

Abstract
Reasoning that overexpression of multiple E2F-responsive genes might be a useful marker for RB1 dysfunction, we compiled a list of E2F-responsive genes from the literature and evaluated their expression in publicly available gene expression microarray data of patients with breast cancer, serous ovarian cancer, and prostate cancer. In breast cancer, a group of tumors was identified, each of which simultaneously overexpressed multiple E2F-responsive genes. Seventy percent of these genes were concerned with cell cycle progression, DNA repair, or mitosis. These E2F-responsive gene overexpressing (ERGO) tumors frequently exhibited additional evidence of Rb/E2F axis dysfunction, were mostly triple negative, and preferentially overexpressed multiple basal cytokeratins, suggesting that they overlapped substantially with the basal-like tumor subset. ERGO tumors were also identified in serous ovarian cancer and prostate cancer. In these cancer types, there was no evidence for a tumor subset comparable to the breast cancer basal-like subset. A core group of about 30 E2F-responsive genes were overexpressed in all three cancer types. Thus, it appears that disorders of the Rb/E2F axis can arise at multiple organ sites and produce tumors that simultaneously overexpress multiple E2F-responsive genes.
Keywords: expression microarray, basal-like, breast cancer, Rb, E2F, gene overexpression
Introduction
Retinoblastoma (pRb) is a multifunctional protein that is frequently lost or mutated in various types of human cancer (cf, Classon and Harlow1). Here we will focus on its role in the inhibition of cell cycle progression.
Activated Rb protein ordinarily binds E2Fs, a family of transcription factors. Rb can be constitutively inactivated by various mechanisms, including phosphorylation, RB1 gene loss, mutation, or by protein sequestration in the hyperphosphorylated state.2 Cyclin D phosphorylates Rb that is complexed with E2Fs, freeing E2Fs to upregulate multiple E2F-responsive genes, including those required for the initiation of DNA replication.3
Early studies suggested that 10–40% of human breast cancers have lost retinoblastoma gene function (reviewed in4). Rb/E2F dysfunctional tumors have been studied using gene expression microarrays.5,6 The dysregulation of genes involved in DNA replication and progression through mitosis has been a common finding.
Over the past decade, several molecular subtypes of breast cancer have been recognized. The basal-like subtype has been of special interest (see7 and reviews by Metzger-Filho et al.8 and Badve et al.9 and references therein). In brief, the major distinctive features of this subtype are that they overexpress basal-like cytokeratins, and they are “triple negative,” ie, they do not overexpress HER2, estrogen receptor (ER), or progesterone receptor (PR). Breast cancers that arise in patients with hereditary BRCA1 mutations are commonly of the basal-like subtype.10 Therapeutic options for patients with these cancers are limited, because they are not candidates for hormonal therapy or targeted therapy against HER2.8 While they may respond to chemotherapy initially, these responses are often of brief duration, and survival times for most patients with these tumors are relatively short.
A link between Rb and the basal-like breast cancer subtype has been recognized. Herschkowitz et al.11 used RB1 loss of heterozygosity, low RNA expression levels of RB1, and high levels of p16ink4a (a functional marker for Rb loss) to show that the loss of Rb was a common occurrence in basal-like and luminal B breast cancers and identified an Rb-gene signature that included many S-phase and mitotic genes. A recently published comprehensive multiplatform genomic analysis of breast cancers12 has confirmed much of what is known about basal-like breast cancers at the molecular level, including loss of RB1 (see below), their association with BRCA1 mutations, and high levels of activity of the PI3K/Akt pathway.13
In view of the difficulties in identifying loss of Rb function directly, we reasoned that Rb/E2F dysfunctional tumors might best be distinguished by the criterion of overexpression of multiple E2F-responsive genes in each tumor. The requirement for the overexpression of multiple genes might provide for a more robust test of Rb/E2F dysfunction, as well as a more direct measure of the functional effects of Rb dysfunction than p16ink4a overexpression or even direct assessment of Rb expression and localization by immunohistochemistry. In addition, characterization of a subset of tumors by a set of multiple genes that they overexpress simultaneously may lead to a better understanding of this subset’s pathobiology and provide insights for developing therapeutic strategies that utilize multiple targeted agents. Finally, we wondered if such a functional subtype would coincide or overlap with recognized subtypes of breast cancer, and with basal-like cancers in particular.
In this paper we have identified a novel subgroup of breast cancers that individually overexpress multiple E2F-responsive genes simultaneously. This subgroup overlaps substantially with the subset of basal-like breast cancers. We also identified subgroups of tumors that simultaneously overexpress multiple E2F-responsive genes in ovarian cancer and prostate cancer. Many of the overexpressed genes in these cancers were often the same overexpressed E2F-responsive genes that were found in breast cancer. Since overexpressed normal cell constituents in cancer cells are potential candidates for targeted therapy, these genes could provide the basis for a rational approach to multitargeted systemic therapy regardless of site of origin.
Materials and Methods
Compilation of E2F-responsive gene list
In prior work,14 we conducted a review of the English literature through 2008 for genes whose expression is altered in response to changes in the expression of Rb or members of the E2F gene family, which yielded 1000 potential E2F1–3 responsive genes. Of these, 325 were confirmed to be E2F responsive by at least two microarray studies or by at least one microarray study plus one alternative method, such as Real-Time Polymerase Chain Reaction (RT-PCR) or Northern blot analysis. These 325 genes (Supplementary Table 1) made up the list of E2F-responsive genes used in the present study.
Microarray and RNA-Seq data sets
The gene expression microarray data set of 117 breast cancers published by van’t Veer et al.,15 available at http://www.rii/com/publications/default.htm, was used to investigate human breast cancer. For this study, 98 primary breast cancers were selected, out of which, 34 developed distant metastases within 5 years, 44 remained disease free after a period of at least 5 years, 18 were hereditary BRCA1 mutation carriers, and 2 were BRCA2 carriers. The samples were analyzed for mRNA expression using an Agilent Hu25K array platform. Two microarray breast cancer data sets were used to investigate the reproducibility of results in the van’t Veer data set. The first data set was published by Dai et al.,16 obtained on a different custom Agilent microarray platform. The Dai data set is available from the National Center for Biotechnology Information’s (NCBI) Gene Expression Omnibus (GEO) data repository as series GSE2845. Among 311 samples studied by Dai et al, there were cases that overlapped with those in the van’t Veer study, and these were excluded prior to analysis in the present study. In the second breast cancer microarray data set by Jönsson et al.,17 available at NCBI GEO as series GSE25307, gene expression profiling was performed on 577 breast tumors, including 73 breast tumors from BRCA1/2 mutation carriers. Thirty-three BRCA1-mutated, 36 BRCA2-mutated, and 48 non–BRCA1/2-mutated breast tumors were analyzed in this study.
