

Abstract
Bone marrow derived endothelial progenitor cells (EPCs) are early precursors of mature endothelial cells which replenish aging and damaged endothelial cells. The authors studied a diabetic swine model to determine if induction of DM adversely affects either bone marrow or circulating EPCs and whether a HMG-CoA reductase inhibitor (statin) improves development and recruitment of EPCs in the absence of cholesterol lowering. Streptozotocin was administered to Yorkshire pigs to induce DM. One month after induction, diabetic pigs were treated with atorvastatin (statin, n = 10), ezetimibe (n = 10) or untreated (n = 10) and evaluated for number of bone marrow and circulating EPCs and femoral artery endothelial function. There was no effect of either medication on cholesterol level. One month after induction of DM prior to administration of drugs, the number of bone marrow and circulating EPCs significantly decreased (P < 0.0001) compared to baseline. Three months after DM induction, the mean proportion of circulating EPCs significantly increased in the atorvastatin group, but not in the control or ezetimibe groups. The control group showed progressive reduction in percentage of flow mediated vasodilatation (no dilatation at 3 months) whereas the atorvastatin group and ezetimibe exhibited vasodilatation, 6% and 4% respectively. DM results in significant impairment of bone marrow and circulating EPCs as well as endothelial function. The effect is ameliorated, in part, by atorvastatin independent of its cholesterol lowering effect. These data suggest a model wherein accelerated atherosclerosis seen with DM may, in part, result from reduction in EPCs which may be ameliorated by treatment with a statin.
Keywords: diabetes mellitus, endothelium, endothelial progenitor cells, statin, inflammation, cytometry
Endothelial dysfunction heralds the development of atherosclerosis and clinical events (1). Patients with diabetes mellitus (DM) have marked endothelial dysfunction, accelerated atherosclerosis and an increased risk of cardiovascular events (2). Endothelial progenitor cells (EPCs) are precursors of mature endothelial cells originating from the bone marrow that presumably replenish aging and damaged endothelial cells that line blood vessels (3,4). EPCs are thought to home to sites of ischemia and participate in neovascularization and collateral development (5–7). The number of circulating EPCs and their migratory activity is reduced in patients with risk factors for ischemic cardiovascular disease and negatively correlated with the Framingham cardiovascular risk factor score (8). Limb ischemia and coronary artery bypass grafting induce a rapid, transient mobilization of EPCs from the bone marrow.
Recent studies suggest that statins may enhance EPC differentiation and migration to sites of vascular injury (9,10). Two independent laboratories published data indicating that statin treatment increases the circulating pool of EPCs by mobilizing them from bone marrow and also inhibiting EPC apoptosis (11,12). It is unknown if statins directly affect bone marrow and circulating EPCs in the setting of cardiac risk factors, such as DM, and whether this effect is independent from an effect on cholesterol level.
The diabetic state is associated with decreased number of circulating EPCs (13). Also, the EPCs of patients with DM are characterized by decreased proliferation capacity and reduction of their adhesiveness and ability to form capillary tubes in vitro (14). One study of diabetic mice found that at sacrifice the number of bone marrow and circulating EPCs was reduced, but the temporal relationship was not evaluated in this small animal model (15). There are no published studies evaluating simultaneously the bone marrow and circulating EPC number in response to the diabetic state over time and/or the response of EPCs to statin treatment.
We previously identified a population of progenitor cells in the porcine bone marrow, called side population cells (SP), which differentiate into endothelial cells (16). Other studies have shown that SP cells are not only present in bone marrow, but they are also observed in the arterial wall where they participate in angiogenesis (17).
In this study, we tested the hypothesis that the diabetic state will reduce EPCs in the bone marrow as well as mobilization of EPCs. Also, we hypothesized that statins promote EPC mobilization from the bone marrow and increase circulating EPCs in the blood in the diabetic state. Although statins have been shown to mobilize EPCs and improve endothelial function (18). It is unknown if other classes of lipid lowering agents such as ezetimibe have similar effects. Accordingly, we conducted a study in a porcine model of DM evaluating the effects of atorvastatin and ezetimibe on bone marrow and circulating EPC number and endothelial function to determine if statins and/or ezetimibe promote mobilization of EPCs and/or improve endothelial function in the setting of DM.
Methods
Study Design
A total of 30 pigs were studied at baseline and 1 month after induction of DM for bone marrow and circulating EPCs, endothelial function and a marker of inflammation, lipoprotein phospholipase A2 (Lp-PLA2). This study was performed following approval of the Institutional Animal Care and Use Committee of the University of Pennsylvania. One month after induction of DM, pigs were randomly assigned to receive atorvastatin 10 mg per day or ezetimibe 10 mg per day or were untreated for a total of 3 months. The aforementioned outcome measures were then repeated at study completion which occurred at 4 months after induction of DM.
Pig Model
Type I DM was induced in 30 Yorkshire pigs (28–30 kg) by infusing a single dose of streptozotocin (Sicor Pharmaceuticals, Irvine, CA, 125 mg/kg) IV over a period of 30 min by a Harvard pump as described in our previous publication (19). All of the pigs, including the controls, were made diabetic using the same protocol. Animals were recovered from anesthesia and returned to cages, followed by feeding that included dextrose powder to avoid hypoglycemic shock due to the excessive blood insulin levels after the lysis of the beta cells. The first blood glucose levels were measured 4 h after induction, followed by daily measurements, using a glucometer (Beyer, Tarrytown, NY) every day before feeding for the first 7 days and then on a weekly basis thereafter.
Bone Marrow Harvest
A sternal bone marrow aspirate (5 ml) was collected at baseline, prior to induction of diabetes, and at 1 month, 4 months after the induction of diabetes in order to assess bone marrow populations of EPCs. The bone marrow aspirate was collected under standard sterile technique while the animal was sedated as per prior published methods (16).
