

Abstract
Matrix metalloprotease-1 (MMP1) is an important mediator of tumorigenesis, inflammation and tissue remodeling through its ability to degrade critical matrix components. Recent studies indicate that stromal-derived MMP1 may exert direct oncogenic activity by signaling through protease-activated receptor-1 (PAR1) in carcinoma cells, however, this has not been established in vivo. We generated a Mmp1a knock-out mouse to ascertain whether stromal-derived Mmp1a affects tumor growth. Mmp1a-deficient mice are grossly normal and born in Mendelian ratios, however, deficiency of Mmp1a results in significantly decreased growth and angiogenesis of lung tumors. Co-implantation of lung cancer cells with wild-type Mmp1a+/+ fibroblasts completely restored tumor growth in Mmp1a-deficient animals, highlighting the critical role of stromal-derived Mmp1a. Silencing of PAR1 expression in the lung carcinoma cells phenocopied stromal Mmp1a-deficiency, thus validating tumor-derived PAR1 as a Mmp1a target. Mmp1a secretion is controlled by the ability of its prodomain to suppress auto-cleavage, whereas human MMP1 is efficiently secreted due to stable pro- and catalytic domain interactions. Together, these data demonstrate that stromal Mmp1a drives in vivo tumorigenesis and provide proof-of-concept that targeting the MMP1-PAR1 axis may afford effective treatments of lung cancer.
Keywords: angiogenesis, lung cancer, Mmp1a, MMP1, PAR1
INTRODUCTION
MMP1 is a zinc-dependent matrix metalloprotease that is frequently overexpressed in a large number of human cancers1–3. High MMP1 levels in patient tumor samples have been associated with metastasis and decreased progression-free survival in melanoma, colorectal and esophageal cancers4–6. Fibroblast MMP1 was originally defined as an interstitial collagenase due to its ability to cleave fibrillar collagens7, but more recent studies found it also cleaves a variety of other substrates8, including the oncogenic receptor, protease-activated receptor-1 (PAR1)9. MMP1 is produced by many sources in tumors, including cancer cells, fibroblasts, inflammatory cells, and the endothelium, and because it is a secreted enzyme, can have both paracrine and autocrine effects in the microenvironment10–12. In an early study of patients with lung cancer, 35% of tumors had MMP1 expressed by the carcinoma cells, whereas 70% had MMP1 overexpression in the stromal cells13. The presence of cancer-associated fibroblasts was recently found to be a powerful independent risk factor to predict recurrence in patients with stage I lung adenocarcinoma14. Fibroblast-derived and cancer cell-derived MMP1 have been shown to be differentially processed, suggesting that there may be functional differences in MMP1 depending on the source15.
In order to effectively study the role of MMP1 and its inhibitors in cancer and other diseases, it is important to develop a relevant in vivo model system. The rodent genome contains an MMP1 gene duplication, resulting in the Mmp1a and Mmp1b genes which are 82% identical16. Mmp1a is the most likely MMP1 homologue because unlike Mmp1a, Mmp1b has no known enzymatic activity16. Mouse Mmp1a, is 58% identical to human MMP1 but is expressed less ubiquitously in healthy mouse tissue than in humans16,17. Elevated Mmp1a expression has been described in a number of inflammatory conditions in mice, including wound healing, lung injury, arthritis, and most recently, sepsis12,18,19. Mmp1a is induced in the mouse stroma of human breast and renal cell carcinoma xenografts9,19, suggesting that like MMP1, Mmp1a is upregulated in the tumor microenvironment.
To investigate the role of stromal-derived MMP1 in tumorigenesis, we generated Mmp1a-deficient mice. Loss of Mmp1a caused significantly decreased lung tumor growth and angiogenesis. This stromal defect was rescued by co-implantation of Mmp1a+/+ fibroblasts with lung cancer cells in the Mmp1a-deficient mice. Biochemical analysis indicated that Mmp1a secretion is controlled by the ability of the Mmp1a prodomain to suppress auto-cleavage, whereas human MMP1 is efficiently secreted due to stable pro- and catalytic domain interactions. Together, these data demonstrate that stromal Mmp1a strongly promotes tumorigenesis in vivo and that MMP1a prodomain has evolved a distinct function from other MMPs by allowing rapid activation and autocleavage. Moreover, the Mmp1a-deficient mouse may serve as a useful tool in studying the role of MMP1 in many other cancers, and various disease states characterized by pathologic tissue remodeling.
RESULTS
Mmp1a-deficient mice exhibit impaired tumorigenesis and angiogenesis
Mmp1a−/− mice were generated by targeted replacement of Mmp1a exon 5 with a PGK-neomycin cassette. Exon 5 encodes the zinc-coordination motif and a major portion of the catalytic domain and is essential for metalloprotease activity. Mmp1a-deficient mice were back-crossed 10 generations into the C57BL/6 background and Mmp1a-null animals were born within range of expected ratios from heterozygote parents (Supplementary Figure S1a–b). Mmp1a+/+ mouse embryonic fibroblasts (MEFs) exhibited Mmp1a protein expression by immunofluorescence which was lost in Mmp1a-deficient MEFs (Supplementary Figure S1c). Adult Mmp1a−/− males and females had no obvious abnormalities and were fertile. This indicates that Mmp1a expression is dispensable for normal mouse development, as has been observed with individual deficiency of the two other secreted mouse collagenases, Mmp8 and Mmp1320–22.