We further examined data sets from two additional tumor types, prostate and ovarian, to test the generality of the mechanism across tissues of origin. An expression microarray data set from the prostate cancer series retrieved from NCBI GEO as series GSE691918,19 was also studied, as well as the ovarian high-grade serous cancer expression microarray data set from series GSE10971,20,21 also found in the NCBI GEO data repository. In the prostate cancer data set, using Affymetrix U95v2 oligonucleotide arrays, gene expression profiles of 24 androgen ablation–resistant metastatic samples obtained from four patients and a previously published data set of 64 primary prostate tumor samples were analyzed. In the ovarian cancer data, gene expression profiles of laser capture microdissected nonmalignant distal fallopian tube epithelium from 12 known BRCA1/2 mutation carriers and 12 control women during the luteal and follicular phase as well as 13 high-grade tubal and ovarian SerCa were collected using the Affymetrix Human Genome U133 Plus 2.0 Array platform.
Finally, we examined reproducibility of the results with respect to the data type by examining RNA-Seq data sets drawn from The Cancer Genome Atlas (TCGA) data sets for breast,12 prostate, and ovarian cancers.22 The number of samples in the TCGA breast, prostate, and ovarian cancer data sets were 779, 419, and 419, respectively.
Identification of E2F-responsive gene-overexpressing tumors
In order to identify E2F-responsive gene-overexpressing tumors, it is necessary to distinguish biologically significant levels of overexpression from normal levels of gene expression. A twofold difference above reference is a commonly used threshold for this purpose. However, tumor cell heterogeneity23 and a variety of technical factors may conspire to alter the appropriate threshold. This issue has been studied extensively in distinguishing HER2 amplification/overexpression for purposes of patient selection for trastuzumab treatment. In this instance, a threshold of 1.8 has been adopted by consensus, below which amplification/overexpression by fluorescence in situ hybridization is excluded.24 Since the diluting effect of tumor cell heterogeneity would be expected to apply to expression microarrays as well, we have adopted a threshold of 1.8-fold expression above reference to distinguish overexpressed from non-overexpressed genes. Its reciprocal was used to distinguish underexpressed genes. Genes that were between these thresholds, ie, neither underexpressed nor overexpressed, were classified as non-overexpressed genes. For RNA-Seq data, a different protocol was required to address the lack of a defined normal control. For those data, we normalized each gene in each data set to mean 0 and standard deviation 1 across all the samples in the data set. We then used a threshold of 1.8 for distinguishing the overexpressed, underexpressed, and non-overexpressed genes.
Setting boundaries for E2F-responsive gene overexpressing tumors and the E2F-responsive genes associated with them
When tumors were sorted and rank ordered with respect to the number of E2F-responsive genes they overexpress, and the E2F-responsive genes were sorted and rank ordered with respect to the number of tumors that overexpress them, it became apparent that there was a group of tumors that overexpressed multiple E2F-responsive genes (Fig. 1A). In order to set optimal boundaries for distinguishing Area 1 from Area 2, we used a strategy that minimizes a weighted version of the Shannon’s entropy25,26 of the classification. Shannon’s entropy for Area 1 in Figure 1 is given by,
| (1) |
Figure 1.

Features of the van’t Veer breast cancer data set (S1). Data for individual genes are thresholded: red boxes representing genes that were overexpressed at least 1.8-fold times the normal reference; green areas represent gene levels of expression less than 0.556-fold the normal reference; white areas represent genes that were between these levels, and were neither overexpressed nor underexpressed. (A) E2F-responsive genes lie along the vertical axis and are sorted by increasing frequency of overexpression (lower to upper) among tumors. Tumors lie along the horizontal axis and are sorted by increasing frequency of overexpression of E2F-responsive genes per tumor (right to left). Vertical and horizontal border lines enclose a subset of tumors that overexpressed a relatively large number of E2F-responsive genes per tumor and a subset of E2F-responsive genes that were simultaneously overexpressed in the largest proportions of these tumors. The remaining tumors were organized by triple-negative, HER2-positive, and ER/PR-positive groups from left to right. (B) Selected gene expression profile for data set S1. Top tier: RB1, CDKN2A; Tier 2, ERBB2, ESR1 PGR, next line, EGFR; Tier 3, KRT5, KRT6A, KRT6B, KRT14, KRT17; Tier 4, E2F1, E2F2, E2F3. Color coding as in Figure 1A.
Entropy for Area 2 in Figure 1 is given by,
| (2) |
and the total weighted entropy E is given by,
| (3) |
where,
f1 = fraction of overexpressed genes in Area 1,
f2 = fraction of overexpressed genes in Area 2,
n1 = total number of gene entries in Area 1,
n2 = total number of gene entries in Area 2.
We search for the tumor and gene boundaries that produce a minimum value for Equation (3) by iteratively stripping away tumor columns and gene rows from Area 1 and adding them to Area 2, recalculating the value of Equation (3), and re-sorting and rank-ordering the tumors and genes remaining in Area 1. The procedure is summarized as follows:
Let boundary = [gene boundary, tumor boundary], where initially gene boundary = total genes – 1 and tumor boundary = total tumors – 1.
Compute E as described in Equation (3), where Area 1 is defined to consist of the entries between rows [1, gene boundary] and columns [1, tumor boundary].
Set minE = E and minBoundary = [gene boundary, tumor boundary].
- Repeat the following until gene boundary or tumor boundary becomes 1:
- Compute E1 with boundary1 = [gene boundary – 1, tumor boundary].
- Compute E2 with boundary2 = [gene boundary, tumor boundary – 1].
- If (E1 < E2), then set gene boundary = gene boundary – 1, else set tumor boundary = tumor boundary – 1.
- Re-sort columns to order columns in decreasing order of number of overexpressed rows in Area 1 and rows in decreasing order of number of overexpressed columns in Area 1.
- If (E1 < E2) and (E1 < minE), then set minE = E1 and minBoundary = boundary1.
- If (E2 < E1) and (E2 < minE), then set minE = E2 and minBoundary = boundary2.
Return minE and minBoundary.
We note that this procedure is a heuristic local optimizer but would not, in general, be guaranteed to find a globally optimal partitioning of tumors and genes.
Principal Component Analysis (PCA), as described in Data analysis and visualization section, was also found to be useful sorting out E2F-responsive gene overexpressing (ERGO) tumors and their associated genes.
Data analysis and visualization
Microsoft Excel was used for data visualization, performing common statistical tests, and data formatting. The entropy minimization routine was implemented in Matlab. PCA was performed using the Multiple Array Viewer software application27 available at http//www. TM4.org. Adobe Illustrator was used to compose figures.
Results
The ERGO tumor subset of human breast cancers
A list of 325 E2F-responsive genes that met quality assurance criteria (see Materials and Methods section) were used with the gene expression microarray data set published by van’t Veer et al.15 to determine which breast cancers overexpressed them, and which E2F-responsive genes they overexpressed. We considered this to be a particularly useful data set, since it contained 18 tumors from patients with known BRCA1 mutations that could serve as additional valuable markers for the basal-like tumor subset.