Side Population Cells
Porcine endothelial progenitors were identified using a modification of the procedure described by Goodell et al. (20,21). A functional assay using Hoechst 33342 was used to identify these progenitors (Fig. 1). In several species, it has been noted that stem cells efflux this dye resulting in a low staining population termed the side population (SP) (22,23). Briefly, red blood cells in porcine bone marrow or peripheral blood were removed using 1× ammonium chloride solution and a single-cell suspension was achieved. Cells were aliquoted (2 × 106 cells per tube) and incubated for 2 h at 37°C with Hoechst 33342 (Sigma-Aldrich) at a concentration of 5 μg/ml. with or without 50 μM verapamil (Sigma-Aldrich). Following two washes with ice cold HBSS + serum, surface staining for CD45 was performed using an indirect method (mouse antiporcine CD45/FITC goat anti-mouse Ig) to identify hematopoietic progenitors. Just prior to analysis, propidium iodide (Molecular Probes, 2 μg/ml) was added as a viability marker. All analyses were performed on a BD LSRII digital high speed analytical flow cytometer (BD Biosciences). After UV excitation at 355 nm, Hoechst blue (collected through a 405/30 filter) and red (670/40 filter) signals were analyzed to detect the SP. One hundred thousand live cells were collected per file and EPCs were defined as propidium iodide negative, singlets (based on light scatter), dim CD45 and positive for SP. Data were analyzed using FlowJo (TreeStar, Inc.) software.
Figure 1.
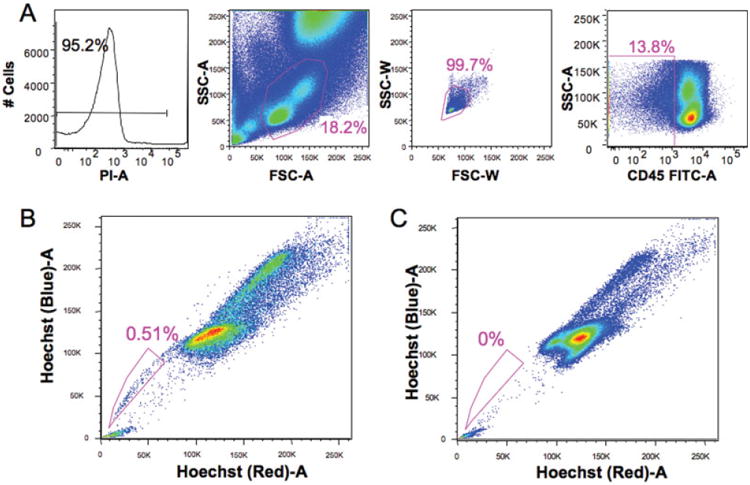
Detection of SP cells in porcine bone marrow. Porcine bone marrow cells were stained with 5 μM of Hoechst 33342 for 120 min at 37°C with or without 50 μM verapamil. SP cells analysis was performed on BD Bioscience LSRΠ with UV laser. Gating was based on live (negative for PI), singlet (based on light scatter) and CD45 negative cells (A). Side population cells (EPCs) were clearly detected after Hoechst 33342 staining, and gated side population cells (EPCs) account for 0.51% of the CD45 negative cells (B). In the presence of verapamil, gated side population cells almost completely disappeared (C).
Endothelial Function
In vivo endothelial function was determined at baseline, as well as 1 month and 4 months after DM induction. The 1-month measurement was done prior to randomization. Flow-mediated dilatation (FMD), the measurement of endothelial function, was evaluated by quantifying changes in femoral artery diameter following a 5-min femoral arterial occlusion with a mechanical clamp. The femoral artery was imaged in the longitudinal plane using an ultrasound system equipped with vascular software for two-dimensional imaging, color and power Doppler, an internal ECG, and a linear array transducer with a minimum frequency of 7 MHz. Shear stress-induced vasodilatation was measured utilizing longitudinal images acquired 1 min after release of occlusive pressure. Analysis of luminal diameter was measured using digital still images captured during end-diastole using specialized imaging software (Medical Imaging Applications, Brachial Analyzer Iowa City, Iowa). The lumen intima was identified automatically by edge detection software, with manual adjustments performed when necessary. Arterial diameters (mm) were calculated as the mean distance between the anterior and posterior wall at the vessel–blood interface. Flow-mediated vasodilatation was expressed as the ratio of diameter change to baseline diameter.
Measurement of Lp-PLA2 Mass and Activity
Measurement of Lp-PLA2 mass was done with the diaDexus “PLAC” immunoassay using two specific monoclonal antibodies described by Caslake et al. (24). The assay system utilizes monoclonal anti-Lp-PLA2 antibody (2C10) directed against Lp-PLA2 for solid phase immobilization on the microwell strips. Plasma samples were added to the plate and incubated for 10 min at 20–26°C. A second monoclonal anti-Lp-PLA2 antibody (4B4) labeled with the enzyme horseradish peroxidase (HRP) was then added and reacted with the immobilized antigen at 20–26°C for 180 min, resulting in the Lp-PLA2 molecules being captured between the solid phase and the enzyme-labeled antibodies. The wells are washed with a supplied buffer to remove any unbound antigen. The substrate, tetramethylbenzidine (TMB), is then added and incubated at 20–26°C for 20 min, resulting in the development of a blue color. Color development is stopped with the addition of Stop Solution, changing the color to yellow. The absorbance of the enzymatic turnover of the substrate is determined spectrophotometrically at 450 nm and is directly proportional to the concentration of Lp-PLA2 present. A set of Lp-PLA2 Calibrators is used to plot a standard curve of absorbance versus Lp-PLA2 concentration from which the Lp-PLA2 concentration in the test sample can be determined.