Given that Mmp1a is often upregulated in the stroma of tumor xenografts9,19, we examined tumor growth in Mmp1a-deficient animals using LLC1 cells, a highly aggressive C57BL/6-derived lung cancer cell line23. LLC1 cells (2×105) were implanted into the abdominal fat pads of Mmp1a−/− and wild-type (WT) mice. The tumors in Mmp1a−/− (KO) mice grew at a significantly slower rate relative to WT mice over the course of the entire experiment (Figure 1a). The tumors from the KO mice also had significantly smaller tumor mass as compared to WT mice at terminal necropsy (Figure 1b), thereby implicating stromal-derived Mmp1a as an important factor in promoting lung cancer tumorigenesis. Conversely, when LLC1 cells were directly inoculated into the venous circulation in an experimental metastasis model, only a slight, non-significant reduction in metastatic foci to lung in the KO vs the WT mice (Supplementary Figure S2), suggesting a more significant role for stromal-derived Mmp1a in primary tumor growth as opposed to metastasis or tumor-take in lung.
Figure 1. Stromal Mmp1a-deficiency attenuates growth of lung tumors.
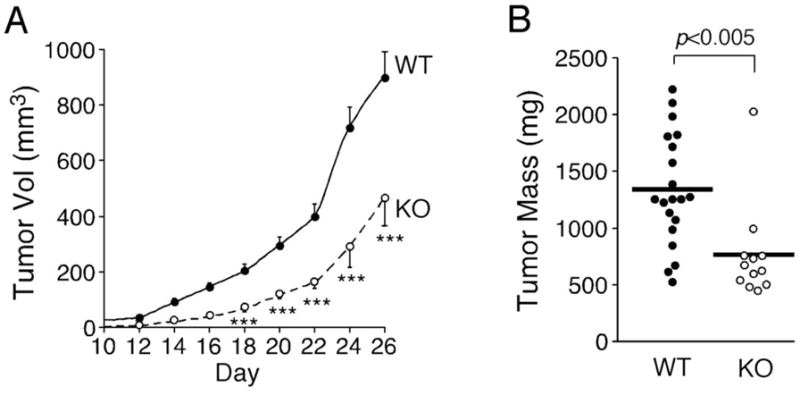
a. Growth of Lewis lung carcinoma (LLC1) cells (2 × 105) implanted subcutaneously into the abdominal fat pad of Mmp1a+/+ (n=20) or Mmp1a−/− (n=12) C57BL/6 female mice.
b. Excised tumor mass at the experiment endpoint, day 26. ***p<0.001 by heteroscedastic T-Test at each time point in both genetic backgrounds.
The human homolog, MMP1 has been shown to be a pro-angiogenic factor by activating endothelial proliferation and migration through PAR124–26. LLC1 tumors were assessed for possible defects in angiogenesis in the Mmp1a-deficient mice. Immunohistochemical staining of the tumors for von Willebrand Factor (vWF), a specific marker of endothelial cells, revealed a significant decrease in angiogenesis in both the tumor edges and tumor centers in the Mmp1a-knockout mice as compared to wild-type mice (Figures 2a,b). When human endothelial cells were stimulated with media from mouse embryonic fibroblasts (MEFs) isolated from wild type or Mmp1a-knockout mice, the Mmp1a-deficient media resulted in significantly less tube formation, and tubes that formed had decreased complexity (Figures 2c–e). To provide further evidence that the observed effects are due to direct involvement of Mmp1a, we performed a reconstitution experiment with recombinant Mmp1a using the endothelial tube formation. Treatment of HUVEC endothelial cells with media from MEFs resulted in a significant 70% decrease in average tube length formed with Mmp1a-KO media vs WT media (Supplemental Figure S3). Supplementation of the KO media with 20 nM recombinant Mmp1a resulted in a significant 2-fold increase in tube length with no effect when added to WT CM. Together, these data indicate that stromal-derived Mmp1a is also a proangiogenic factor in tumors and in nascent vessel formation.
Figure 2. Tumor angiogenesis is suppressed in Mmp1a-deficient animals.

a. Von Willebrand Factor (vWF) immunohistochemistry on LLC1 subcutaneous tumors from wild type or Mmp1a−/− mice (40X magnification).
b. Number of vWF-positive blood vessels as determined by the sum of 50 fields (40X) per tumor, n=10 per cohort.
c–e. Tube formation of primary human endothelial cells (HUVECs) following 6 h stimulation with media isolated from the embryonic fibroblasts (MEFs) of wild-type or Mmp1a-deficient mice. 4X phase contrast micrographs of representative (c) cultures with corresponding quantification of (d) tubal length and (e) branch point complexity (arbitrary units). All P values were determined by heteroscedastic T-Test.