Initially, the tumors in the van’t Veer data set were rank ordered by frequency of overexpressed E2F-responsive genes per tumor, and the E2F-responsive genes were rank ordered by their frequency of overexpression across tumors (Fig. 1A). It is clear from the data that there was a subset of 25–30% of tumors where each member overexpressed multiple E2F-responsive genes simultaneously (ERGO tumors). It is also notable that the most abundantly overexpressed genes were frequently underexpressed in non-ERGO tumors (Fig. 1A).
In order to examine the characteristics of the ERGO tumor subset in greater detail, an optimal classification of the ERGO tumor subset was determined by entropy minimization (see Methods section). The ERGO tumor subset was found to include 35 tumors and 121 E2F-responsive genes. This gene list is provided in Supplementary Table 2. The ERGO subset of tumors overexpressed a minimum of 20 E2F-responsive genes per tumor, a maximum of 73, and a mean of 41 E2F-responsive genes per tumor.
A gene expression profile for ERGO tumors consisting of ERGO and non-ERGO genes of particular interest is shown in Figure 1B. RB1 was preferentially underexpressed in ERGO tumors in comparison with non-ERGO tumors (Table 1). CDKN2A and CCNE1 or CCNE2, both markers for Rb loss,2 were preferentially overexpressed in ERGO tumors, as was E2F1 overexpression. All these results were highly significant statistically (P values < 0.003). With regard to basal-like markers, the ERGO tumor subset was mostly triple negative (30/35, or 88%), and ERGO tumors preferentially overexpressed at least two basal cytokeratins; these results were also highly significant statistically (P values < 0.0001; Table 1).
Table 1.
Comparison of properties of data sets S1, S2, S3 and S4.
| S1 | S2 | S3 | S4 | |
|---|---|---|---|---|
| Number of tumor samples | 117 | 262 | 577 | 779 |
| E2F genes | 325 | 315 | 165 | 250 |
| ERGO tumors | 35 | 53 | 142 | 125 |
| ERGO E2F-responsive genes | 121 | 128 | 61 | 156 |
| Common ERGO E2F-responsive genes | 121 | 115 | 86 | 89 |
| Matched common ERGO E2F-responsive genes | 121 | 95 | 45 | 77 |
| P value | P < 0.0001 | P = 0.0339 | P < 0.0001 | |
| >1 basal Cytokeratins overexpressed in ERGO tumors | 17 | 11 | N/A | 15 |
| >1 basal Cytokeratins overexpressed in non-ERGO tumors | 5 | 16 | N/A | 9 |
| P value | P < 0.0001 | P = 0.0278 | P < 0.0001 | |
| Rb underexpressed in ERGO tumors | 14 | 17 | 25 | 7 |
| Rb underexpressed in non-ERGO tumors | 7 | 18 | 11 | 1 |
| P value | P = 0.0028 | P < 0.0005 | P < 0.0001 | P < 0.0001 |
| CDKN2A overexpressed in ERGO tumors | 22 | 22 | 21 | 27 |
| CDKN2A overexpressed in non-ERGO tumors | 6 | 5 | 2 | 12 |
| P value | P < 0.0001 | P < 0.0001 | P < 0.0001 | P < 0.0001 |
| CCNE1 or CCCIME2 overexpressed in ERGO tumors | 14 | 20 | 85 | 19 |
| CCNE1 or CCNE2 overexpressed in non-ERGO tumors | 1 | 6 | 20 | 10 |
| P value | P < 0.0001 | P < 0.0001 | P < 0.0001 | P < 0.0001 |
| E2F1 overexpressed in ERGO tumors | 9 | 14 | 2 | 35 |
| E2F1 overexpressed in non-ERGO tumors | 1 | 7 | 1 | 12 |
| P value | P < 0.0005 | P < 0.0001 | P = 0.1538 | P < 0.0001 |
aChi-squared test, or Fisher’s exact test for n < 6; statistically significant at <0.05.
Seventeen of the 18 BRCA1 tumors were triple negative. Twelve of 18 BRCA1 tumors (67%) were ERGO tumors, and 8/12 (67%) overexpressed at least two basal cytokeratins. The remaining six BRCA1 tumors were non-ERGO triple negatives, and 2/6 (33%) overexpressed at least two basal-like markers. Thus, most BRCA1 tumors were members of the ERGO tumor subset, and most expressed at least two basal-like markers. EGF receptor overexpression was more common in the ERGO tumor subset (4/35, or 11%) than in non-ERGO tumors (4/82, or 5%), but the difference was not significant statistically, and EGFR expression was not otherwise helpful in identifying basal-like tumor patterns.
Thus, we have identified a group of ERGO breast cancers, where each tumor in the group simultaneously overexpressed multiple E2F-responsive genes. This is a novel finding. This group of tumors also shows statistically significant differences in Rb expression and markers for Rb dysfunction, as well as specific markers for the basal-like tumor subset, including hereditary BRCA1 mutations, in keeping with known properties of the basal-like tumor subset. These findings suggest that the ERGO tumor subset overlaps with the basal-like subset, based on similar immunohistochemical criteria for basal-like tumors.28 There were basal-like tumors that did not overexpress multiple E2F-responsive genes simultaneously and were clearly excluded from the ERGO tumor subset. There were also ERGO tumors that were triple negative but did not overexpress cytokeratins, which appears to exclude them from the basal-like subset. However, other histologic markers for the basal-like subset did mark cytokeratin-negative ERGO tumors and cytokeratin-negative non-ERGO BRCA1 tumors (see below).
Of the E2F-responsive genes that were overexpressed in ERGO tumors from the van’t Veer data set, 60% were involved in DNA synthesis, DNA repair, or mitosis, based on associated Gene Ontogeny (GO) terms. The list of mitotic E2F-responsive genes overexpressed in ERGO tumors was significantly enriched (2.2-fold) over that of all E2F-responsive genes (chi-squared, P < 0.0001). The degree of enrichment of E2F-responsive genes involved in DNA repair was not statistically significant, and for E2F-responsive genes involved in DNA synthesis, the degree of enrichment was of borderline significance (P < 0.0496). The results thus show that the category of mitosis is significantly enriched and the category of DNA synthesis weakly enriched within the ERGO-associated gene set relative to the full 325 E2F-responsive genes. We performed a further unbiased analysis with the GOrilla tool for measuring statistical association of GO terms with a gene set, comparing the ERGO gene set to the full E2F-responsive gene set. The term “cell cycle process” showed up as the most significant (P value 9 × 10−8).