The plasma Lp-PLA2 activity was measured with a colorimetric activity method (CAM) provided by diaDexus, Inc. (South San Francisco, CA, USA). Samples, standards, or controls are added to wells of a non-binding 96-well microplate, followed by addition of CAM reaction buffer containing substrate. In the presence of Lp-PLA2 enzyme, the substrate is converted upon hydrolysis by the phospholipase enzyme. The change in absorbance is immediately measured at 405 nm over 60–180 s. The level of Lp-PLA2 activity in nmol/min/mL is calculated from the slope (OD405/min), based on a standard conversion factor from a p-nitrophenol calibration curve.
Statistical Analysis
For each treatment group, changes in glucose, cholesterol and weight over time were analyzed using paired t-tests. The FMD data were analyzed using a mixed effects model where correlations between repeated measurements were considered on the same animals across time (25). Terms in the model included age of animal, treatment group (control, atorvastatin, ezetimibe), side (left versus right artery) and time of measurement (1 min or 2 min) after initiation of hyperemia. Differences in the effect of treatment over time were examined using an interaction between treatment group and age of animal using a global F-test to test for differences in the effects of animal age among the different treatment groups, followed by Wald tests to examine specific changes (e.g., baseline versus 1 month).
For EPC experiments, linear regressions were constructed using the individual paired data time points (baseline versus 1 month and 1 versus 4 months. For the analysis of 1 month versus 4 months, the baseline percentage of EPCs was added to the model in an analysis of covariance to account for variability among animals. Linear regression analysis was used to examine the association between the mean FMD change and EPC change between baseline and 1 month. For this analysis, mean FMD was determined across both sides and times of measurement for each animal.
For analysis of Lp-PLA2, differences between time points were examined using paired Wilcoxon signed-rank tests using only those animals with measurements at both time points of interest. To ensure maximum power to detect effects, all of the treatment groups were pooled for the comparison of animals from baseline to 1 month. For the comparison of animals from 1 month to post-treatment, treatment groups were analyzed separately (26).
Data were analyzed using R 2.6; a Type I error of 0.05 was used to declare significance for hypothesis tests and any P value (P) less than 0.10 was reported. All hypothesis tests were two-sided except those for the comparison of EPC at baseline versus 1 month, and for 4 months versus 1 month. Here a priori hypotheses indicated that we should test only for a decrease in EPCs between baseline and 1 month and an increase between 1 month and 4 months. No corrections for multiple comparisons were performed.
The statistical methods were chosen with the goal of balancing the competing need to maximize power and protect against false positives/invalid type I error rates in our hypothesis tests. The experiment was initially designed to look at within animal differences, and with the available sample sizes we anticipated that we would have insufficient statistical power to make meaningful comparisons between groups. Thus rather than carry out an ANOVA-type analysis comparing time, treatment and treatment by time effects, we focused our analyses on a limited number of within-subject analyses, specifically comparing the combined baseline versus 1 month and treatment-specific 1 month versus 4 month time points. This analysis suggested that EPCs significantly increased in the atorvastatin group, but the P value did not initially achieve strict statistical significance. We adjusted statistically for baseline variability in EPCs in the analysis using an analysis of covariance. Our study may have been limited somewhat by statistical power; however, the findings are unique and we have reported both P values and estimates of the effect, with the hope of stimulating further research and hypotheses in this area.
Results
Laboratory Data
The animal weight and laboratory data is listed in Table 1. There were three noteworthy changes over time in these data. The mean glucose level was significantly elevated above baseline in all three groups 1 month after induction of diabetes (P < 0.0001). When analyzed separately by group, changes in cholesterol levels declined from baseline to 1 month but not significantly. Since the animals were not yet randomized to treatment at 1 month, we also used a more powerful analysis, comparing cholesterol levels at 1 month versus baseline in a pooled comparison of all animals. Here the reduction in cholesterol was statistically significant (P = 0.005). Lastly, the animals from all three groups gained weight in the final 4 weeks of the study compared to week 1 of the study (P < 0.05 for each group).
Table 1. Mean (SEM) glucose, cholesterol and weight over timea.
| DM ONLY | DM + ATORVASTATIN | DM + EZETIMIBE | |
|---|---|---|---|
| Glucose (mg/dL) | |||
| 0 month | 100.0 (7.2) | 115.9 (5.5) | 81.6 (11.4) |
| 1 months | 323.9 (21.3) | 319.3 (24.8) | 359.8 (21.8) |
| 4 months | 307.3 (54.0) | 339.4 (25.3) | 306.6 (59.0) |
| P value | |||
| 0 vs. 1 month | <0.0001 | <0.0001 | <0.0001 |
| P value | |||
| 1 vs. 4 month | NS | NS | NS |
| Cholesterol (mg/dL) | |||
| 0 month | 110.7 (11.5) | 85.9 (5.2) | 89.3 (4.7) |
| 1 months | 74.6 (6.7) | 72.4 (3.9) | 66.5 (5.4) |
| 4 months | 86.4 (3.2) | 78.6 (4.6) | 60.5 (4.7) |
| P value | |||
| 0 vs. 1 month | NS | NS | 0.06 |
| P value | |||
| 1 vs. 4 month | NS | NS | NS |
| Weight (kg) | |||
| 0 month | 35.2 (2.3) | 34.3 (3.3) | 34.2 (3.7) |
| 1 months | 36.1 (3.2) | 33.0 (3.5) | 31.9 (2.8) |
| 4 months | 56.1 (7.6) | 49.1 (3.6) | 42.7 (4.8) |
| P value | |||
| 0 vs. 1 month | NS | NS | NS |
| P value | |||
| 1 vs. 4 months | 0.004 | 0.001 | 0.017 |
NS indicates a P value greater than 0.10.
