like many other carboxy-metallopeptidases. Therefore, our crystallized enzyme with a calcium ion in the active site is a noncompetent variant.
The N-terminal CPase domain of Csd4 shows structural similarity to M14 metallopeptidases such as pig CPase B (PDB entry 2jew; Bunnage et al., 2007 ▶) and human CPase A4 (PDB entry 2bo9; Pallarès et al., 2005 ▶), both of which are members of the M14A subfamily. To date, four subfamilies (M14A–M14D) have been identified among the M14 metallopeptidase family (MEROPS peptidase database; Rawlings et al., 2014 ▶). Similarly to both human CPase B and CPase A4, Csd4 cleaves the C-terminal residue. However, despite its structural similarity to M14 metallopeptidases, Csd4 appears to be unique among M14 family members regarding substrate specificity and active-site residues, including the metal-binding motif, as discussed below in detail. We therefore propose that Csd4 belongs to a new metallo-CPase M14 subfamily distinct from the known M14 subfamilies M14A–M14D.
Firstly, Csd4 is active without proteolytic processing owing to the lack of a pro-domain for a zymogenic form (Figs. 2 ▶ c–2 ▶ e), whereas both pig CPase B and human CPase A4 require proteolytic cleavage of their precursor forms for activation (Bunnage et al., 2007 ▶; Pallarès et al., 2005 ▶). The substrate specificity of Csd4 is also different from those of the known M14 subfamilies: M14A subfamily CPases favour residues with aromatic or branched side chains and M14B and M14D subfamily CPases prefer basic amino acids. While Csd4 cleaves the peptide bond of d-Glu2-mDAP3 derived from peptidoglycan with concurrent recognition of its C-terminus, M14C family members carry out hydrolysis at the same cleavage site of peptidoglycan but they behave as endopeptidases. In addition, the overall three-domain architecture of H. pylori Csd4, consisting of an N-terminal CPase domain, a central β-barrel domain and a C-terminal Ig-like domain, is conserved in Csd4 homologues from other bacteria (Fig. 3 ▶), but is absent in other CPases with known structures. Interestingly, Csd4 homologues are mainly found in spiral-shaped bacteria (Sycuro et al., 2012 ▶; Frirdich et al., 2012 ▶).
Figure 3.

Sequence alignment of four Csd4 homologues. Sequence alignment of Csd4 from H. pylori strain 26695 (HP_Csd4; SWISS-PROT accession code O25708), Pgp1 from Campylobacter jejuni (CJ_Pgp1; A1W0W1), Csd4 from Wolinella succinogenes (WS_Csd4; Q7MSQ3) and Csd4 from H. pylori strain J99 (JHP_Csd4; Q9ZM72) was performed and presented using ClustalX (Larkin et al., 2007 ▶) and ESPript (Gouet et al., 2003 ▶; http://espript.ibcp.fr). Red and blue triangles indicate the metal-binding site motif (Gln46, Glu49 and His128) and the two key Arg residues (Arg86 and Arg94) showing conformational changes in the active site of the CPase domain. Green circles indicate the Ca2+-binding motif in the C-terminal Ig-like domain.
Next, the CPase domain (Met22–Asn267) of H. pylori Csd4 shows relatively large r.m.s. deviations (2.2 Å for 185 equivalent Cα positions and 2.5 Å for 187 equivalent Cα positions, respectively) and low sequence conservation (sequence identities of 15 and 14%, respectively) when compared with pig CPase B and human CPase A4. The central β-sheet of the Csd4 CPase domain has essentially the same topology as in other CPases. However, it consists of only seven strands, lacking a β-strand equivalent to β0′′ of pig CPase B and human CPase A4. In addition, an extra α-helix (α0′′) is present at the N-terminus of pig CPase B and human CPase A4 (Supplementary Fig. S3). In the Csd4 structure this α-helix is missing and is substituted by the central β-barrel domain (Supplementary Fig. S3). Another α-helix of pig CPase B and human CPase A4 is replaced by a mini two-stranded β-sheet (β3 and β11) in Csd4. Large structural differences also exist in the loops around the active-site cleft. The four loops (α2–α3, β5–β8, β8–α4 and β9–α5) contribute to the unique shape of the Csd4 active-site cleft and determine its substrate specificity (Supplementary Fig. S3). The Csd4 β5–β8 loop contains an extra two-stranded β-sheet formed by β6–β7 that is absent in other CPases.
