

Abstract
Summary: The application of protein–protein docking in large-scale interactome analysis is a major challenge in structural bioinformatics and requires huge computing resources. In this work, we present MEGADOCK 4.0, an FFT-based docking software that makes extensive use of recent heterogeneous supercomputers and shows powerful, scalable performance of >97% strong scaling.
Availability and Implementation: MEGADOCK 4.0 is written in C++ with OpenMPI and NVIDIA CUDA 5.0 (or later) and is freely available to all academic and non-profit users at: http://www.bi.cs.titech.ac.jp/megadock.
Contact: akiyama@cs.titech.ac.jp
Supplementary information: Supplementary data are available at Bioinformatics online
1 INTRODUCTION
Protein–protein interactions can provide valuable insights for understanding the principles of biological systems and for elucidating causes of incurable diseases. Although many structures of interacting proteins have been determined by X-ray crystallography and Nuclear Magnetic Resonance spectroscopy, the structures of many protein complexes have still not been determined experimentally because of cost and technical limitations. Protein–protein docking, a computational method for predicting the structure of a protein complex from known component structures, is a powerful approach that facilitates the discovery of otherwise unattainable protein complex structures.
A number of fast Fourier transform (FFT)-based rigid-body initial protein–protein docking tools have been developed for predicting protein complex structures (Cheng et al., 2007; Pierce et al., 2011; Ritchie and Venkatraman, 2010). However, faster docking tools are still required to perform large-scale interactome predictions. Some applications also require a huge number of dockings, such as ensemble docking techniques using multiple conformations for flexible docking (Grünberg et al., 2004; Król et al., 2007), cross-docking for identification of protein interaction partners (Lopes et al., 2013; Matsuzaki et al., 2009; Wass et al., 2011; Zhang et al., 2014) and multiple docking (Karaca and Bonvin, 2011). To achieve these large-scale analyses, use of the supercomputing environment has become absolutely necessary.
On the other hand, 35% of computing performance of supercomputers ranked in top500.org (June 2014) is currently achieved by hardware accelerators, such as graphics processing units (GPUs), and this percentage is increasing. Therefore, tools that can be used with such ‘heterogeneous’ supercomputers are necessary. While some docking tools are accelerated by GPUs on a node (Ritchie and Venkatraman, 2010; Sukhwani and Herbordt, 2009), ‘heterogeneous’ supercomputers, which have massive numbers of nodes including multiple CPU cores and GPU cards, have not yet been used for acceleration of docking tool performance.
Here, we present ultra–high-performance docking software, ‘MEGADOCK 4.0’, which makes extensive use of supercomputers equipped with GPUs.
2 IMPLEMENTATION
2.1 MEGADOCK scheme
MEGADOCK uses a Katchalski-Katzir algorithm known as a traditional FFT-based rigid-docking scheme (Katchalski-Katzir et al., 1992). Its original scoring function, based on shape complementarity, electrostatics and desolvation free energy, is calculated by only one correlation function (Ohue et al., 2012, 2014). This is advantageous for faster calculation because multiple correlation functions and thus multiple FFT calculations are used to evaluate multiple effects in previous methods (Kozakov et al., 2006; Pierce et al., 2011). (see Supplementary Text S1 for details)
2.2 GPU implementation
MEGADOCK has been implemented on multiple GPUs using the CUDA library (Shimoda et al., 2013). A previous study (Sukhwani and Herbordt, 2009) mapped only FFT processes onto a GPU, and its implementation could not use multiple GPUs. We mapped the whole docking process (voxelization, ligand rotation, FFTs and finding solutions) onto GPUs, and our implementation was able to use multiple GPUs and CPU cores (Shimoda et al., 2013).