Restoration of fibroblast Mmp1a rescues tumor formation in Mmp1a-knockout mice
Tumor stroma is a complex tissue composed of multiple cell types that could produce Mmp1a, especially stromal fibroblasts and other mesenchymal cells11. The human homolog, stromal-derived MMP1, is a potent inducer of cancer cell migration, invasion and mitogenesis, potentially through its activation of the PAR1 oncogene9,27. To determine whether stromal fibroblasts expressing Mmp1a could rescue tumorigenesis in Mmp1a-null animals, we isolated MEFs at embryonic day 12.5 from WT and Mmp1a-deficient mice (Supplementary Figure S1c). Media produced from KO MEFs resulted in >2-fold less chemoinvasion of LLC1 cells as compared to media from wild-type MEFs (Figure 3a). Likewise, an almost identical reduction in the migration of A549 human lung cancer cells towards media from Mmp1a-deficient MEFs was observed (Figure 3b). Migration of MCF7 breast cancer cells ectopically expressing PAR1 was four-fold higher towards media from WT MEFs as compared to Mmp1a-KO MEFs and inhibition of PAR1 with a small molecule antagonist, RWJ-58259, caused a decrease in migration by 50% towards WT MEF media (Figure 3c). The PAR1 antagonist did not completely reduce migration to the level observed with Mmp1a-KO conditioned media, suggesting PAR1-independent effects for Mmp1a in migration.
Figure 3. Stromal Mmp1a promotes proliferation, migration, and tumorigenesis of lung cancer.

a. LLC1 chemoinvasion through type I collagen towards Mmp1a+/+ (WT) or Mmp1a−/− (KO) MEF conditioned media (CM).
b. Migration of the human lung cancer cell line A549 toward WT or KO MEF conditioned media.
c. Migration of MCF7 breast cancer cells ectopically expressing PAR1 towards MEF conditioned media in the absence (black) or presence (white) of the small molecule PAR1 antagonist, RWJ-58259 (3 μM).
d. 96 h MTT proliferation of LLC1 cells in response to 10% FBS or MEF conditioned media.
e. Tumor growth in Mmp1a+/+ (WT) or Mmp1a−/− (KO) mice co-implanted with 2 × 105 LLC1 and 1 × 105 Mmp1a+/+ (WT MEF) or Mmp1a−/− (KO MEF) fibroblasts (n=12–16 per cohort). * p<0.05, **p<0.005, ***p<0.001 by heteroscedastic T-Test or ANOVA followed by T-test at each time point.
Given the differences we observed in tumor growth in vivo, we also examined the ability of MEF conditioned media to induce LLC1 proliferation in vitro. Wild type MEF conditioned media induced a significant 2.2-fold induction of LLC1 proliferation (Figure 3d). Conditioned media from Mmp1a-KO mice MEFs resulted in a significant reduction in proliferation of LLC1 cells as compared to wild type MEF conditioned media. These data demonstrate that MEFs, like cancer-associated fibroblasts, can induce cancer cell migration, invasion, and proliferation and that these tumor-promoting functions are reduced in MEFs from Mmp1a-KO mice.
To confirm that the reduced tumorigenesis seen in Mmp1a-KO mice is due to a stromal defect, we next performed co-implantation experiments with LLC1 cells and MEFs. WT and Mmp1a-KO mice were injected in their abdominal fat pads with 2×105 LLC1 cells mixed with 1×105 wild type or Mmp1a-KO MEFs. LLC1/Mmp1a-KO MEF co-implants in Mmp1a-KO mice formed significantly smaller tumors over the entire growth period (Figure 3e). However, this phenotype was completely rescued by co-implantation of LLC1 cells with WT Mmp1a MEFs in the Mmp1a-KO mice, thus demonstrating the importance of a stromal source of Mmp1a in promoting tumorigenesis. Co-implantation with WT Mmp1a MEFs did not result in supergrowth of the LLC1 tumors in WT mice, suggesting a maximal effect of stromal Mmp1a on tumor growth.
Stromal Mmp1a promotes tumorigenesis through PAR1 in LLC1 lung cancer
Lung carcinoma cells from patients and tumors from mice, including LLC1, express high levels of the oncogenic receptor PAR128,29. Like human MMP1, recombinant Mmp1a directly cleaved the N-terminal exodomain of PAR1 in T7-PAR1 cleavage assays, demonstrating a direct interaction between Mmp1a and PAR1 (Supplementary Figure S4). We assessed whether the observed protumorigenic properties of stromal-produced Mmp1a were dependent on PAR1 expression in the lung carcinoma cells. Tumor genograft experiments in WT and Mmp1a-KO mice were performed with LLC1 cells that were stably transduced with a short hairpin-RNA that silenced PAR1 expression (shPAR1) or control (shLuc), as characterized previously29. In WT mice, shPAR1 knockdown tumors grew slower at all time points (Figure 4a) and weighed less at terminal necropsy (Figure 4b) than control shLuc LLC1 tumors, indicating that the growth rate of the lung tumors was dependent at least in part on the expression of PAR1. LLC1 cells silenced for PAR1 expression were also injected into the abdominal fat pads of Mmp1a-KO mice. The relative decreases in lung tumor growth rates and mass were essentially identical between tumors that had PAR1 silenced on the lung carcinoma cells and those that grew in mice that lacked Mmp1a expression in the stroma (Figure 4a–b). Double loss of Mmp1a in the stroma and PAR1 in the LLC1 cells (KO+shPAR1) did not result in further decreases in lung tumor growth and mass (Figures 4a,b), indicating that stromal-Mmp1a and PAR1 on the carcinoma cells act on the same, e.g. paracrine, pathway in promoting tumorigenesis.
Figure 4. Stromal Mmp1a promotes the growth of lung cancer through PAR1.

a. Tumor growth following subcutaneous implantation of 200,000 shLuc control (n=10–20) versus shPAR1 (n=10) transduced LLC1 cells in Mmp1a+/+ (WT) or Mmp1a−/− (KO) mice.
b. Mass of excised LLC1 tumors at the day 26 endpoint. # p=0.06, *p<0.05 by one-way ANOVA followed by T-test at each time point.