Reproducibility of the list of overexpressed E2F-responsive genes in ERGO tumors
To determine if the findings in the van’t Veer data set (S1) are unique to that data set, we used three breast cancer data sets reported by Dai et al.16 (referred to here as S2), Jönsson et al,17 (S3), and the TCGA RNA-Seq data set (S4). Cases in the Dai set that overlapped with the van’t Veer data set were excluded from analysis of the Dai data set. Thus, analysis of the S1, S2, S3, and S4 data sets were performed on entirely different patients and on different platforms. ERGO tumor subsets were also identified in S2, S3, and S4, and the boundaries for partitioning tumors and genes in S2, S3, and S4 were determined by the entropy minimization method. Fifty-three ERGO tumors and 128 ERGO E2F-responsive genes were identified on S2. Similarly, 142 ERGO tumors, 61 ERGO E2F-responsive genes, 125 ERGO tumors, and 156 ERGO E2F-responsive genes were identified on S3 and S4, respectively. When the lists of ERGO E2F-responsive genes in data sets S1 and S2 were compared, 115 of the ERGO genes were shared between S1 and S2 and 95 (82.6%) of them were identified by the entropy minimization method on S2. In many other respects, the two data sets were similar (Table 1). Both supported preferential overexpression of cytokeratins in ERGO tumors, and both indicated a preferential defect in the Rb/E2F axis in ERGO tumors. A similar observation can be made about the other two breast cancer data sets, S3 and S4 (see also Table 1). There was significant overlap in the ERGO genes identified between S1 and both S3 and S4. As with S1 and S2, there was preferential overexpression of CDKN2A and CCNE1 in the ERGO tumors for both S3 and S4. S4 exhibited a statistically significant defect in the Rb/E2F axis in ERGO tumors, similar to S1 and S2. GO analysis of the TCGA ERGO genes using the E2F-responsive genes as background resulted in 56.7% of the genes being associated with DNA replication, DNA repair, or mitosis. The “cell cycle process” GO category that had showed up as most significant for S1 also showed up as significant for S2, S3, and S4 (P value < 4 × 10−6 in each case) and was the most significant category for all but S4 (where “DNA metabolic process” was most significant).
Principal component analysis and the basal-like subset
PCA can cluster data subsets based on their distinctive features, but the results of PCA alone can often be difficult to interpret. It was hoped that insights gained from partitioning the tumors by entropy minimization might inform the interpretation of the PCA findings. It was also hoped that PCA might, in turn, shed more light on the relationship between the ERGO tumor subset and the basal-like tumor subset.
PCA of the van’t Veer data set revealed a tumor cluster (Group 1) consisting of 38 samples (Fig. 2A, red spheres), all of which underexpressed ER, and 35/38 (92%) of which were triple negative. Twenty of 38 cluster members overexpressed at least two basal-like cytokeratins (53%), compared with only 2/79 tumors not in the cluster (3%). Conversely, 91% of all tumors that overexpressed at least two basal-like cytokeratins were found in the cluster. In addition, 17 of 18 BRCA1 tumors were found in the Group 1 cluster.
Figure 2.

PCA results for breast cancer data set S1. (A) PCA results using input of all genes and all tumors and output of tumor clustering. Red spheres represent a distinct tumor cluster that consists mostly of basal-like tumors, including 28/35 ERGO tumors and 17/18 BRCA1 tumors (Group 1); Group 2 contains the non–basal-like tumors. (B1) A comparison of gene expression patterns in Group 1 and Group 2 using published alternative basal-like markers; markers and literature references are found in Supplementary Table 3. (B2) A comparison of gene expression patterns in Group 1 and Group 2 for basal-like cytokeratins. (C) PCA results using input of 325 E2F-responsive genes only and all tumors and output of tumor clustering. Red spheres represent a distinct tumor cluster that contains 27/28 of the ERGO tumors found in the basal-like cluster (A), and 12/17 of the BRCA1 tumors found in A.
Others have found that additional biomarkers for basal-like breast cancer either clustered with the basal cytokeratins in expression microarray studies29,30 or have been shown to be statistically significantly more frequent in basal-like tumors in immunohistochemical studies. It has been suggested that triple-negative basal cytokeratin-negative tumors can still be identified as basal-like tumors using such immunohistochemical markers.7,31 Figure 2B1 compares the patterns of overexpression in Group 1 versus Group 2 of 10 such alternative markers gathered from the literature. These markers and their references are listed in Supplementary Table 3. The differences in frequencies of the alternative marker overexpression between Group 1 and Group 2 were all statistically significant, with all P values less than 0.0001 (Fisher’s exact test). Figure 2B2 compares the patterns of overexpression in Group 1 versus Group 2 of the basal cytokeratins. Eighteen of 20 (90%) tumors in Group 1 that overexpressed at least two basal cytokeratin markers also overexpressed at least three of the alternate markers. However, the number of tumors overexpressing at least three alternative markers identified seven additional Group 1 tumors as potential basal-like tumors, bringing the total to 27/38 (71%). Six of the seven (86%) were triple negative. In Group 2, of the 2/79 (3%) of tumors that overexpressed at least two cytokeratins, only one overexpressed at least three alternative markers, and in Group 2 there were no tumors that overexpressed at least three alternative markers without overexpressing at least two cytokeratins.
It appears that the Group 1 PCA cluster contained a mixture that consisted mostly of basal-like tumors (~70%), plus a minor component of tumors that did not meet criteria of basal cytokeratin or alternative marker overexpression. Furthermore, Group 1 contained almost all the tumors in the data set that did meet the criteria for basal-like tumors. Therefore, we shall treat Group 1 tumors as the PCA basal-like subset.
PCA and the ERGO tumor subset
The Group 1 PCA basal-like cluster included 28/35 ERGO tumors that were identified by the entropy minimization method. Again, this would indicate that the ERGO tumors overlapped substantially with the basal-like tumor subset.
Since the unique feature of the ERGO tumors was the simultaneous overexpression of multiple E2F-responsive genes, one might expect that by restricting the PCA input to E2F-responsive genes, PCA tumor clustering would distinguish the ERGO tumors from the non-ERGO tumors in the more inclusive basal-like cluster and perhaps identify ERGO tumors that were not in the cluster. The red cluster shown in Figure 2C contained 27 of the 28 ERGO tumors that were included in the Group 1 PCA basal-like subset. Twelve of the 18 BRCA1 tumors were included in this cluster. This reinforces the conclusion that the ERGO tumor subset overlaps substantially with the basal-like tumor subset but does not account for the ERGO tumors not identified by PCA. Of the 28 ERGO tumors identified both by PCA and entropy minimization, 19 overexpressed basal cytokeratins and/or alternative basal markers. Of the six cases identified as ERGO tumors by entropy minimization that were not identified by PCA, none overexpressed multiple basal cytokeratins or alternative basal-like markers. Comparison by Fisher’s exact test produced a P value of <0.003, suggesting that the latter group does not share identifiable basal-like features with the former and that the overlap between ERGO tumors and basal-like tumors is not complete.
For the 28 ERGO tumors found by PCA, we examined the top 100 overexpressed E2F-responsive genes (by frequency of overexpression across tumors). The differences in mean expression levels between ERGO and non-ERGO tumors were statistically significant for 94 genes, with P values ranging from 1 × 10−3 to <1 × 10−16 (Student’s t test). For the 121 overexpressed E2F-responsive genes identified by the entropy minimization method, the differences in mean expression levels between ERGO and non-ERGO tumors were statistically significant for 96 genes, with P values ranging from 2 × 10−3 to <1 × 10−16. Ninety-two overexpressed E2F-responsive genes were common to both lists (Supplementary Fig. 1). Thus, with either method, substantially the same commonly overexpressed E2F-responsive genes were more highly overexpressed in ERGO tumors.