2.3 Hybrid CUDA, MPI and OpenMP parallelization
For extensive execution of docking jobs, an implementation that can be performed among many computing nodes is required. We previously parallelized the calculation of each docking processes using MPI and OpenMP with the master/worker model (Matsuzaki et al., 2013). On cluster computers, a master process acquires a list of protein pairs and distributes the docking jobs to worker processes on available nodes. This implementation guarantees fault tolerance in that the master process surveys all docking jobs.
The proposed software, MEGADOCK 4.0, is implemented by hybrid CUDA, MPI and OpenMP parallelization. Reducing the usage of memory space is important with systems that have many CPU cores, multiple GPUs per node and relatively little memory (e.g. there is only 6 GB memory on an NVIDIA Tesla K20X GPU). We assigned one docking job to each node and then distributed the calculations of ligand rotation by thread parallelization with CPU cores and GPUs. This implementation model manages one node as the master and the other nodes as workers. The master node distributes the docking jobs to worker nodes, and a worker node executes distributed docking jobs with multiple GPUs by CUDA and all CPU cores by OpenMP thread parallelization. This implementation also guarantees fault tolerance similar to the CPU version.
3 RESULTS AND DISCUSSION
To check the performance of MEGADOCK 4.0, we used the ZLAB benchmark 4.0 dataset (Hwang et al., 2010). Speed measurement experiments were conducted on the TSUBAME 2.5 supercomputing system (Tokyo Institute of Technology, Japan). We used its ‘thin nodes’ with a reservation service of exclusive use (up to 420 nodes). Each ‘thin’ node contained two Intel Xeon X5670 (six cores, 2.93 GHz) and three NVIDIA Tesla K20X (GK110) GPUs. The specifications of the environment are shown in Supplementary Text S2 and Table S1.
Figure 1 shows the average of five measurements of computation time and the parallel scalability of MEGADOCK 4.0 on 30 976 protein pairs from combinations between 176 receptors and 176 ligands, assuming a cross-docking study. The observed calculation acceleration was close to ideal. Strong scaling values from 35 nodes were >97% for all numbers of nodes measured here (Supplementary Table S2). Notably, a high scalability (98%) was obtained with the largest number of nodes (420 nodes).
Fig. 1.
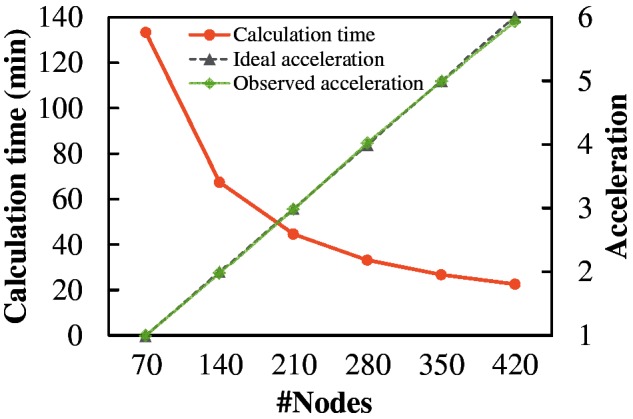
Calculation time and acceleration by parallelization among nodes on 30 976 docking jobs
We also measured docking time on a half million and a million protein pairs for simulation of large-scale interactome analyses using averaged-sized proteins (FFT size of 108, see Supplementary Table S3). In this simulation, a half million docking jobs required 5.71 h, while a million jobs required 11.51 h. The epidermal growth factor receptor-related pathway, which we are studying in non–small-cell lung cancer, required approximately a quarter million dockings. This analysis could be completed in only 3 h with MEGADOCK 4.0 using 420 nodes, whereas solving the same problem requires several days with an older version of MEGADOCK.
4 CONCLUSIONS
MEGADOCK 4.0 is a docking software for heterogeneous supercomputing environments and shows excellent scalability. Heterogeneous supercomputers equipped with hardware accelerators, such GPUs, will become common in the future. Fully using such computers is crucial for bioinformatics research, which must analyze massive amounts of data. MEGADOCK 4.0 can serve as a tool to promote analysis of the whole interactome within a reasonable time.