Bone Marrow and Circulating EPCs
The number of bone marrow EPCs was measured at baseline, 1 month and 4 months after induction of DM, based on identification of SP (Fig. 1). One month following the induction of DM, the mean levels of both bone marrow-derived and circulating EPCs were 44.7% (P = 0.001) and 26.9% (P < 0.0001) respectively below baseline (Fig. 2). In the atorvastatin group at 4 months, the mean levels of bone marrow-derived and circulating EPC's increased 181% (P = 0.065) and 203% (P = 0.080) above the level observed at 1 month. While statistical significance was not achieved for this direct comparison, the result for circulating cells (P = 0.043), but not marrow-derived cells (P = 0.065) achieved significance after adjusting for variation in baseline levels of EPCs. In the ezetimibe group at 4 months, the mean levels of bone marrow-derived and circulating EPC's were 76% and 94%, respectively, of the levels observed at 1 month (P > 0.05, NS). In the control group at 4 months, the mean levels of bone marrow-derived and circulating EPC's were 63% and 68%, respectively, of the levels observed at 1 month (P > 0.05, NS). Taken together, these data demonstrate that treatment of diabetic pigs with atorvastatin, but not ezetimibe, significantly increased circulating EPCs.
Figure 2.
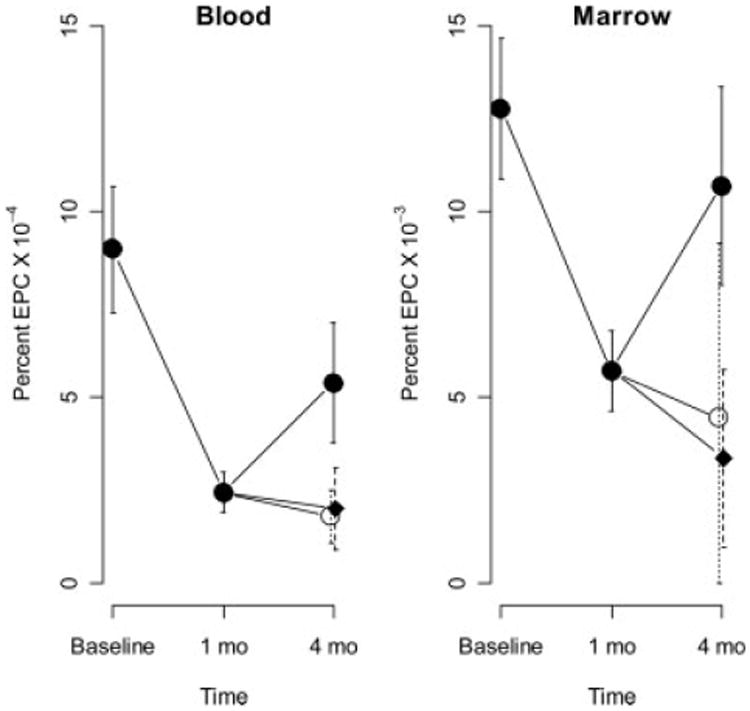
Mean EPC levels across time in blood and marrow. Vertical lines indicate standard errors for the combined groups at baseline and 1 month, and for control: rectangles, atorvastatin closed circles, ezetimibe open circles at 4 months.
Endothelial Function
Next, we examined the effect of treatment on endothelial function. Prior to induction of DM, femoral arteries demonstrated a flow-mediated vasodilatory response of 11.7%; comparable in magnitude to that reported in humans (8). The mean FMD did not differ significantly among the right or left femoral artery or at 1 min or 2 min post-hyperemia. One month after induction, the mean FMD decreased significantly (P < 0.0001, Fig. 3) demonstrating the effect of diabetes. There was also strong evidence of differences in FMD over time that varied among the control and treated groups (P < 0.0001, Fig. 3). At 4 months, the mean FMD was lower than the 1-month (P = 0.027) for control group, however, in contrast, the atorvastatin group mean FMD was significantly higher (P = 0.003, Table 2). In the ezetimibe treatment group at 4 months, the FMD was significantly lower than baseline (P = 0.007, results not shown) and also significantly lower than 1 month (P < 0.0001, Fig. 3). However, the mean FMD for the ezetimibe group was higher than the control group at 4 months (P = 0.004, Table 2).
Figure 3.
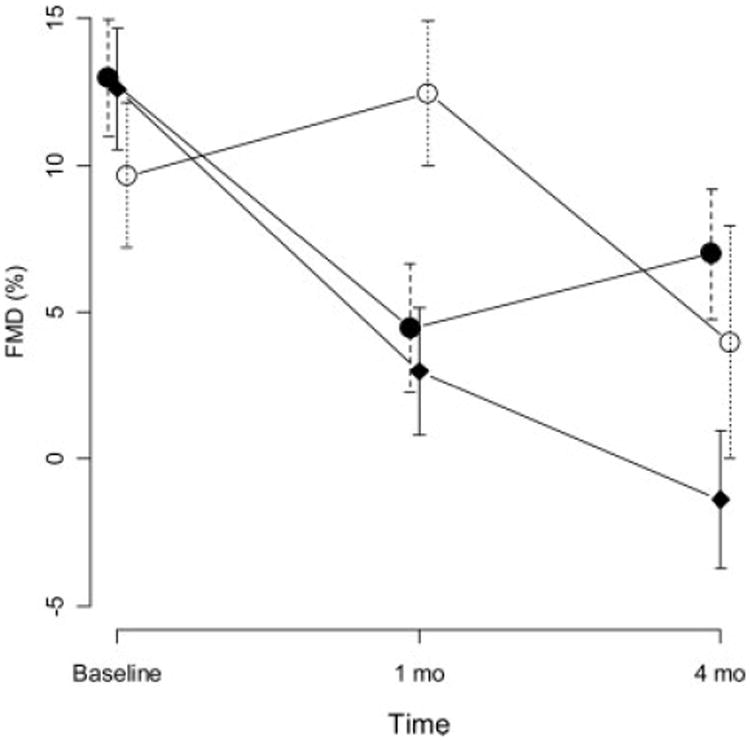
Mean percent flow mediated vasodilatation (FMD) time points during the study. Vertical lines indicate standard errors. Control: Rectangles, Atorvastatin closed circles, Ezetimibe open circles.