ERGO tumors arise at other organ sites
Here we show that ERGO tumors are found at organ sites other than breast cancer, namely, high-grade serous ovarian cancer and prostate cancer.
Ovarian cancer is of particular interest because, like breast cancer, it arises in large proportions of patients with a hereditary BRCA1 mutation (~50% of cases by age 70). Serous carcinoma is the predominant histological type of cancer arising in such patients. Also of interest, BRCA1 mutation carriers undergoing prophylactic surgery are frequently found to have occult serous cancers of the fallopian tube, suggesting that this is a preferred site of tumor formation in BRCA1 mutation carriers. Tone et al.20 studied normal fallopian tube epithelium in BRCA1 mutation carriers and high-grade serous fallopian tube and ovarian carcinomas in both BRCA1 gene mutation carriers and noncarriers using expression microarrays. We have analyzed these data, and our findings are presented in Figure 3A.
Figure 3.

High-grade serous ovarian cancer and prostate cancer are ERGO tumors. (A) Fallopian tube (FT) samples of patients with BRCA1 mutations (controls), compared with samples from 13 patients with high-grade serous ovarian cancer (horizontal axis). Genes lie along the vertical axis. Thresholds and color coding as in Figure 1A. Top tier, RB1 and CDKN2A; Tier 2, E2F1-3; Tier 3, basal cytokeratins; Tier 4, top 100 overexpressed E2F-responsive genes. (B) PCA results using 325 E2F-responsive genes and only primary prostate cancers as inputs, and tumor clustering as output. Pattern 1, red spheres represent a cluster of primary tumors in which the patterns of most frequently overexpressed E2F-responsive genes are similar to those of metastatic tumors (Pattern 1, see Fig. 3C). Pattern 2 represents the remainder of the primary tumors. (C) Comparison of patterns of gene overexpression in metastatic tumors (left), primary tumors with PCA Pattern 1 (middle) and PCA Pattern 2 (right). Genes lie along the vertical axis. Line 1, CDKN2A; Tier 1, E2F1-3; Tier 2, basal cytokeratins; Tier 3, top 100 overexpressed E2F-responsive genes. Thresholds and color coding as in Figure 1A.
High-grade serous carcinomas from fallopian tube and ovary of both BRCA1 and non-BRCA1 mutation carriers were all advanced ERGO tumors. Nearly all the top 50 ERGO tumor-associated E2F-responsive genes were overexpressed in 12 of the 13 tumors. Twelve of 13 tumors overexpressed E2F3, but none underexpressed RB1 or overexpressed p16ink4a. Thus, there appears to be an abnormality in the Rb/E2F axis, but there is no clear indication from these data that RB1 itself was affected. Twelve of 13 tumors overexpressed at least one basal cytokeratin, and six overexpressed at least two basal cytokeratins, raising the possibility of a connection to the basal-like phenotype, but this phenotype does not appear to be as well developed in high-grade serous ovarian cancers as in breast cancers. Among the top 100 overexpressed genes in high-grade serous ovarian cancers (by frequency of overexpression per tumor), 60% were shared with ERGO tumor-associated E2F-responsive genes in breast cancer.
None of the fallopian tube samples underexpressed RB1 or overexpressed p16ink4a. Only one of the 12 fallopian tube samples from BRCA1 carriers overexpressed E2F3, and only one overexpressed two cytokeratins. Ten of 12 fallopian tube samples showed a paucity of overexpressed E2F-responsive genes in comparison with the high-grade serous cancers. In the remaining two fallopian tube samples, approximately half of the top 50 ERGO tumor-associated genes were overexpressed. The findings in these two samples may be transitional evolutionary patterns on the way to aggressive cancer in BRCA1 carriers.
When we applied our entropy minimization method on the TCGA ovarian cancer RNA-Seq data set, we again found broad agreement in identified ERGO genes. Eighty-five percent of the ERGO E2F-overexpressed genes from the van’t Veer data set that were also present in the TCGA data set were identified as ERGO associated. Furthermore, CDKN2A, CCNE1, and E2F1 were preferentially overexpressed in ERGO tumors and RB1 was preferentially underexpressed. Thus, this subset of ERGO ovarian cancer samples overexpress the same set of basal-like markers as were noted for the breast cancer case.
We conclude that these high-grade serous ovarian and fallopian tube cancers, regardless of whether they were from BRCA1, are advanced ERGO tumors. Our data generally support the findings of the recently published comprehensive breast cancer analysis showing that basal-like breast cancers and high-grade serous ovarian cancers are closely related at the molecular level.12
Published studies have indicated that E2F3 overexpression plays a role in the development of prostate cancer, suggesting the possibility that at least some might be ERGO tumors. The study of Olsson et al.32 suggested that E2F3 overexpression together with loss of RB1 function increased the rate of proliferation of prostate cancer cell lines. Sharma et al.33 have further found indications of a specific role for Rb dysfunction in progression of prostate cancers and development of resistant forms with poor clinical prognosis acting via dysregulation of androgen receptor mediated by E2F-responsive genes. We therefore repeated our analyses using the GSE6919 data set, available from the NCBI GEO site, which contained gene expression microarray data on metastatic and primary prostate cancer samples, as well as on data on normal prostate, which was used for normalization. The data were published originally in a paper by Chandran et al.19
The data set includes multiple samples from each of three cases of metastatic, androgen ablation–resistant prostate cancer, and a single sample from a fourth metastatic tumor. All metastatic samples showed fully developed ERGO tumor-associated gene expression patterns (Fig. 3C). Rb was not underexpressed in any of the tumor samples (data not shown). Although the total number of patients is relatively small, the fact that every one of these cases of metastatic prostate cancer exhibited overexpression of large numbers of overexpressed ERGO tumor-associated genes suggests that E2F3-driven ERGO tumor development is a feature of advanced prostate cancers. In the absence of any RB1 underexpression, it is unclear whether RB1 was directly involved or whether a defect occurred distal to Rb in the Rb/E2F pathway. None of the metastatic samples overexpressed multiple cytokeratins, suggesting that this feature is not a necessary component of the ERGO tumor phenotype.
When PCA was applied to the cases of primary prostate cancers without metastases in this data set using the subset of 325 E2F-responsive genes as input, a separate subset of primary tumors (red spheres, Fig. 3B) was identified that exhibits many of the same features present in metastatic tumor samples (PCA Pattern 1, Fig. 3C). In particular, 8/19 (42%) cases overexpressed CDKN2A, and 5/19 (26%) overexpressed E2F3. This compares with 10/46 (21%) cases with overexpression of CDKN2A with PCA Pattern 2 (Fig. 3C), and 2/46 (4%) cases overexpressing E2F3. Cytokeratin overexpression did not play a prominent role in PCA Pattern 1 or 2, with only 1/15 (7%) cases showing two overexpressed cytokeratins in PCA Pattern 1, and 5/46 (11%) in PCA Pattern 2.