Supplementary Material
ACKNOWLEDGEMENTS
This research used computational resources of the TSUBAME 2.5 supercomputer provided by Tokyo Institute of Technology through the HPCI System Research project (Project ID: hp140173).
Funding: this work was supported by a Grant-in-Aid for JSPS Fellows (238750 and 2630002) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).
Conflict of interest: none declared.
REFERENCES
- Cheng TM-K, et al. pyDock: electrostatics and desolvation for effective scoring of rigid-body protein–protein docking. Proteins. 2007;68:503–515. doi: 10.1002/prot.21419. [DOI] [PubMed] [Google Scholar]
- Grünberg R, et al. Complementarity of structure ensembles in protein-protein binding. Structure. 2004;12:2125–2136. doi: 10.1016/j.str.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Hwang H, et al. Protein–protein docking benchmark version 4.0. Proteins. 2010;78:3111–3114. doi: 10.1002/prot.22830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Bonvin AMJJ. A multidomain flexible docking approach to deal with large conformational changes in the modeling of biomolecular complexes. Structure. 2011;19:555–565. doi: 10.1016/j.str.2011.01.014. [DOI] [PubMed] [Google Scholar]
- Katchalski-Katzir E, et al. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. USA. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D, et al. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006;65:392–406. doi: 10.1002/prot.21117. [DOI] [PubMed] [Google Scholar]
- Król M, et al. Flexible relaxation of rigid-body docking solutions. Proteins. 2007;68:159–169. doi: 10.1002/prot.21391. [DOI] [PubMed] [Google Scholar]
- Lopes A, et al. Protein–protein interactions in a crowded environment: an analysis via cross-docking simulations and evolutionary information. PLOS Comput. Biol. 2013;9:e1003369. doi: 10.1371/journal.pcbi.1003369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki Y, et al. In silico screening of protein-protein interactions with all-to-all rigid docking and clustering: an application to pathway analysis. J. Bioinform. Comput. Biol. 2009;7:991–1012. doi: 10.1142/s0219720009004461. [DOI] [PubMed] [Google Scholar]
- Matsuzaki Y, et al. MEGADOCK 3.0: a high-performance protein–protein interaction prediction software using hybrid parallel computing for petascale supercomputing environments. Source Code Biol. Med. 2013;8:18. doi: 10.1186/1751-0473-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohue M, et al. Improvement of the protein–protein docking prediction by introducing a simple hydrophobic interaction model: an application to interaction pathway analysis. Lect. Notes Comput. Sci. 2012;7632:178–187. [Google Scholar]
- Ohue M, et al. MEGADOCK: An all-to-all protein–protein interaction prediction system using tertiary structure data. Protein Pept. Lett. 2014;21:766–778. doi: 10.2174/09298665113209990050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce BG, et al. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLOS ONE. 2011;6:e24657. doi: 10.1371/journal.pone.0024657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie DW, Venkatraman V. Ultra-fast FFT protein docking on graphics processors. Bioinformatics. 2010;26:2398–2405. doi: 10.1093/bioinformatics/btq444. [DOI] [PubMed] [Google Scholar]
- Shimoda T, et al. Proceedings of the ACM-BCB’13. New York, NY: ACM Press; 2013. MEGADOCK-GPU: Acceleration of protein–protein docking calculation on GPUs; pp. 883–889. [Google Scholar]
- Sukhwani B, Herbordt MC. Proceedings of the GPGPU-2. New York, NY: ACM Press; 2009. GPU acceleration of a production molecular docking code; pp. 19–27. [Google Scholar]
- Wass MN, et al. Towards the prediction of protein interaction partners using physical docking. Mol. Syst. Biol. 2011;7:469. doi: 10.1038/msb.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, et al. Discovery of binding proteins for a protein target using protein–protein docking-based virtual screening. Proteins. 2014;82:2472–2482. doi: 10.1002/prot.24611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.