Finally, the metal-binding motif in the CPase domain of Csd4 differs from those in other CPases. In the Csd4-unbound, Csd4–muramyltripeptide and Csd4–mDAP structures a calcium ion is coordinated in an octahedral fashion by Gln46, Glu49 and His128 from the metal-binding motif (Q xx E x n H), which is strictly conserved in Csd4 homologues (Fig. 3 ▶), and three water molecules, with metal–ligand atom distances of 2.3–2.6 Å (Fig. 2 ▶ a). x stands for any amino acid and the strictly conserved residue is in bold. In contrast, most other known M14 metallo-CPases have a different metal-binding motif, H xx E x n H (Supplementary Fig. S3).
3.4. The Csd4–muramyltripeptide and Csd4–mDAP structures reveal the substrate-bound and product-bound states
To understand the structural basis for the specific interactions of Csd4 with the peptidoglycan-derived muramyltripeptide substrate and the product mDAP, we solved the Csd4–muramyltripeptide and Csd4–mDAP structures. mDAP is the C-terminal residue of the substrate muramyltripeptide and is one of the products produced by the reaction catalyzed by Csd4. Therefore, our Csd4–muramyltripeptide and Csd4–mDAP structures represent the substrate-bound and product-bound states prior to and after the cleavage reaction by Csd4, respectively. Clear electron densities were observed both for l-Ala1-γ-d-Glu2-mDAP3 in the Csd4–muramyltripeptide structure and for mDAP in the Csd4–mDAP structure (Fig. 4 ▶). In both structures, extensive interactions exist between mDAP and Csd4. They are highly conserved between the two structures, highlighting the importance of these interactions in determining substrate specificity.
Figure 4.

Ligand interactions in the Csd4–muramyltripeptide and Csd4–mDAP structures. (a, c) Ribbon diagrams of the active site in the Csd4–muramyltripeptide (a) and Csd4–mDAP (c) structures. The tripeptide and mDAP are shown as stick models. Since the sugar moiety of the NAM-tripeptide was invisible, only the tripeptide portion has been modelled into the electron-density map. Residues interacting with the tripeptide or mDAP are shown as stick models. The OMIT mF o − DF c maps for the tripeptide (contoured at 1.5σ) and mDAP (contoured at 2.5σ) are coloured green. The red and blue dotted lines indicate the interactions of the tripeptide or mDAP with the metal ion and two key Arg residues (Arg86 and Arg94), respectively. (b, d) Schematic diagrams of interactions of the bound muramyltripeptide (b) and mDAP (d) with Csd4. The red and grey labels represent electrostatic and hydrophobic residues interacting with these ligands, respectively.