The prodomains of Mmp1a and MMP1 have divergent effects on protein secretion and autocatalysis
As human and mouse collagenases exert their tumorigenic effects as secreted factors, we next compared the efficiency of protein secretion of human MMP1 with mouse Mmp1a, Mmp1b and the two other related collagenases from mice, Mmp8 and Mmp13. Three heterologous expression systems, HEK293T, CHO-K1, and COS7, were used to compare protein levels of all the secreted collagenases. Surprisingly, secreted Mmp1a protein levels were markedly lower than human MMP1 or mouse Mmp1b, Mmp8, and Mmp13 across the three expression systems, despite comparable mRNA levels (Supplementary Figure S5a–b). We tested the possibility that the Mmp1a signal peptide was responsible for its low secretion efficiency. Mmp1b, which was highly secreted across all the tested expression systems, has a signal peptide identical to that of Mmp1a except for a serine residue at position 2 instead of proline. However, mutation of the Mmp1a signal peptide to that of Mmp1b (P2S Mmp1a) had no effect on secretion of Mmp1a (Supplementary Figure S5c), indicating that the signal peptide of Mmp1a was not responsible for its low levels of secretion.
We next examined the prodomains of mouse Mmp1a and human MMP1, which are 53% identical to each other (Figure 5a–c). A hPro-Mmp1a chimera was generated in which the prodomain of Mmp1a (residues 1-95) was exchanged with the prodomain of human MMP1 (residues 1-98). We also generated the complementary mPro-MMP1 chimera for human MMP1, with residues 1-96 of Mmp1a replacing human MMP1 residues 1-99. Replacement of the mouse Mmp1a prodomain with that of human MMP1 resulted in massive secretion of soluble hPro-Mmp1a (Figure 5d). Conversely, replacement of the human prodomain with mouse prodomain eliminated secretion of the mPro-MMP1 chimera. These data indicate that the mouse and human prodomains have widely divergent and dominant effects on secretion of MMP1 and Mmp1a from cells.
Figure 5. Mmp1a and MMP1 prodomain-catalytic domain interactions regulate secretion and autocatalysis.

a. Structure of Mmp1a as predicted by homology modeling with human proMMP1 showing the prodomain (yellow), catalytic domain (blue), linker (green), and hemopexin domain (orange). Black arrow-zymogen activating cleavage site; arrow head-linker cleavage site resulting in loss of hemopexin domain.
b. Docking of the Mmp1a prodomain (yellow) onto the catalytic domain/active site region (blue) of Mmp1a. Residues A58 and L67 in helix 2 (H2) are highlighted in red while the corresponding human residues V61 and F70, respectively are depicted in green.
c. Alignment of human MMP1, Mmp1a, and Mmp1b depicting the structural motifs within the prodomain; H=helix, L=linker. Point mutations are highlighted in black.
d. Secretion of Mmp1a and MMP1 prodomain mutants into the media (40 μL) of transfected HEK293T cells as determined by anti-Myc Western blot.
e. MMP expression levels in cell lysates (40 μg) of transfected HEK293T cells, showing proMMP (56 kDa), active MMP (48 kDa), and hemopexin degradation product (26 kDa).
We next tested the hypothesis that the prodomain of Mmp1a might associate less stably with its catalytic domain and thereby be less effective in maintaining Mmp1a in the zymogen or proteolytic-’off’ state as compared to human MMP1. A structural model of proMmp1a (Figures 5a,b) was created based on the highly homologous proMMP1 structure30. The cysteine-lock motif, which acts as the fourth coordination residue for the catalytic zinc effectively sealing off the catalytic pocket, is critical for the auto-inhibitory effects of MMP prodomains on their catalytic domains31. The proximal residues and geometry of the cysteine-lock were completely conserved between human MMP1 and Mmp1a (Figures 5b,c). However, Mmp1a residue L67 located at the C-terminal end of helix 2 (H2) at the interface between the prodomain and catalytic domains presents a much small hydrophobic surface than the analogous F70 in human proMMP1, suggesting a potentially weaker interaction with the catalytic domain surface in the active site cleft than the corresponding human counterpart (Figures 5b,c). In the human proMMP1 crystal structure, F70 interacts with the critical H228S229T230 of the catalytic domain30 and this phenylalanine is conserved in the other mouse collagenases and mammalian homologues of MMP1.