Table 2. Mean (SEM) for percent FMD for atorvastatin and ezetimibe groups compared to control at same time pointa.
| AGE | ATORVASTATIN | EZETIMIBE |
|---|---|---|
| Baseline | 12.96 (1.99) | 9.64 (2.47) |
| 1 month | 4.45 (2.11) | 12.44 (2.47)* |
| 4 months | 6.98 (2.23)* | 3.96 (2.55)* |
Asterisks (*) indicate P < 0.05. Data are compared to control at same time point.
When the number of EPCs were compared to the change in FMD between baseline and 4 weeks, animals with higher circulating EPCs at baseline had less reduction in FMD (P = 0.027, Fig. 4). However the difference between FMD at 4 weeks and baseline was significantly positively associated with baseline EPCs. Thus, animals with higher baseline EPC's experienced less of a decrease in FMD, and in some cases actually increased FMD at 4 weeks versus baseline (Fig. 4). No significant trends were observed between FMD changes and EPC levels in the marrow or when the 1-month and 4-month time points were compared.
Figure 4.
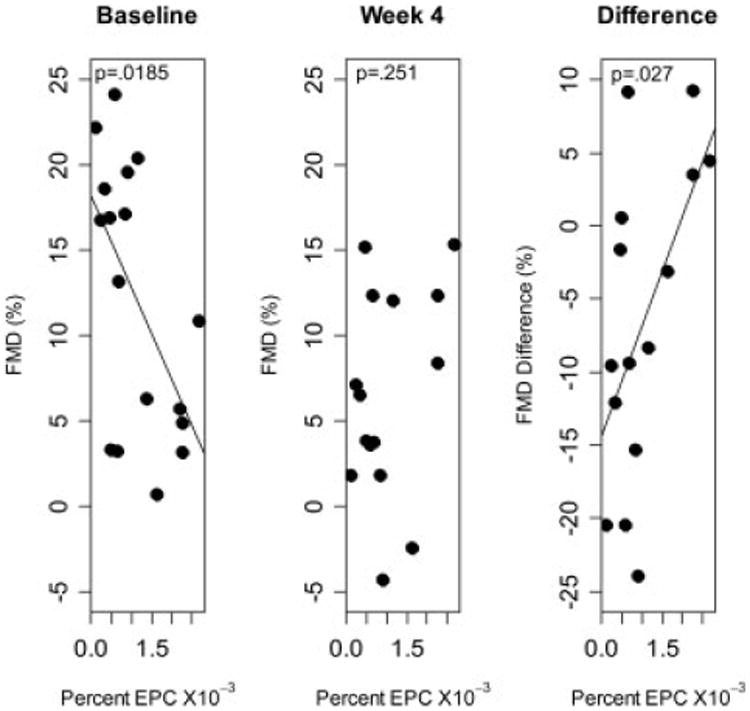
Association between baseline EPC and FMD at baseline, 4 weeks or the difference in FMD between 4 weeks and baseline.
Lp-PLA2
In order to characterize the state of oxidative stress Lp-PLA2, a marker of inflammation (27), was measured at various time points (Table 3). In association with induction of diabetes Lp-PLA2 activity was significantly higher compared to baseline for the three groups combined (P = 0.003). A strong trend in reduction of Lp-PLA2 activity was observed in both the atorvastatin (P = 0.055) and ezetimibe (P = 0.063) treatment groups at 4 months compared to baseline. The Lp-PLA2 mass was also evaluated at pre-determined time points. There was a trend in elevation of mass at 1 month when all groups were pooled. There was no significant change in Lp-PLA2 mass at the 1 month or 4 month time points compared to control (Table 4).
Table 3. Lp-PLA2 activity (nmol/min/mL) for animals with paired measurements at each set of time pointsa.
| BASELINE | 1 MONTH | 4 MONTHS | P VALUE | |
|---|---|---|---|---|
| Control | 112.7 (104, 121) | 126.9 (123, 149) | 131.6 (94, 169) | 0.689 |
| Atorvastatin | 117.2 (109, 120) | 157.5 (143, 167) | 137.5 (128, 150) | 0.055 |
| Ezetimibe | 112.2 (109, 128) | 149.9 (134, 154) | 93.5 (73, 138) | 0.063 |
Entries show median (inter-quartile range). P = 0.0003 for 1 month compared to baseline when pooled among all groups.
Table 4. Lp-PLA2 mass (mcg/L) for animals with paired measurements at each set of time pointsa.
| BASELINE | 1 MONTH | 4 MONTHS | P VALUE | |
|---|---|---|---|---|
| Control | 1.1 (0.9, 1.3) | 1.7 (1.3, 2.3) | 1.4 (1.1, 2.1) | 0.25 |
| Atorvastatin | 1.3 (0.8, 1.5) | 1.2 (1.1, 1.3) | 1.0 (1.0, 2.1) | 1.00 |
| Ezetimibe | 1.0 (1.0, 2.0) | 1.6 (1.0, 2.1) | 2.1 (1.7, 2.5) | 1.00 |
Entries show median (inter-quartile range). P = 0.079 for 1 month compared to baseline when pooled among all groups.