In the Csd4–muramyltripeptide structure, the bulk of the interactions are centred around the scissile peptide bond, the mDAP moiety and the active-site metal ion (Figs. 4 ▶ a and 4 ▶ b). In this structure, the linear tripeptide part of the substrate lies antiparallel to the β5 strand of Csd4 (Fig. 4 ▶ a) and is surrounded by four loops (β2–α1, α2–α3, β7–β8 and β9–α5; Fig. 5 ▶ b). The sugar moiety is not well defined in the electron-density map, likely because it protrudes from the active site and does not make a specific interaction with the active-site residues. While the l-Ala1-d-Glu2 segment of the peptide is largely exposed to the solvent, it is well defined by the electron density as it makes specific interactions with Csd4. The mDAP3 part sits deep in the active-site cleft and is enclosed by the loops (Fig. 5 ▶ b and 5 ▶ c), thus making extensive interactions with Csd4. The main-chain carbonyl O atom of l-Ala1 makes a polar interaction with the side chain of Arg86 (Figs. 4 ▶ a and 4 ▶ b). The aliphatic portions of d-Glu2 and mDAP3 make nonpolar interactions with hydrophobic parts of the Trp148, Ile153, Met203, Ala220 and His128 side chains (Figs. 4 ▶ a and 4 ▶ b). The scissile carbonyl O atom of d-Glu2 interacts with the side chain of Arg86. The carboxylate group of d-Glu2 is bonded to Gly131N and a water molecule, which is hydrogen-bonded to Arg147. Notably, the terminal mDAP3 is involved in tight interactions with Csd4. The side-chain amine N atom of mDAP3 interacts with Met203δ and Thr208γ. The side-chain carboxylate O atoms of mDAP3 make contacts with Asn93δ2, His126δ1, His128∊2, Leu207N and Thr208N.
Figure 5.

Open and closed conformations of the flap. Electrostatic surface diagrams of the mobile flap (left panels) and surface diagrams showing the active-site cleft (right panels) in the Csd4-unbound (a), Csd4–muramyltripeptide (b) and Csd4–mDAP (c) structures.
The mDAP-dependency of selectivity is revealed in the Csd4–mDAP structure as well as in the Csd4–muramyltripeptide structure (Figs. 4 ▶ c and 4 ▶ d). The binding of mDAP alone induces ligand interactions similar to the case of Csd4–muramyltripeptide (Fig. 4 ▶). The shape of the active-site cleft of Csd4–mDAP is nearly identical to that of Csd4–muramyltripeptide; mDAP of Csd4–mDAP is well ensconced in the cleft (Figs. 4 ▶ b and 4 ▶ c), suggesting that the presence or absence of the mDAP segment is one of the key factors in determining the suitable shape of the active-site cleft for catalysis and is an important factor in substrate selectivity.
The Csd4–muramyltripeptide structure shows that the tripeptide moiety and the metal ion are suitably predisposed for interaction during the course of catalysis. The metal ion is coordinated by Gln46∊1, Glu49∊1,∊2 and His128δ1 and two water molecules (Wat1 and Wat2). However, a third water molecule (Wat3), one of the six metal-binding ligands in the Csd4-unbound structure, is substituted by the carbonyl O atom of the scissile peptide bond between d-Glu2 and mDAP3 in the Csd4–muramyltripeptide structure, thereby enabling a direct interaction with the metal ion to polarize the peptide carbonyl bond. The carbonyl O atom of the scissile peptide bond is also hydrogen-bonded to Arg86η1 with a distance of 2.8 Å to stabilize the transient oxyanion species of the polarized carbonyl bond (Fig. 4 ▶). This species is the product of the addition of Wat1 to the carbonyl carbon, which would be promoted by Glu222 as a general base. Furthermore, the scissile amide N atom interacts with the carboxyl group of Glu222∊1, rendering it susceptible to nucleophilic attack. The water molecule Wat1, interacting with both Glu222∊1,∊2 and the metal ion, is well positioned for nucleophilic attack at the carbonyl C atom of the scissile bond with a distance of 3.2 Å (Fig. 4 ▶). In addition, Csd4 is set to recognize the C-terminal carboxylate of mDAP3. The Csd4–muramyltripeptide structure shows that Arg94 is the key residue for recognition of the C-terminal mDAP residue of the substrate. The free C-terminal carboxylate group of mDAP3 is salt-bridged to Arg94η1 and Arg94η2 in a bidentate manner, accounting for the specificity of Csd4 as a ‘carboxy’ peptidase. The C-terminal carboxylate group is also bound to Asn93δ2 and the metal ion via the water Wat2, demonstrating that the metal ion serves to hold key elements for catalyzing the reaction.