To determine whether the L67 residue contributed to the low secretion levels of Mmp1a, we generated a L67F point mutant along with a non-interacting substitution at the opposite end of helix 2, A58V (Figure 5b–c). The humanized L67F mutant exhibited increased Mmp1a expression, whereas the A58V substitution had no enhancing effect on Mmp1a protein secretion (Figure 5d). The increased Mmp1a expression with L67F was still less than that observed with Mmp1b (Supplementary Figure S5a), which also retains the L67 residue. However, unlike Mmp1a, Mmp1b does not have any identified enzymatic activity, suggesting that the proteolytic activity (e.g. autocleavage) of Mmp1a may reduce Mmp1a secretion. Consistent with this notion, catalytically inactive Mmp1a (E216A) was expressed at much higher levels than WT Mmp1a (Figure 5d). To determine whether autocatalysis of Mmp1a in the cells may be reducing secretion, we examined cell lysates of WT Mmp1a and the point mutants. As shown in Figure 5e, WT Mmp1a had significant zymogen activation (48 kDa active form) and cleavage at the linker separating the catalytic domain from the hemopexin domain as evidenced by appearance of the 26 kDa C-terminal hemopexin domain in the cell lysates. The cleaved hemopexin domain was completely absent in the inactive E216A mutant and the highly secreted hPro-Mmp1a chimera as well as WT hMMP1. Likewise, the humanized Mmp1a L67F point mutant had nearly no hemopexin domain cleavage product and much less 48 kDa active form in the cell lysates, further suggesting that stable prodomain-catalytic domain interactions suppress autocatalysis prior to secretion (Figure 5e). To further compare the stability of the human and mouse MMP1, we performed a pulse-chase experiment in which transfected HEK293T cells were treated with cycloheximide to inhibit protein synthesis at 24 h (time 0) after transfection and expression levels of hMMP1 and Mmp1a in the lysates were followed for 48 h. Human MMP1 levels were maintained in the zymogen state over 48 h whereas Mmp1a underwent accelerated protein activation and degradation, consistent with decreased Mmp1a protein stability (Supplementary Figure S5d).
Based on the inherent instability/autocatalysis of Mmp1a, we hypothesized that Mmp1a would have high activity in the absence of exogenous activation despite its low expression levels while the hyper-stable hpro-Mmp1a chimera would show the opposite phenomenon. We have previously shown that CYR61 is a proangiogenic factor that is strongly induced by both MMP1 and Mmp1a in a PAR1-dependent manner29,32. Treatment of Mmp1a-null C57MG cells with conditioned media from MMP1-expressing Cos7 cells resulted in a 2.3-fold induction of CYR61 mRNA, as compared with conditioned media from vector control Cos7 cells (Figure 6). Media from mpro-MMP1 or Mmp1a-expressing cells caused a slightly higher 2.7-fold induction in CYR61 mRNA in the C57MG cells. However, the stabilized hpro-Mmp1a chimera had reduced CYR61 transcription, similar to the catalytically-dead E216AMmp1a mutant (Figure 6). These data suggest that intrinsic Mmp1a instability/autocatalysis leads to increased angiogenesis activity and conversely, over-stabilization of the Mmp1a prodomain interactions with its catalytic domain lead to decreased signaling due to difficulty in activating the pro-enzyme.
Figure 6. Effect of Mmp1a and MMP1 prodomains on expression of the angiogenesis factor, Cyr61.

Induction of CYR61 mRNA in mouse epithelial cells (C57MG) following 24 h treatment with MMP-transfected Cos7 CM as described in the Methods. Data represent means ± SE of triplicate experiments.
DISCUSSION
Using mice deficient in the collagenase Mmp1a, we show that Mmp1a is a functional MMP1 homologue in driving lung tumorigenesis with significantly less tumor growth and angiogenesis in Mmp1a-deficient animals. Loss of stromal-produced Mmp1a from fibroblasts isolated from Mmp1a-deficient mice resulted in decreased invasion, migration, and proliferation of lung carcinoma cells, and defects in angiogenesis. Co-implantation of wild-type Mmp1a fibroblasts with lung cancer cells completely restored tumor growth in Mmp1a-deficient animals. The decrease in tumor growth observed in Mmp1a-deficient mice is consistent with observations in patient samples that stromal MMP1 is upregulated in aggressive human cancers13,27,33,34. Together these results demonstrate the importance of stromal Mmp1a in driving lung tumorigenesis and angiogenesis and suggest that the Mmp1a-deficient mouse will be a valuable tool in further interrogating the (patho)physiologic functions of MMP1 in the mouse.
Despite the lack of obvious developmental abnormalities, the Mmp1a-deficient mice or media produced from Mmp1a-deficient fibroblasts exhibited significant defects in tumor angiogenesis and endothelial tube formation. This is in agreement with a recent study that indicates that Mmp1a triggers PAR1-dependent endothelial cell activation12. Endothelial PAR1 has been shown to be critical for angiogenesis in the developing mouse35 and is an important mediator of tube formation of endothelial cells in vitro upon activation by MMP126. MMP1-PAR1 induces an increase in intracellular calcium and vWF exocytosis from HUVECs24 and causes production of other pro-angiogenic chemokines including growth-regulated oncogene-α, IL-8, and monocyte chemoattractant protein-125. Pharmacologic blockade of MMP1 activity (e.g. Mmp1a) in septic mice suppressed endothelial barrier disruption, disseminated intravascular coagulation, multi-organ failure, systemic cytokine production, and improved survival which was lost in Par1-deficient mice12. As MMP1 small molecule inhibitors have exhibited antagonist effects on related collagenases36 including negating potentially protective effects by MMP837, this new Mmp1a-deficient mouse strain may prove to be a useful tool in studying the specific effects of Mmp1a on endothelial biology in various pathological states such as sepsis.
Interestingly, stromal-deficiency of Mmp1a had striking effects on primary tumorigenesis, despite autocrine Mmp1a expression in the LLC1 cells. Previous work has demonstrated that autocrine Mmp1a modulates the invasive and metastatic phenotype29. Knockdown of LLC1 Mmp1a expression results in a 40% reduction in experimental metastasis29, whereas stromal Mmp1a-deficiency did not affect experimental metastasis to lung. This may indicate that autocrine (cancer cell-produced) MMP1/Mmpla may play a more prominent role in metastasis, whereas stromal MMP1/Mmp1a may support primary tumor growth and angiogenesis. Although the interaction between stromal and cancer cell-secreted MMPs in the tumor microenvironment has not been extensively characterized, these results suggest that the varied sources of MMPs in the microenvironment may play differential and non-complementary roles in tumor biology15. The Mmp1a-deficient mouse presents the opportunity to characterize these functional roles for tumor and stromal-derived Mmp1a.