Among the top 100 E2F-responsive genes (by frequency of overexpression per tumor), mean frequency of overexpression was 50 for metastatic tumors, 46 for PCA Pattern 1 tumors, and 19 for PCA Pattern 2 tumors. The difference between metastatic and PCA Pattern 1 tumors was not statistically significant. The difference between PCA Pattern 1 tumors and PCA Pattern 2 tumors was highly significant (P < 2 × 10−7). Thus, it appears that among primary tumors, there is a distinct subset that represents ~30% of all primary tumors and is similar in its patterns of overexpression of E2F-responsive genes to those of metastatic samples.
Among the top 100 overexpressed genes in primary prostate cancers in the GSE6919 data set, 44 were shared with the top ERGO tumor-associated E2F-responsive genes in breast cancer and 44 were shared with the top 100 ERGO tumor-associated E2F-responsive genes in ovarian cancer. Thirty-one overexpressed E2F-responsive genes appeared in each of the top 100 lists for breast cancer, ovarian cancer, and prostate cancer.
We further applied the entropy minimization algorithm on the TCGA RNA-Seq data set and it identified 87.6% of the ERGO genes identified in the van’t Veer data set. Similar to the other cancer types, the ERGO tumors showed preferential overexpression of CDKN2A, CCNE1, and E2F and underexpression of RB1.
Together, these findings on multiple data sets from breast, ovarian, and prostate tumors would suggest that there may be a similar mechanism for ERGO tumor development that is active at multiple organ sites.
Discussion
Cell cycle–related genes have been identified in previous publications as being associated with cell proliferation and tumor aggressiveness. Tabach et al.34 identified a group of 167 genes, which they called “the proliferation cluster,” because the cluster was significantly enriched for cell cycle–related genes. At least 66 of these genes appear on our list of E2F-responsive genes that are overexpressed in ERGO cancers, and most of these are involved in progression through G1/S or G2M cell cycle phase transit. Carter et al.35 identified a 70-gene signature of chromosomal instability that includes 41 cell cycle–related genes.35 At least 37 of these appear on our list of E2F-responsive genes. Mosley and Keri36 showed that cell cycle–related genes correlated with the prognostic power of breast cancer gene lists from multiple studies. At least 24 of these appear on our list of E2F-responsive genes. It had previously been shown that E2F upregulates clusters of genes that are involved in both the G1/S and in the G2/M transition.11,12
Here we show for the first time that groups of genes that contribute to the G1/S, DNA repair, and G2/M progression make up the bulk of a core group of ~100 E2F-responsive genes, many of which are often overexpressed simultaneously in individual members of a restricted subset of ERGO human breast cancers. Many of these genes are also simultaneously overexpressed in other solid tumors. These genes are not systematically overexpressed in non-ERGO breast cancer subtypes. BRCA1 breast cancers were shown to be nearly all basal-like cancers (17/18 samples) that were either ERGO or non-ERGO tumors. These findings are in keeping with those of Foulkes et al.,37 who showed that BRCA1 tumors are associated with the basal-like phenotype.
Figure 4 presents a model for breast cancer evolution consistent with the various findings of this study and intended to summarize their possible implications for breast cancer development and progression. Basal-like tumors are presumed to arise mostly from HER2 nonamplified and ER-negative precursors, and ERGO tumors develop from basal-like precursors. This is based on the findings by PCA that the tumor subset that consists mostly of basal-like tumors also includes most ERGO tumors and that PCA using E2F-responsive genes only separates the ERGO tumors from the non-ERGO basal-like tumors (see Fig. 2). A small group of ERGO tumors identified only by the entropy minimization method do not overexpress cytokeratins or alternative basal-like markers and is presumed not to overlap with the basal-like subset. Two-thirds of BRCA1-mutated breast cancers are ERGO tumors, and almost all the remaining BRCA1 tumors are grouped with the basal-like tumors. Since ERGO tumors represent a more advanced evolutionary state than basal-like tumors, it is reasonable to suppose that BRCA1-mutated tumors undergo the transition from basal-like to ERGO cancers as they evolve. This remains to be shown directly. Preliminary data in ovarian cancer and prostate cancer suggest that the basal-like to ERGO transition might not be a prominent feature of ERGO tumors that arise at other organ sites.
Figure 4.
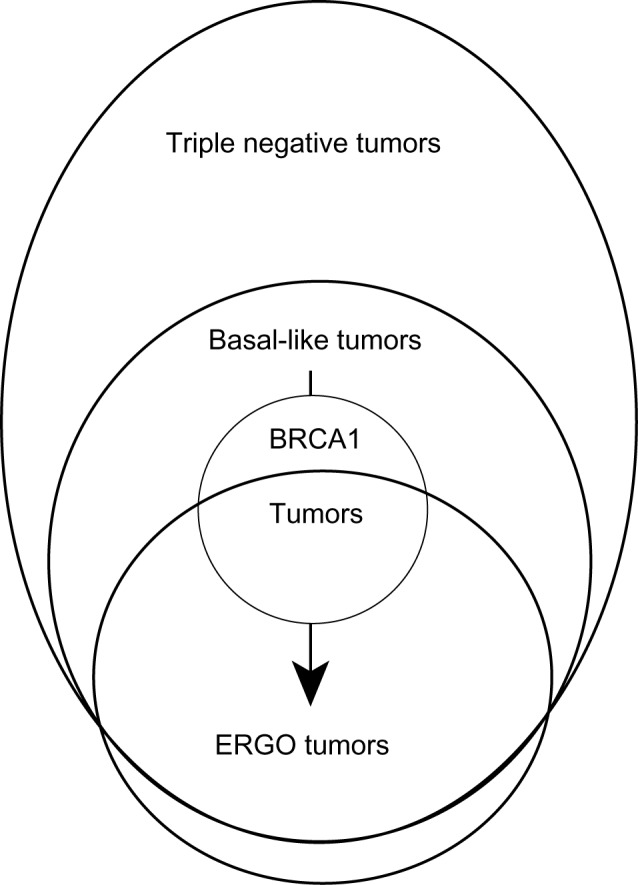
A conceptual model for the evolutionary development of ERGO tumors qualitatively describing a governing hypothesis to explain the results of the paper in terms of relationships between the related but distinct categories of ERGO, triple-negative, basal-like, and BRCA1 tumors.
Our data indicate that many ERGO breast cancers show signs of Rb/E2F1 dysregulation at the functional level, in agreement with Herschkowitz et al.11 These include underexpression of RB1 in approximately 40% of cases and overexpression of p16ink4a and/or Cyclin E, both established markers for Rb dysfunction.38 E2F1 itself is overexpressed in 25% of ERGO breast cancers. These abnormal patterns of expression are also present in non-ERGO basal-like tumors, but are less common, and are rarely observed in the other subtypes of breast cancer. E2F3 overexpression is commonly observed in ERGO tumors that arise in ovarian and prostate cancer, indicating a derangement in the Rb/E2F axis, but in the absence of39 widespread RB1 underexpression, it is not clear from our findings that the molecular lesion involves RB1 itself. However, in the recently published comprehensive analysis of breast cancer, Rb loss was identified as a feature of high-grade serous ovarian cancers.12
The fact that many of the ERGO tumor-associated E2F-responsive genes are “simultaneously” overexpressed in different tumor samples, and in different tumor types, raises interesting questions regarding the mechanisms underlying this phenomenon. Did these changes occur sequentially, one gene at a time in each tumor, or did they occur as a consequence of the activation of a multigene expression program? The latter possibility is speculative, but it is appealing, if only because it is parsimonious. Moreover, Rb is known to recruit histone deacetylase 1, a histone-modifying enzyme.40,41 Both Rb and E2F participate in multiprotein complexes that alter chromatin and can affect E2F-dependent gene expression42–44 at multiple sites. The examination of patterns of E2F-responsive gene expression in multiple contiguous samples from individual tumors may shed light on this issue.