3.5. Ligand binding modulates the conformation of the CPase domain
Among the three structures of Csd4, the β9–α5 loop region (residues Thr196–Ala206) containing two 310-helices exhibits the largest structural variation (Supplementary Fig. S2). This region is moved towards the active site in the Csd4–muramyltripeptide and Csd4–mDAP structures when we compare them with Csd4-unbound (Fig. 5 ▶, left panels). This flexible region works as a flap and undergoes a conformational change upon binding of the substrate or the product. In the substrate-free state of Csd4-unbound, the flap is positioned away from the active site (Fig. 5 ▶ a). Upon binding of the muramyltripeptide, it moves closer to the active site and its surface shape and electrostatic potential are altered (Fig. 5 ▶ b). As shown in Fig. 5( ▶ b), the flap forms the edge of the active-site cleft and is responsible for recognition of the mDAP portion of the muramyltripeptide. In the Csd4–mDAP structure the flap has a similar conformation as in the Csd4–muramyltripeptide structure, indicating that the conformation of the mobile flap is primarily influenced by the mDAP moiety.
Depending on the ligand binding, we observe interesting differences in the orientation of the side chains of two key arginine residues on the α2–α3 loop, i.e. Arg86 and Arg94 (Fig. 5 ▶). Their side chains are well defined by a clear electron density. In the Csd4–muramyltripeptide structure, the side chain of Arg86 is directed towards the active site to interact with the O atom of the scissile peptide bond of the bound muramyltripeptide (Fig. 5 ▶ b). In comparison, it points away from the active site in the Csd4-unbound and Csd4–mDAP structures (Figs. 5 ▶ a and 5 ▶ c). Compared with Csd4-unbound and Csd4–mDAP, the Nη atom of Arg86 in Csd4–muramyltripeptide is moved by about 8 Å so that Arg86 is recruited for binding d-Glu2-mDAP3 within the substrate. Arg94, which is positioned between the active site and the mobile flap, plays an interesting role in the flap movement by interacting with both the free C-terminal carboxylate group of the mDAP part and Glu202 of the flap. The side chain of Arg94 interacts with the flap via a strong salt bridge with Glu202 in all three structures. It points towards the active site in both the Csd4–muramyltripeptide and Csd4–mDAP structures, whereas it points away from the active site in the Csd4-unbound structure (Fig. 5 ▶). In other words, Arg94 pulls the flap towards the active site via a strong salt bridge with Glu202 in both the Csd4–muramyltripeptide and Csd4–mDAP structures when its side chain recognizes the free C-terminal carboxylate group of mDAP. Furthermore, this structural change induces an additional interaction between the side-chain amine group of mDAP and Met203 (Fig. 4 ▶). Therefore, we suggest that Arg94 is the key determinant of selectivity in the ‘carboxy’-peptidase activity of Csd4.
The conformations of the flap (β9–α5 loop) and the two key Arg residues (Arg86 and Arg94) on the α2–α3 loop affect the shape and size of the active site, as they form the wall of the active-site cleft. Loops forming the surface of the active site define the front and the bottom of the active site and the specificity of the active-site pocket (Fig. 5 ▶, right panels). The Csd4-unbound structure reveals an open active-site cleft in the substrate-free state, thus exposing the metal ion and the inside of the cleft to the bulk solvent (Fig. 5 ▶ a). In this structure, the flap is in the open conformation and the two Arg residues point towards the outside of the active site, predisposing the enzyme to binding of the substrate. In the Csd4–muramyltripeptide structure the flap is in the closed conformation and the tripeptide moiety of the substrate fits nicely into the active site, thus interacting extensively with the loops around the active site (Fig. 5 ▶ b). In the Csd4–mDAP structure the flap is also in the closed conformation and the inner side of the active-site cleft is occupied by mDAP and is covered by the flap, while the outer side of the cleft is open towards the surface, exposing the metal site to the bulk solvent (Fig. 5 ▶ c).