Another unexpected finding of the present work was the identification of divergent effects of the human MMP1 versus mouse Mmp1a prodomains in suppressing autocatalysis and protein secretion. Substitution of the prodomain of Mmp1a with that of human MMP1 resulted in major increases in protein secretion of Mmp1a and loss of auto-activation inside the cell. Conversely, the efficient secretion of human MMP1 was ablated upon substitution of its prodomain with that of mouse Mmp1a. Analysis of the amino acids located at the interface between the pro- and catalytic domains revealed a key pro- and catalytic domain interaction site in which a phenylalanine in human MMP1 is substituted for a leucine in Mmp1a. Modification of this residue with the human side-chain resulted in increased Mmp1a protein secretion and improved proteolytic stability of Mmp1a in cell lysates, suggesting that prodomain instability may regulate constitutive Mmp1a secretion. Alternatively, our data indicate that the intrinsic instability of its prodomain may prime mouse Mmp1a for facile activation and heightened signaling activity, making it more difficult to negatively regulate the activity of mouse Mmp1a in the tissue or tumor microenvironment.
Lastly, we found that silencing of PAR1 in the lung carcinoma cells phenocopied stromal Mmp1a-deficiency, underscoring the importance of cancer cell-expressed PAR1 as a stromal Mmp1a target in the tumor microenvironment. In addition to activating PAR1, Mmp1a may modulate tumor-stromal interactions, angiogenesis and inflammation through cleavage of other bioactive molecules. For example, MMP1 and several other MMPs cleave membrane bound pro-tumor necrosis factor-alpha (proTNF-α) into its active, soluble form38. MMP1 degrades insulin-like growth factor binding proteins (IGFBP) thereby increasing bioavailability of IGF and inducing fibroblast proliferation39. MMP1 cleavage inactivates stromal cell-derived factor 1 alpha (SDF-1α), leading to decreased leukocyte and hematopoietic stem cell chemotaxis40. Interleukin 1β (IL1β), which is a potent inducer of MMP transcription, is itself a target for both activation and degradation by MMP1, suggesting a regulatory mechanism by which IL1β induces MMP expression and thereby downregulates IL1β signaling once sufficient MMP levels have been reached41,42. Thus, it is anticipated that the Mmp1a-deficient mouse will be a highly useful tool to help delineate and validate the many in vivo functions ascribed to MMP1 in both tumor biology and other disease processes.
MATERIALS AND METHODS
Plasmid DNA
C-terminal Myc-His tagged MMP1, Mmp1a, E216A Mmp1a, Mmp1b, Mmp8, and Mmp13 in pCMV6-Entry (Origene) were made as previously described29. The Mmp1a point mutations, A58V and L67F were generated by QuickChange site directed mutagenesis (Agilent). The Mmp1a and MMP1 prodomain chimeras were generated by utilizing naturally occurring NdeI restriction sites in Mmp1a at nucleotide 87 and in pCMV6Entry located 5′ to the open reading frame. The prodomain of MMP1 was generated by PCR with an exogenous NdeI site and ligated into NdeI-digested Mmp1a to generate hpro-Mmp1a. A NdeI site was introduced in MMP1 and mpro-MMP1 was generated by ligation of NdeI digested MMP1 vector and Mmp1a prodomain.
Cell Culture
LLC1, Cos7, and HEK293T cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. A549 and MCF7+PAR1 (N55) cells9 were maintained in 10% FBS RPMI. CHOK1 cells were maintained in F12 media with 10% FBS. To generate mouse embryonic fibroblasts (MEFs), embryos were harvested from Mmp1a heterozygote crosses at embryonic day 12.5 and maintained in DMEM supplemented with 10% FBS.
Mmp1a-deficient mice
Mmp1a−/− were generated in the laboratory of Dr. Lopez Otin. Exon 5 of Mmp1a was replaced by PGK-Neo cassette using standard techniques20. Chimeric males were mated to C57BL/6 females and pups were screened by PCR. Heterozygotes were backcrossed for 10 generations into the C57BL/6 background. Homozygotes and wild type controls were obtained by crossing heterozygous littermates. Genotyping PCR was performed with a three primer strategy using genomic DNA isolated from tail fragments. The primer sequences used were: Wild type allele primer (5′-3′): acgcattctgcctactgcaagg; Knockout allele primer (5′-3′): tgaccgcttcctcgtgcttta; Common primer (5′-3′): gcagaccatggtgacaacaacc.
Tumor xenografts & Experimental Metastasis
All mouse experiments were conducted in accordance with the National Institutes of Health guidelines and were approved by the Tufts University Institutional Animal Care and Use Committee. Six to nine week old female C57BL/6 wild type (bred in house or obtained from Charles River Laboratories) and Mmp1a−/− animals were injected with 2×105 LLC1 cells or LLC1 stably transduced with shLuc or shPAR129 in sterile PBS in the abdominal fat pad (2 inoculations per mouse). Starting at day 12, palpable tumors were measured every other day and tumor volume was calculated using the equation (LxW2)/2. At the day 26 endpoint, tumors were harvested, weighed, and formalin-fixed for histology.