Discussion
Evaluation of endothelial progenitor numbers and function are important in many pathologic conditions. As with many studies of progenitors, flow cytometry offers a useful approach to detect these rare cells. This is the first prospective study, using a large animal model, showing that DM reduces both the concentration of bone marrow as well as circulating EPCs. Flow cytometric measurement of EPCs, using the SP approach revealed that in diabetic animals, administration of atorvastatin enhanced EPC mobilization to the circulation. Interestingly, ezetimibe did not induce a significant increase in bone marrow or circulating EPCs strongly suggesting that the mobilization of EPCs following treatment with atorvastatin is not wholly related to its cholesterol lowering effects. This effect was observed in pigs and may not be seen with other animals or humans.
One major difficulty in studying the effect of endothelial progenitors is that the definition in both humans and animals is controversial. Unfortunately the cell surface markers available in pigs are significantly limited compared to humans and mice. For example, there is no available CD34 pig surface maker for flow cytometry. Therefore, we chose to evaluate a progenitor population of cells in the bone marrow that has been well defined SP cells. This cell population was reported by Goodell et al. almost a decade ago (20,22,23). In a previous publication we have reported that SP cells differentiate into endothelial cells (16). In humans, by flow cytometry endothelial progenitors lie in the CD45 dim to negative population (as compared to the CD45 bright for hematopoietic progenitors) so we chose to further limit our study to this population. As such, the SP population provides for study of EPC phenotype cells (and potentially other progenitor cells) that may be affected by diabetes and play a role in the phenotypic manifestations of diabetes.
As anticipated, the induction of DM was accompanied by endothelial dysfunction and evidence of inflammation as measured by Lp-PLA2. Treatment with either atorvastatin or ezetimibe improved endothelial dysfunction over time and attenuated the increase in inflammation. These data suggest that the potential benefits of statins and ezetimibe in the setting of diabetes are not solely dependent upon mobilization of EPCs from the bone marrow.
The clinical data examining the mechanism(s) of action of ezetimibe compared with statin drugs are poorly defined at present. One study of 20 patients with chronic heart failure showed FMD improved after simvastatin, but not ezetimibe (18). Another study that included 20 patients with rheumatoid arthritis revealed that both of these classes of drugs improved FMD (28). The results from our study indicate that in this large animal model of DM, statin treatment as well as ezetimibe improved endothelial function in the setting of normal cholesterol levels. However, the absolute value for atorvastatin group was higher at 4 months compared to ezetimibe group.
Diabetic vasculopathy is characterized by early endothelial dysfunction and a subsequent marked increased risk of morbidity and mortality (2,29). A prospective study of patients with type II DM revealed that statin administration is efficacious in reducing the risk of first cardiovascular disease events, including stroke (30). Previous studies indicate that patients with DM have reduced levels of circulating EPCs (31,32) as well as abnormal function (33). Rivard et al. evaluated the effect of diabetes on angiogenesis and found that diabetes impairs endogenous neovascularization of ischemic tissues resulting from reduced expression of vascular endothelial growth factor (VEGF) (34). Prior to the current study a cause-effect relationship between EPCs and a biologic response, or therapeutic effect, on endothelial function was not discerned. Our results indicate that reduced circulating EPCs in the diabetic state are due to decreased bone marrow production of cells as well as decreased EPCs in the peripheral circulation. The potential mechanisms for increased circulating EPCs in response to atorvastatin are increased mobilization from marrow, increased survival (known effect in vitro), and reduced homing due to reduced endothelial damage. As such, the results indicate that the systemic effects of DM extend to the bone marrow microenvironment resulting in endothelial dysfunction. Furthermore, the role of DM in causing vasculopathy may be caused, at least in part, by endothelial injury coupled with a reduced endothelial response mechanism(s).
Statin drugs are reported to have pleiotropic effects beyond cholesterol lowering (35,36). The treatment of dyslipidemia with a statin increases the circulating pool of EPCs and inhibits EPC apoptosis in vitro (11,12). Clinically stable angina pectoris patients treated for 4 weeks with atorvastatin demonstrated an increase in the number of circulating EPCs and increased migratory capacity ex vivo (37). However, these studies were in the setting of elevated cholesterol and it was not clear if the reduction in low density lipoprotein (LDL) or a non-cholesterol related effect was responsible for the beneficial effect on EPC mobilization. The present results indicate that DM reduces both bone marrow reserves and circulating EPCs with accompanying endothelial dysfunction, and this effect is partially reversed by statin administration in the setting of normal cholesterol.
A consequence of DM is increased oxidative stress and inflammation (2). A previous investigation showed that DM increases oxidative stress and inflammation in coronary arteries of normocholesterolemic pigs (38) and the elevated Lp-PLA2 activity noted in this study confirms this finding. The EPC is thought to reside in the marrow and release into the circulation due to increased nitric oxide (NO) levels (39). The high oxidative state of DM likely results in uncoupling endothelial derived nitric oxide synthase (eNOS) and development of reactive oxygen species that impair NO availability that is important for EPC mobilization (4). It is likely that the reduction of circulating EPCs secondary to DM and the impaired endothelial dependent vasodilatation observed in this study are related to reduced NO availability. Statin administration has been shown to enhance EPC differentiation via PI 3-kinase/akt pathway (12) and this may be one pleiotropic effect that improves the bone marrow EPC niche and ultimately reduction in cardiovascular events in the diabetic population. Furthermore, HMG-CoA reductase inhibition normalizes endothelial function and reduces oxidative stress in the diabetic state by inhibiting activation of NADPH oxidase and by preventing eNOS uncoupling (40).