Taken together, these findings enable us to propose a catalytic mechanism for H. pylori Csd4 as a d,l-CPase. After access through an open entrance to the active-site pocket (Fig. 5 ▶ a), the substrate carbonyl O atom replaces a metal-bound water molecule (Wat3) to interact with the metal ion directly. By abstracting a proton from a water molecule (Wat1), the active-site Glu222 promotes the water for nucleophilic attack of the sessile carbonyl, en route to formation of the reactive tetrahedral species. Glu222 shuttles the proton to the departing amide N atom of the sissile amide bond. The metal ion serves to stabilize Wat1 and the scissile carbonyl of the substrate. Arg86 is then recruited to interact with the scissile carbonyl O atom to both position the carbonyl O atom and to stabilize the tetrahedral species. In addition, Arg94 fixes the position of the C-terminal carboxylate of the bound substrate, inducing the flap movement to cover mDAP3. Protonation of the amide N atom is the committed step in bond scission leading to the two products.
3.6. Csd4 interacts physically with Csd5
Previously, the csd4csd5 double mutant was reported to resemble the csd4 mutant both in global peptidoglycan changes and by having a straighter shape than the csd5 mutant, suggesting that Csd4 acts upstream of Csd5 (Sycuro et al., 2012 ▶). This suggests that Csd4 may interact physically with Csd5, another cell-shape determinant. To date, no such interaction between Csd4 and Csd5 has been demonstrated. Therefore, we investigated whether Csd4 could interact directly with Csd5. The Csd4–Csd5 interaction was detected by SPR using immobilized Csd4 with Csd5 as the analyte at concentrations between 0.25 and 2.00 µM. The dissociation constant (K d) was calculated as ∼4.5 µM (Fig. 6 ▶ a). This shows that Csd5 can bind saturably to Csd4, which is indicative of an evolved function of the formation of a complex between the two proteins. Interestingly, the observed interaction between Csd4 and Csd5 disappeared upon treatment of the Csd4-immobilized chip with 20 mM EDTA to remove intrinsically bound metal ions (Fig. 6 ▶ b). Moreover, retreatment of the EDTA-treated Csd4-immobilized chip with 5 mM calcium chloride recovered the binding between the two proteins, yielding a similar K d value (∼4.3 µM; Fig. 6 ▶ b). Therefore, we conclude that Csd4 physically interacts with Csd5 in a Ca2+-dependent manner. Upon showing that Csd5 interacts physically with Csd4, we hypothesized that Csd5 may also interact with the peptidoglycan-derived dipeptide which is the product of the enzymatic reaction catalyzed by Csd4. An FITC-labelled dipeptide (FITC-l-Ala-d-Glu) was synthesized and was used to carry out fluorescence polarization experiments. The FITC-labelled dipeptide was found to bind to Csd5 with a K d value of ∼1.69 µM (Fig. 6 ▶ c). Therefore, we suggest that Csd5 is capable of recognizing both Csd4 and the dipeptide product of Csd4 downstream of the transformation catalyzed by Csd4.
Figure 6.

Biophysical studies of the Csd4–Csd5 or the Csd5–dipeptide interaction in solution. (a) SPR experiments with immobilized Csd4 and Csd5 as an analyte at different concentrations (0.25, 0.50, 1.00 and 2.00 µM) are shown as grey-coloured traces. Black traces show the corresponding binding-model curves. (b) At the same concentration of Csd5 (2.00 µM), the interaction between Csd4 and Csd5 (‘no treatment’) was lost upon treatment of the Csd4-immobilized chip with 20 mM EDTA to remove intrinsic metal ions (‘after EDTA treatment’). Retreatment of the EDTA-treated Csd4-immobilized chip with 5 mM calcium chloride recovered the binding between two proteins (‘after CaCl2 treatment’). All sensorgrams were obtained by subtracting the nonspecific binding of the analyte to the BSA-immobilized chip. (c) Fluorescence polarization binding assay using an FITC-labelled dipeptide against increasing concentrations of Csd5.