The findings of the present study have potential clinical applications in diagnosis, prognosis, and therapy. With regard to therapeutic targeting, overexpressed E2F-responsive genes that are associated with ERGO tumors are an unusually well-suited group of potential candidate genes for targeted therapy. Their provenance as defining genes for the ERGO tumor subset, and the recurring patterns of their derangement in different tumors at the same organ site and at a different organ site would assign them substantial importance as potential drivers of malignant progression. The fact that most of these overexpressed genes are involved in DNA synthesis, DNA repair, or mitosis, and especially the last, makes these genes especially attractive as cancer therapeutic targets.
Our novel finding that multiple members of a group of E2F-responsive genes are overexpressed simultaneously in “each” ERGO tumor makes it possible to focus on appropriate targets, consider new strategies for combining targeted therapies, and identify the specific subsets of patients who are most likely to benefit from specific multitarget treatment regimens. This focuses attention on a circumscribed, but relatively long list of potential target genes that share similar critical functions required for cell proliferation. While the ERGO phenotype itself is restricted to a relatively small subset of human breast cancers (20–30%), multiple therapeutic targets of interest would be expected to occur together in the same tumors. Therefore, it might be feasible to develop optimized combinations of targeted agents in advance, with the expectation that substantial numbers of patients could still benefit from a particular combination. Similarly, the substantial overlap in overexpression of ERGO tumor–associated E2F-responsive genes across organ sites of tumor origin (~30%) might be expected to further increase the number of potential candidates that might respond to a particular multitargeted drug combination.
The patterns observed in primary prostate cancer offer a promising lead with regard to the potential “prognostic” value of patterns of E2F-responsive gene overexpression in ERGO tumors. Primary prostate cancers were shown here to produce two major patterns of E2F-responsive gene overexpression, one of which was similar to the pattern found in metastatic tumors (Fig. 3C, PCA Pattern 1) and the other of which was not (Fig. 3C, PCA Pattern 2). We speculate that cases with PCA Pattern 1 may be more likely to develop micrometastases prior to diagnosis and later develop clinical recurrences than those with PCA Pattern 2. This distinction has practical therapeutic consequences in that it might help to clearly separate patients who do not require surgery for their primary tumors from those who might benefit from intensive systemic adjuvant therapy. One must be cautious in deriving conclusions that depend on small numbers of patients, however. While we have shown through a variety of statistical analyses that there is a meaningful and reproducible subset of E2F-responsive genes that serve as ERGO markers, that result does not guarantee that these genes will define a signature of clinically useful prognostic value. These issues remain to be clarified in future studies.
Supplementary Data
Supplementary Figure 1. A comparison of patterns of overexpression of the PCA list of top overexpressed E2F-responsive genes and the list of the top overexpressed E2F-responsive genes derived from the entropy minimization method (EM). For each gene in each list, the differences between levels of E2F-responsive gene overexpression between Group 1 and Group 2 were subjected to Student’s t test, and in each list, the genes were sorted by P values in ascending order (top down). For the EM list, each successive group of 25 genes was color-coded from red to yellow, and each color-coded gene that was shared by both lists was identified by color in the PCA list. Genes not shared by both lists were color coded as white.
Supplementary Table 1. List of E2F responsive genes compiled from the literature.
Supplementary Table 2. List of overexpressed E2F responsive genes identified using the entropy minimization method on the van’t Veer15 dataset.
Supplementary Table 3. List of alternative markers for basal-like breast cancers with associated references.
Footnotes
Author Contributions
Conceived and designed the experiments: SES, SAC, RS. Analyzed the data: SES, SAC. Wrote the first draft of the manuscript: SES. Contributed to the writing of the manuscript: SES, SAC, RS. Agree with manuscript results and conclusions: SES, SAC, RS. Jointly developed the structure and arguments for the paper: SES, SAC, RS. Made critical revisions and approved final version: SAC, RS. SES passed away while the manuscript was in final revision and was therefore unable to review the final manuscript or to provide a full disclosure of his potential competing interests. The other authors reviewed and approved of the final manuscript.
ACADEMIC EDITOR: JT Efird, Editor in Chief
FUNDING: This work was supported in part by U.S. National Institutes of Health R01 Awards CA140214 and AI076318. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Dr. Shackney was an employee of Intelligent Oncotherapeutics. The other authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties.