Experimental metastasis to lung was performed by tail vein injection of 1×106 LLC1 in 200 μl PBS in six to 8 week old female WT or KO mice as before29. Lungs were harvested and fixed at 28 days. Number of metastatic nodules was determined by counting 3 coronal H&E sections per mouse in a blinded fashion.
Tumor-MEF co-implantation experiments were performed by injection of 2×105 LLC1 and 1×105 wild type or knockout MEFs in sterile PBS into the abdominal fat pad of six to nine week old wild type or Mmp1a knockout females. Tumor growth was measured every other day starting at day 12. The experiment was terminated at day 22 due to protocol limits being reached.
Histology
Parafin-embedding, sectioning, and immunohistochemistry for Von Willebrand Factor were performed by the Tufts Medical Center Pathology department. Quantification of the number of vWF-positive blood vessels was performed in a blinded fashion by counting the number of blood vessels per 50 40X fields, in viable/non-necrotic regions of the tumors.
Endothelial tube formation
MatTek plates were chilled and coated with 100 μl Matrigel. HUVEC cells (3.5 × 104, p2–5) in EBM2 media with 0.5% BSA were plated on top. The cells were stimulated with MEF conditioned media (diluted 1:2, from 1 × 106 MEFs following 24 h conditioning in 0.5% BSA DMEM). Endothelial tubes were observed after 5–6 hours under phase contrast inverted microscopy (4X magnification). ImageJ software was used to quantify tubal length and branch complexity from digital images (n=4).
Migration and invasion assays
All migration and invasion assays were performed using Boyden chambers with 8 μm pores as described before29. For migration, A549 or MCF7+PAR1 cells were serum starved and then 5 × 104 cells were migrated towards MEF conditioned media (harvested from 1 × 106 cells in 0.1% FBS over 24 h) for 18h. Chemo-invasion was performed by crosslinking 50 μg of type I rat tail collagen (BD Biosciences) atop a Boyden membrane. LLC1 cells (2 × 104) were then allowed to invade for 48 h toward MEF conditioned media (harvested from 1 × 106 cells in 0.1% FBS over 24 h).
Proliferation Assays
LLC1 cells (2 × 103) were plated in 96-well plates and allowed to adhere overnight. Cells were starved for 4 hours and then stimulated for 72 h with WT or KO MEF conditioned media, made by incubating MEFs (1 × 106) cells per 10 cm plate for 72 h in 10% FBS DMEM. MTT activity was read as absorbance at 570 nm following 4 h incubation with MTT reagent.
Heterologous expression systems and protein purification
HEK293T cells were passaged 1–3 h before transfection with C-terminal Myc-His tagged MMP constructs in pCMV6-Entry via the calcium phosphate method. CHOK1 and Cos7 cells were transfected with polyethylenimine (PEI) using a 1:3 DNA:PEI ratio. The following day, the transfection media was removed and replaced with 0.1% FBS F12 (CHOK1) or 0.1% FBS DMEM (Cos7). After 48 h, conditioned media and cells were harvested. Protein expression was determined by western blotting of 40 μl conditioned media or 40 μg lysate for Myc-tag expression (Cell Signaling).
RNA levels were compared between expression constructs using semi-quantitative PCR with primers designed against the C-terminal construct tag to recognize all pCMV6 plasmids. A standard PCR reaction was performed for 40 cycles. Primers (5′-3′) were: ACGCGTACGCGGCCG (pCMV6-F), GCTTACAGATCCTCTTCTGA (pCMV6-R), GGCTCTTCCAGCCTTCCTTCCT (actin-F), CACAGAGTACTTGCGCTCAGGAGG (actin-R).
His-tagged Mmp1a or human MMP1 was affinity purified from 293T conditioned media using Ni-IDA resin (Biorad) according to the manufacturer’s protocol. The amount of enzyme was quantified using DQ-collagenase activity as compared to a standard curve of collagenase activity from commercially available MMP1 (EMD Millipore).
T7-PAR1 Cleavage
PAR1 cleavage was measured by loss of surface expression of an N-terminal T7 epitope as described previously9. Briefly, Cos7 cells transiently expressing T7-tagged PAR1 were treated with 10 nM thrombin, APMA-activated MMP1 or Mmp1a, or PBS-buffer control for 30 min at 37°C. Cells were than stained on ice using mouse anti-T7 antibody (Novagen) with FITC secondary and analyzed via flow cytometry.
CYR61 Induction
Cos7 cells were polyethylenimine (PEI) transfected with Myc-tagged MMP constructs in pCMV6-Entry (Origene) at a DNA to PEI ratio of 1:3. Following transfection, serum free DMEM was left to condition for 48 h. C57MG cells (100,000 cells per 6 cm plate) were serum starved overnight and then treated with transfected Cos7 CM for 24 h. RNA was harvested from C57MG cells using the RNeasy kit with on-column DNase digestion (Qiagen). cDNA was prepared using a standard MLV-RT reaction and real time PCR for CYR61 and GAPDH mRNA was performed using SYBR green (Qiagen) using the primers (5′-3′): TAAGGTCTGCGCTAAACAACTC (CYR61F), CAGATCCCTTTCAGAGCGGT (CYR61R), AGGTCGGTGTGAACGGATTTG (GAPDHF), TGTAGACCATGTAGTTGAGGTCA (GAPDHR). Data presented are the mean of 2ΔΔCT.