4. Conclusions
The non-invasive Gram-negative pathogen H. pylori is recognized by epithelial cells via the Nod1 receptor (Viala et al., 2004 ▶). The substrate of Csd4, mDAP-containing muramyltripeptide, is an agonist for the cytosolic innate immune receptor Nod1 (Girardin et al., 2003 ▶; Chamaillard et al., 2003 ▶), and thus a decreased amount of muramyltripeptide as a result of catalysis by Csd4 is likely to affect Nod1 activation and ultimately NF-κB activity. Δpgp1, the mutant of the Csd4 homologue in C. jejuni, produces a significantly higher interleukin-8 (IL-8) response in human epithelial cells (INT407) than does wild-type C. jejuni (Frirdich et al., 2012 ▶; Sycuro et al., 2012 ▶), even though the Δcsd4 mutant is comparable to wild-type H. pylori in pro-inflammatory cytokine induction of gastric epithelial AGS cells (Sycuro et al., 2012 ▶). Therefore, it is suggested that d,l-carboxypeptidation by Csd4 homologues could affect the overall amount of released mDAP-containing muramyltripeptides, affect the helical shape and influence immune detection (Frirdich et al., 2012 ▶; Wyckoff et al., 2012 ▶).
In this work, we report the crystal structure of Csd4 in three different states. The N-terminal CPase domain has unique features among members of the M14 metallopeptidase family. Our Csd4–muramyltripeptide and Csd4–mDAP structures represent the substrate-bound and product-bound states, showing how the mDAP-containing muramyltripeptide could be cleaved to the dipeptide variant by Csd4. In the two complex structures extensive interactions between Csd4 and ligands are centred on the mDAP moiety, highlighting the important role of mDAP in specific substrate recognition. Moreover, we also show that the shift of the flap (β9–α5 loop) and two key Arg residues (Arg86 and Arg94) upon binding the muramyltripeptide or mDAP affects the shape and the size of the active-site cleft.
H. pylori Csd4 catalyzes the final step in the generation of the shortest muropeptide from the peptidoglycan, so that its catalytic process and localization should be tightly regulated to control the cell shape, the host immune response and even new peptidoglycan synthesis. H. pylori Csd5 has no known enzymatic activity, exerts little effect on the global peptidoglycan composition and is not required for Csd4 activity. Csd4 was suggested to act upstream of Csd5, raising the possibility that Csd5 could directly interact with Csd4 or the muramyldipeptide product of the Csd4-catalyzed reaction. Here, we have demonstrated that Csd5 interacts physically with both Csd4 and the dipeptide product of the reaction catalyzed by Csd4. We suggest that Csd5 may play a regulatory role in modulating the function of Csd4. This study reports the first structural insights into the events that are influenced by Csd4 of H. pylori, which will serve as a point of departure for deeper understanding of the regulatory events leading to the proper cell shape.
Supplementary Material
PDB reference: Csd4, Zn-bound form I, 4q6p
PDB reference: Zn-bound form II, 4q6q
PDB reference: apo, 4q6m
PDB reference: tripeptide-bound form, 4q6n
PDB reference: mDAP-bound form, 4q6o
Supporting Information.. DOI: 10.1107/S1399004714018732/mh5144sup1.pdf
Acknowledgments
We thank the beamline staff for assistance during X-ray diffraction experiments at the Pohang Light Source (beamlines BL-5C and BL-7A), SPring-8 (beamline BL-26B1) and the Photon Factory (beamlines BL-1A, BL-5A, BL-17A, NE3A and NW12A). We also thank Professor Hyun Kyu Song of Korea University for providing the equipment for the fluorescence polarization binding assay. The work in the laboratory of SWS was supported by the Korea Ministry of Science, ICT and Future Planning, National Research Foundation (NRF) of Korea (2013R1A2A1A05067303) and the Innovative Drug Research Center for Metabolic and Inflammatory Disease, the Korea Ministry of Health, Welfare and Family Affairs (Korea Healthcare Technology R&D Project A092006). The work in the laboratory of BIL was supported by the Mid-career Researcher Program (NRF-2011-0029294) through the NRF of Korea funded by the Ministry of Education. BWH acknowledges support from the Korea Healthcare Technology R&D Project funded by the Ministry of Health and Welfare. HSK and HJY were supported by NRF grants funded by the Korea Ministry of Education (2012-039930 and 2011-0013663). The work in the USA was supported by a grant from the National Institutes of Health (AI090348).