In conclusion, DM results in reduced levels of both bone marrow and circulating EPCs and was accompanied by impaired endothelial function. These detrimental effects are partially ameliorated by statin administration and as such demonstrate beneficial cholesterol-independent effects of statins on EPCs and endothelial function in the setting of DM. The pathological mechanism(s) resulting in reduced bone marrow EPCs deserves further investigation.
Acknowledgments
We thank Alexandra Sibley, BS, for her helpful comments and Harilla Profka for his expertise in animal care.
E.R. Mohler reports receiving honoria from Merck and BMS/Sanofi and research funding from GSK. R.L. Wilensky received research funding from GSK, Astra Zeneca and Boston Scientific and has equity interest in J&J. M. Yoder reports being co-founder and paid consultant for EndGenitor Technologies.
Grant sponsor: Pfizer Corporation.
Footnotes
Conflict of interest: None of the other authors report a potential conflict of interest.
Literature Cited
- 1.Kinlay S, Libby P, Ganz P. Endothelial function and coronary artery disease. Curr Opin Lipidol. 2001;12:383–389. doi: 10.1097/00041433-200108000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Mohler ER., III Therapy insight: Peripheral arterial disease and diabetes—From pathogenesis to treatment guidelines. Nat Clin Pract Cardiovasc Med. 2007;4:151–162. doi: 10.1038/ncpcardio0823. [DOI] [PubMed] [Google Scholar]
- 3.Fadini GP, Agostini C, Avogaro A. Characterization of endothelial progenitor cells. Biochem Biophys Res Commun. 2005;336:1–2. doi: 10.1016/j.bbrc.2005.07.119. [DOI] [PubMed] [Google Scholar]
- 4.Khakoo AY, Finkel T. Endothelial progenitor cells. Ann Rev Med. 2005;56:79–101. doi: 10.1146/annurev.med.56.090203.104149. [DOI] [PubMed] [Google Scholar]
- 5.Asahara T, Murohara T, Sullivan A, Silver M, van der ZR, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 6.Hristov M, Erl W, Weber PC. Endothelial progenitor cells: mobilization, differentiation, and homing. Arterioscler Thromb Vasc Biol. 2003;23:1185–1189. doi: 10.1161/01.ATV.0000073832.49290.B5. [DOI] [PubMed] [Google Scholar]
- 7.Möbius-Winkler S, Höllriegel R, Schuler G, Adams V. Endothelial progenitor cells: Implications for cardiovascular disease. Cytometry A. 2009;75A doi: 10.1002/cyto.a.20669. in press. this issue. [DOI] [PubMed] [Google Scholar]
- 8.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 9.Walter DH, Rittig K, Bahlmann FH, Kirchmair R, Silver M, Murayama T, Nishimura H, Losordo DW, Asahara T, Isner JM. Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105:3017–3024. doi: 10.1161/01.cir.0000018166.84319.55. [DOI] [PubMed] [Google Scholar]
- 10.Landmesser U, Engberding N, Bahlmann FH, Schaefer A, Wiencke A, Heineke A, Spiekermann S, Hilfiker-Kleiner D, Templin C, Kotlarz D, Mueller M, Fuchs M, Hornig B, Haller H, Drexler H. Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation. 2004;110:1933–1939. doi: 10.1161/01.CIR.0000143232.67642.7A. [DOI] [PubMed] [Google Scholar]
- 11.Llevadot J, Murasawa S, Kureishi Y, Uchida S, Masuda H, Kawamoto A, Walsh K, Isner JM, Asahara T. HMG-CoA reductase inhibitor mobilizes bone marrow-derived endothelial progenitor cells. J Clin Invest. 2001;108:399–405. doi: 10.1172/JCI13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M, Rutten H, Fichtlscherer S, Martin H, Zeiher AM. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–397. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fadini GP, Miorin M, Facco M, Bonamico S, Baesso I, Grego F, Menegolo M, de Kreutzenberg SV, Tiengo A, Agostini C, Avogaro A. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. 2005;45:1449–1457. doi: 10.1016/j.jacc.2004.11.067. [DOI] [PubMed] [Google Scholar]
- 14.Fadini GP, Sartore S, Albiero M, Baesso I, Murphy E, Menegolo M, Grego F, Vigili de KS, Tiengo A, Agostini C, Avogaro A. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler Thromb Vasc Biol. 2006;26:2140–2146. doi: 10.1161/01.ATV.0000237750.44469.88. [DOI] [PubMed] [Google Scholar]
- 15.Gallagher KA, Liu ZJ, Xiao M, Chen H, Goldstein LJ, Buerk DG, Nedeau A, Thom SR, Velazquez OC. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest. 2007;117:1249–1259. doi: 10.1172/JCI29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohler ER, 3rd, Fang Y, Shaffer RG, Moore J, Wilensky RL, Parmacek M, Levitan I. Hypercholesterolemia suppresses Kir channels in porcine bone marrow progenitor cells in vivo. Biochem Biophys Res Commun. 2007;358:317–324. doi: 10.1016/j.bbrc.2007.04.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sainz J, Al Haj Zen A, Caligiuri G, Demerens C, Urbain D, Lemitre M, Lafont A. Isolation of “side population” progenitor cells from healthy arteries of adult mice. Arterioscler Thromb Vasc Biol. 2006;26:281–286. doi: 10.1161/01.ATV.0000197793.83391.91. [DOI] [PubMed] [Google Scholar]
- 18.Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N, Schulz S, Manes C, Fischer D, de GK, Fliser D, Fauler G, Marz W, Drexler H. Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation. 2005;111:2356–2363. doi: 10.1161/01.CIR.0000164260.82417.3F. [DOI] [PubMed] [Google Scholar]