REFERENCES
- 1.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2(12):910–7. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- 2.Gray-Bablin J, Zalvide J, Fox MP, Knickerbocker CJ, DeCaprio JA, Keyomarsi K. Cyclin E, a redundant cyclin in breast cancer. Proc Natl Acad Sci USA. 1996;93(26):15215–20. doi: 10.1073/pnas.93.26.15215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60(14):3689–95. [PubMed] [Google Scholar]
- 4.Shackney SE, Silverman JF. Molecular evolutionary patterns in breast cancer. Adv Anat Pathol. 2003;10(5):278–90. doi: 10.1097/00125480-200309000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Ishida S, Huang E, Zuzan H, et al. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol. 2001;21(14):4684–99. doi: 10.1128/MCB.21.14.4684-4699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black EP, Huang E, Dressman H, et al. Distinct gene expression phenotypes of cells lacking Rb and Rb family members. Cancer Res. 2003;63(13):3716–23. [PubMed] [Google Scholar]
- 7.Livasy CA, Karaca G, Nanda R, et al. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol. 2006;19(2):264–71. doi: 10.1038/modpathol.3800528. [DOI] [PubMed] [Google Scholar]
- 8.Badve S, Dabbs DJ, Schnitt SJ, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol. 2011;24(2):157–67. doi: 10.1038/modpathol.2010.200. [DOI] [PubMed] [Google Scholar]
- 9.Metzger-Filho O, Tutt A, de Azambuja E, et al. Dissecting the heterogeneity of triple-negative breast cancer. J Clin Oncol. 2012;30(15):1879–87. doi: 10.1200/JCO.2011.38.2010. [DOI] [PubMed] [Google Scholar]
- 10.Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene. 2006;25(43):5846–53. doi: 10.1038/sj.onc.1209876. [DOI] [PubMed] [Google Scholar]
- 11.Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008;10(5):R75. doi: 10.1186/bcr2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koboldt DC, Fulton RS, McLellan MD, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez-Knowles E, O’Toole SA, McNeil CM, et al. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int J Cancer. 2010;126(5):1121–31. doi: 10.1002/ijc.24831. [DOI] [PubMed] [Google Scholar]
- 14.Park Y, Shackney S, Schwartz R. Network-based inference of cancer progression from microarray data. IEEE/ACM Trans Comput Biol Bioinform. 2009;6(2):200–12. doi: 10.1109/TCBB.2008.126. [DOI] [PubMed] [Google Scholar]
- 15.van’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 16.Dai H, van’t Veer L, Lamb J, et al. A cell proliferation signature is a marker of extremely poor outcome in a subpopulation of breast cancer patients. Cancer Res. 2005;65(10):4059–66. doi: 10.1158/0008-5472.CAN-04-3953. [DOI] [PubMed] [Google Scholar]
- 17.Jönsson G, Staaf J, Vallon-Christersson J, et al. The retinoblastoma gene undergoes rearrangements in BRCA1-deficient basal-like breast cancer. Cancer Res. 2012;72(16):4028–36. doi: 10.1158/0008-5472.CAN-12-0097. [DOI] [PubMed] [Google Scholar]
- 18.Yu YP, Landsittel D, Jing L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22(14):2790–9. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 19.Chandran UR, Ma C, Dhir R, et al. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tone AA, Begley H, Sharma M, et al. Gene expression profiles of luteal phase fallopian tube epithelium from BRCA mutation carriers resemble high-grade serous carcinoma. Clin Cancer Res. 2008;14(13):4067–78. doi: 10.1158/1078-0432.CCR-07-4959. [DOI] [PubMed] [Google Scholar]
- 21.Tone AA, Virtanen C, Shaw PA, Brown TJ. Decreased progesterone receptor isoform expression in luteal phase fallopian tube epithelium and high-grade serous carcinoma. Endocr Relat Cancer. 2011;18(2):221–34. doi: 10.1530/ERC-10-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Network CGAR. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shackney SE, Pollice AA, Smith CA, et al. Intracellular coexpression of epidermal growth factor receptor, Her-2/neu, and p21ras in human breast cancers: evidence for the existence of distinctive patterns of genetic evolution that are common to tumors from different patients. Clin Cancer Res. 1998;4(4):913–28. [PubMed] [Google Scholar]
- 24.Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med. 2007;131(1):18–43. doi: 10.5858/2007-131-18-ASOCCO. [DOI] [PubMed] [Google Scholar]
- 25.Shannon CE. A mathematical theory of communication. ACM SIGMOBILE Mob Comput Commun Rev. 2001;5(1):3–55. [Google Scholar]
- 26.Li H, Zhang K, Jiang T. Minimum entropy clustering and applications to gene expression analysis. Proc IEEE Comput Syst Bioinform Conf. 2004:142–51. doi: 10.1109/csb.2004.1332427. [DOI] [PubMed] [Google Scholar]
- 27.Saeed AI, Sharov V, White J, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34(2):374–8. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 28.Nielsen TO, Hsu FD, Jensen K, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10(16):5367–74. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 29.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 30.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U SA. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sousa B, Paredes J, Milanezi F, et al. P-cadherin, vimentin and CK14 for identification of basal-like phenotype in breast carcinomas: an immunohistochemical study. Histol Histopathol. 2010;25(8):963–74. doi: 10.14670/HH-25.963. [DOI] [PubMed] [Google Scholar]
- 32.Olsson AY, Feber A, Edwards S, et al. Role of E2F3 expression in modulating cellular proliferation rate in human bladder and prostate cancer cells. Oncogene. 2007;26(7):1028–1037. doi: 10.1038/sj.onc.1209854. [DOI] [PubMed] [Google Scholar]
- 33.Sharma A, Yeow WS, Ertel A, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010;120(12):4478–92. doi: 10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tabach Y, Milyavsky M, Shats I, et al. The promoters of human cell cycle genes integrate signals from two tumor suppressive pathways during cellular transformation. Mol Syst Biol. 2005;2005;1:0022. doi: 10.1038/msb4100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38(9):1043–8. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- 36.Mosley JD, Keri RA. Cell cycle correlated genes dictate the prognostic power of breast cancer gene lists. BMC Med Genomics. 2008;1:11. doi: 10.1186/1755-8794-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foulkes WD, Brunet JS, Stefansson IM, et al. The prognostic implication of the basal-like (cyclin E high/p27 low/p53+/glomeruloid-microvascular-proliferation+) phenotype of BRCA1-related breast cancer. Cancer Res. 2004;64(3):830–5. doi: 10.1158/0008-5472.can-03-2970. [DOI] [PubMed] [Google Scholar]
- 38.Parry D, Bates S, Mann DJ, Peters G. Lack of cyclin D-Cdk complexes in Rb-negative cells correlates with high levels of p16INK4/MTS1 tumour suppressor gene product. EMBO J. 1995;14(3):503–11. doi: 10.1002/j.1460-2075.1995.tb07026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herrera RE, Sah VP, Williams BO, Mäkelä TP, Weinberg RA, Jacks T. Altered cell cycle kinetics, gene expression, and G1 restriction point regulation in Rb-deficient fibroblasts. Mol Cell Biol. 1996;16(5):2402–7. doi: 10.1128/mcb.16.5.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magnaghi-Jaulin L, Groisman R, Naguibneva I, et al. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391(6667):601–5. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- 41.Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92(4):463–73. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- 42.Gunawardena RW, Siddiqui H, Solomon DA, et al. Hierarchical requirement of SWI/SNF in retinoblastoma tumor suppressor-mediated repression of Plk1. J Biol Chem. 2004;279(28):29278–85. doi: 10.1074/jbc.M400395200. [DOI] [PubMed] [Google Scholar]
- 43.Schmit F, Korenjak M, Mannefeld M, et al. LINC, a human complex that is related to pRB-containing complexes in invertebrates regulates the expression of G2/M genes. Cell Cycle. 2007;6(15):1903–13. doi: 10.4161/cc.6.15.4512. [DOI] [PubMed] [Google Scholar]
- 44.Trouche D, Le Chalony C, Muchardt C, Yaniv M, Kouzarides T. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci USA. 1997;94(21):11268–73. doi: 10.1073/pnas.94.21.11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. A comparison of patterns of overexpression of the PCA list of top overexpressed E2F-responsive genes and the list of the top overexpressed E2F-responsive genes derived from the entropy minimization method (EM). For each gene in each list, the differences between levels of E2F-responsive gene overexpression between Group 1 and Group 2 were subjected to Student’s t test, and in each list, the genes were sorted by P values in ascending order (top down). For the EM list, each successive group of 25 genes was color-coded from red to yellow, and each color-coded gene that was shared by both lists was identified by color in the PCA list. Genes not shared by both lists were color coded as white.
Supplementary Table 1. List of E2F responsive genes compiled from the literature.
Supplementary Table 2. List of overexpressed E2F responsive genes identified using the entropy minimization method on the van’t Veer15 dataset.
Supplementary Table 3. List of alternative markers for basal-like breast cancers with associated references.