Footnotes
Supporting information has been deposited in the IUCr electronic archive (Reference: MH5144).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. (2001). Proc. Natl Acad. Sci. USA, 98, 10037–10041. [DOI] [PMC free article] [PubMed]
- Berg, H. C. & Turner, L. (1979). Nature (London), 278, 349–351. [DOI] [PubMed]
- Bonis, M., Ecobichon, C., Guadagnini, S., Prévost, M. C. & Boneca, I. G. (2010). Mol. Microbiol. 78, 809–819. [DOI] [PubMed]
- Bunnage, M. E. et al. (2007). J. Med. Chem. 50, 6095–6103. [DOI] [PubMed]
- Cha, S.-S., An, Y. J., Jeong, C.-S., Kim, M.-K., Lee, S.-G., Lee, K.-H. & Oh, B.-H. (2012). Acta Cryst. D68, 1253–1258. [DOI] [PMC free article] [PubMed]
- Chamaillard, M., Hashimoto, M., Horie, Y., Masumoto, J., Qiu, S., Saab, L., Ogura, Y., Kawasaki, A., Fukase, K., Kusumoto, S., Valvano, M. A., Foster, S. J., Mak, T. W., Nuñez, G. & Inohara, N. (2003). Nature Immunol. 4, 702–707. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Costa, K., Bacher, G., Allmaier, G., Dominguez-Bello, M. G., Engstrand, L., Falk, P., de Pedro, M. A. & García-del Portillo, F. (1999). J. Bacteriol. 181, 3710–3715. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Frirdich, E., Biboy, J., Adams, C., Lee, J., Ellermeier, J., Gielda, L. D., Dirita, V. J., Girardin, S. E., Vollmer, W. & Gaynor, E. C. (2012). PLoS Pathog. 8, e1002602. [DOI] [PMC free article] [PubMed]
- Girardin, S. E., Boneca, I. G., Carneiro, L. A., Antignac, A., Jéhanno, M., Viala, J., Tedin, K., Taha, M. K., Labigne, A., Zähringer, U., Coyle, A. J., DiStefano, P. S., Bertin, J., Sansonetti, P. J. & Philpott, D. J. (2003). Science, 300, 1584–1587. [DOI] [PubMed]
- Gouet, P., Robert, X. & Courcelle, E. (2003). Nucleic Acids Res. 31, 3320–3323. [DOI] [PMC free article] [PubMed]
- Grizot, S., Fauré, J., Fieschi, F., Vignais, P. V., Dagher, M. C. & Pebay-Peyroula, E. (2001). Biochemistry, 40, 10007–10013. [DOI] [PubMed]
- Holm, L. & Rosenström, P. (2010). Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed]
- Kabsch, W. & Sander, C. (1983). Biopolymers, 22, 2577–2637. [DOI] [PubMed]
- Koch, M. K., McHugh, C. A. & Hoiczyk, E. (2011). Mol. Microbiol. 80, 1031–1051. [DOI] [PMC free article] [PubMed]
- Kühn, J., Briegel, A., Mörschel, E., Kahnt, J., Leser, K., Wick, S., Jensen, G. J. & Thanbichler, M. (2010). EMBO J. 29, 327–339. [DOI] [PMC free article] [PubMed]
- Kusters, J. G., van Vliet, A. H. & Kuipers, E. J. (2006). Clin. Microbiol. Rev. 19, 449–490. [DOI] [PMC free article] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Lee, M., Zhang, W., Hesek, D., Noll, B. C., Boggess, B. & Mobashery, S. (2009). J. Am. Chem. Soc. 131, 8742–8743. [DOI] [PMC free article] [PubMed]