- 19.Mohler ER, 3rd, Sarov-Blat L, Shi Y, Hamamdzic D, Zalewski A, Macphee C, Llano R, Pelchovitz D, Mainigi SK, Osman H, et al. Site-specific atherogenic gene expression correlates with subsequent variable lesion development in coronary and peripheral vasculature. Arterioscler Thromb Vasc Biol. 2008;28:850–855. doi: 10.1161/ATVBAHA.107.154534. [DOI] [PubMed] [Google Scholar]
- 20.Goodell MA, Rosenzweig M, Kim H, Marks DF, DeMaria M, Paradis G, Grupp SA, Sieff CA, Mulligan RC, Johnson RP. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med. 1997;3:1337–1345. doi: 10.1038/nm1297-1337. [DOI] [PubMed] [Google Scholar]
- 21.Challen GA, Boles N, Lin KYK, Goodell MA. Mouse hematopoietic stem cell identification and analysis. Cytometry A. 2009;75A doi: 10.1002/cyto.a.20674. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodell MA. Multipotential stem cells and ‘side population’ cells. Cytotherapy. 2002;4:507–508. doi: 10.1080/146532402761624638. [DOI] [PubMed] [Google Scholar]
- 24.Caslake MJ, Packard CJ, Suckling KE, Holmes SD, Chamberlain P, Macphee CH. Lipoprotein-associated phospholipase A(2), platelet-activating factor acetylhydrolase: a potential new risk factor for coronary artery disease. Atherosclerosis. 2000;150:413–419. doi: 10.1016/s0021-9150(99)00406-2. [DOI] [PubMed] [Google Scholar]
- 25.Fitzmaurice GM, Laird NM, Ware JH. Applied Longitudinal Analysis. New York: Wiley-IEEE; 2004. [Google Scholar]
- 26.Hollander M, Wolfe DA. Nonparametric Statistical Methods. New York: John Wiley; 1999. [Google Scholar]
- 27.Macphee CH, Nelson JJ, Zalewski A. Lipoprotein-associated phospholipase A2 as a target of therapy. Curr Opin Lipidol. 2005;16:442–446. doi: 10.1097/01.mol.0000174155.61307.5f. [DOI] [PubMed] [Google Scholar]
- 28.Maki-Petaja KM, Booth AD, Hall FC, Wallace SM, Brown J, McEniery CM, Wilkinson IB. Ezetimibe and simvastatin reduce inflammation, disease activity, and aortic stiffness and improve endothelial function in rheumatoid arthritis. J Am Coll Cardiol. 2007;50:852–858. doi: 10.1016/j.jacc.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 29.Creager MA, Luscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy. Part I. Circulation. 2003;108:1527–1532. doi: 10.1161/01.CIR.0000091257.27563.32. [DOI] [PubMed] [Google Scholar]
- 30.Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ, Thomason MJ, Mackness MI, Charlton-Menys V, Fuller JH. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364:685–696. doi: 10.1016/S0140-6736(04)16895-5. [DOI] [PubMed] [Google Scholar]
- 31.Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, Levine JP, Gurtner GC. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–2786. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 32.Avogaro A, Fadini GP, Gallo A, Pagnin E, de KS. Endothelial dysfunction in type 2 diabetes mellitus. Nutr Metab Cardiovasc Dis. 2006;16(Suppl 1):S39–S45. doi: 10.1016/j.numecd.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 33.Loomans CJ, de Koning EJ, Staal FJ, Rookmaaker MB, Verseyden C, de Boer HC, Verhaar MC, Braam B, Rabelink TJ, van Zonneveld AJ. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53:195–199. doi: 10.2337/diabetes.53.1.195. [DOI] [PubMed] [Google Scholar]
- 34.Rivard A, Silver M, Chen D, Kearney M, Magner M, Annex B, Peters K, Isner JM. Rescue of diabetes-related impairment of angiogenesis by intramuscular gene therapy with adeno-VEGF. Am J Pathol. 1999;154(2):355–363. doi: 10.1016/S0002-9440(10)65282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poli A, Pujia A. Pleiotropic effects of statins and early benefit in the PROVE IT-TIMI-22 study. J Am Coll Cardiol. 2006;48:852–853. doi: 10.1016/j.jacc.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 36.Fichtlscherer S, Schmidt-Lucke C, Bojunga S, Rossig L, Heeschen C, Dimmeler S, Zeiher AM. Differential effects of short-term lipid lowering with ezetimibe and statins on endothelial function in patients with CAD: clinical evidence for ‘pleiotropic’ functions of statin therapy. Eur Heart J. 2006;27:1182–1190. doi: 10.1093/eurheartj/ehi881. [DOI] [PubMed] [Google Scholar]
- 37.Vasa M, Fichtlscherer S, Adler K, Aicher A, Martin H, Zeiher AM, Dimmeler S. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103:2885–2890. doi: 10.1161/hc2401.092816. [DOI] [PubMed] [Google Scholar]
- 38.Zhang L, Zalewski A, Liu Y, Mazurek T, Cowan S, Martin JL, Hofmann SM, Vlassara H, Shi Y. Diabetes-induced oxidative stress and low-grade inflammation in porcine coronary arteries. Circulation. 2003;108:472–478. doi: 10.1161/01.CIR.0000080378.96063.23. [DOI] [PubMed] [Google Scholar]
- 39.Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441:1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- 40.Wenzel P, Daiber A, Oelze M, Brandt M, Closs E, Xu J, Thum T, Bauersachs J, Ertl G, Zou MH, Forstermann U, Munzel T. Mechanisms underlying recoupling of eNOS by HMG-CoA reductase inhibition in a rat model of streptozotocin-induced diabetes mellitus. Atherosclerosis. 2008;198:65–76. doi: 10.1016/j.atherosclerosis.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]