- Lowe, E. C., Baslé, A., Czjzek, M., Firbank, S. J. & Bolam, D. N. (2012). Proc. Natl Acad. Sci. USA, 109, 7298–7303. [DOI] [PMC free article] [PubMed]
- Malfertheiner, P., Selgrad, M. & Bornschein, J. (2012). Curr. Opin. Gastroenterol. 28, 608–614. [DOI] [PubMed]
- Meroueh, S. O., Bencze, K. Z., Hesek, D., Lee, M., Fisher, J. F., Stemmler, T. L. & Mobashery, S. (2006). Proc. Natl Acad. Sci. USA, 103, 4404–4409. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Ottemann, K. M. & Lowenthal, A. C. (2002). Infect. Immun. 70, 1984–1990. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pallarès, I., Bonet, R., García-Castellanos, R., Ventura, S., Avilés, F. X., Vendrell, J. & Gomis-Rüth, F. X. (2005). Proc. Natl Acad. Sci. USA, 102, 3978–3983. [DOI] [PMC free article] [PubMed]
- Petersen, T. N., Brunak, S., von Heijne, G. & Nielsen, H. (2011). Nature Methods, 8, 785–786. [DOI] [PubMed]
- Rawlings, N. D., Waller, M., Barrett, A. J. & Bateman, A. (2014). Nucleic Acids Res. 34, D270–272. [DOI] [PMC free article] [PubMed]
- Read, R. J. & Schierbeek, A. J. (1988). J. Appl. Cryst. 21, 490–495.
- Sycuro, L. K., Pincus, Z., Gutierrez, K. D., Biboy, J., Stern, C. A., Vollmer, W. & Salama, N. R. (2010). Cell, 141, 822–833. [DOI] [PMC free article] [PubMed]
- Sycuro, L. K., Rule, C. S., Petersen, T. W., Wyckoff, T. J., Sessler, T., Nagarkar, D. B., Khalid, F., Pincus, Z., Biboy, J., Vollmer, W. & Salama, N. R. (2013). Mol. Microbiol. 90, 869–883. [DOI] [PMC free article] [PubMed]
- Sycuro, L. K., Wyckoff, T. J., Biboy, J., Born, P., Pincus, Z., Vollmer, W. & Salama, N. R. (2012). PLoS Pathog. 8, e1002603. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. (2003). Acta Cryst. D59, 38–44. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Viala, J. et al. (2004). Nature Immunol. 5, 1166–1174. [DOI] [PubMed]
- Vollmer, W. & Bertsche, U. (2008). Biochim. Biophys. Acta, 1778, 1714–1734. [DOI] [PubMed]
- Vollmer, W., Blanot, D. & de Pedro, M. A. (2008). FEMS Microbiol. Rev. 32, 149–167. [DOI] [PubMed]
- Vollmer, W., Joris, B., Charlier, P. & Foster, S. (2008). FEMS Microbiol. Rev. 32, 259–286. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wyckoff, T. J., Taylor, J. A. & Salama, N. R. (2012). Trends Microbiol. 20, 540–547. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Csd4, Zn-bound form I, 4q6p
PDB reference: Zn-bound form II, 4q6q
PDB reference: apo, 4q6m
PDB reference: tripeptide-bound form, 4q6n
PDB reference: mDAP-bound form, 4q6o
Supporting Information.. DOI: 10.1107/S1399004714018732/mh5144sup1.pdf