Mmp1a structural modeling
Mmp1a was homology modeled using SWISS-MODEL (swissmodel.expasy.org)43. The human pro-MMP1 structure was used as the template structure (PDB code 1su3). Images were generated using the PyMOL software package.
Statistical analysis
Significance was determined by two-tailed, heteroscedastic T test with significance defined as p<0.05. If multiple cohorts were compared, one-way ANOVA was performed initially followed by T test using Kaleidagraph and PRISM software.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants F30-HL104835 (to CJF), CA122992, HL64701 (to AK) and CA104406 and United Against Lung (to LC).
We are grateful to Sheida Sharifi for her expertise in quantifying tumor angiogenesis, and Rutika V. Pradhan and Namrata Nammi for analysis of endothelial tube formation. This work was supported in part by NIH grants F30-HL104835 (to CJF), CA122992, HL64701 (to AK) and CA104406 (to LC).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Boström P, Söderström M, Vahlberg T, Söderström K-O, Roberts PJ, Carpén O, et al. MMP-1 expression has an independent prognostic value in breast cancer. BMC Cancer. 2011;11:348. doi: 10.1186/1471-2407-11-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith V, Wirth GJ, Fiebig HH, Burger AM. Tissue microarrays of human tumor xenografts: characterization of proteins involved in migration and angiogenesis for applications in the development of targeted anticancer agents. Cancer Genomics Proteomics. 2008;5:263–273. [PMC free article] [PubMed] [Google Scholar]
- 3.Kanamori Y, Matsushima M, Minaguchi T, Kobayashi K, Sagae S, Kudo R, et al. Correlation between expression of the matrix metalloproteinase-1 gene in ovarian cancers and an insertion/deletion polymorphism in its promoter region. Cancer Research. 1999;59:4225–4227. [PubMed] [Google Scholar]
- 4.Nikkola J, Vihinen P, Vlaykova T, Hahka-Kemppinen M, Kähäri V-M, Pyrhönen S. High expression levels of collagenase-1 and stromelysin-1 correlate with shorter disease-free survival in human metastatic melanoma. Int J Cancer. 2002;97:432–438. doi: 10.1002/ijc.1636. [DOI] [PubMed] [Google Scholar]
- 5.Murray GI, Duncan ME, O’Neil P, Melvin WT, Fothergill JE. Matrix metalloproteinase-1 is associated with poor prognosis in colorectal cancer. Nat Med. 1996;2:461–462. doi: 10.1038/nm0496–461. [DOI] [PubMed] [Google Scholar]
- 6.Murray GI, Duncan ME, O’Neil P, McKay JA, Melvin WT, Fothergill JE. Matrix metalloproteinase-1 is associated with poor prognosis in oesophageal cancer. J Pathol. 1998;185:256–261. doi: 10.1002/(SICI)1096-9896(199807)185:3<256::AID-PATH115>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 7.Goldberg GI, Wilhelm SM, Kronberger A, Bauer EA, Grant GA, Eisen AZ. Human fibroblast collagenase. Complete primary structure and homology to an oncogene transformation-induced rat protein. J Biol Chem. 1986;261:6600–6605. [PubMed] [Google Scholar]
- 8.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 9.Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 10.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 11.Eck SM, Blackburn JS, Schmucker AC, Burrage PS, Brinckerhoff CE. Matrix metalloproteinase and G protein coupled receptors: co-conspirators in the pathogenesis of autoimmune disease and cancer. J Autoimmun. 2009;33:214–221. doi: 10.1016/j.jaut.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tressel SL, Kaneider NC, Kasuda S, Foley C, Koukos G, Austin K, et al. A matrix metalloprotease-PAR1 system regulates vascular integrity, systemic inflammation and death in sepsis. EMBO Mol Med. 2011;3:370–384. doi: 10.1002/emmm.201100145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolon I, Gouyer V, Devouassoux M, Vandenbunder B, Wernert N, Moro D, et al. Expression of c-ets-1, collagenase 1, and urokinase-type plasminogen activator genes in lung carcinomas. Am J Pathol. 1995;147:1298–1310. [PMC free article] [PubMed] [Google Scholar]
- 14.Ito M, Ishii G, Nagai K, Maeda R, Nakano Y, Ochiai A. Prognostic impact of cancer-associated stromal cells in stage I lung adenocarcinoma patients. Chest. 2012;142:151–158. doi: 10.1378/chest.11-2458. [DOI] [PubMed] [Google Scholar]
- 15.Saarinen J, Welgus HG, Flizar CA, Kalkkinen N, Helin J. N-glycan structures of matrix metalloproteinase-1 derived from human fibroblasts and from HT-1080 fibrosarcoma cells. Eur J Biochem. 1999;259:829–840. doi: 10.1046/j.1432-1327.1999.00105.x. [DOI] [PubMed] [Google Scholar]
- 16.Balbín M, Fueyo A, Knäuper V, López JM, Alvarez J, Sánchez LM, et al. Identification and enzymatic characterization of two diverging murine counterparts of human interstitial collagenase (MMP-1) expressed at sites of embryo implantation. J Biol Chem. 2001;276:10253–10262. doi: 10.1074/jbc.M009586200. [DOI] [PubMed] [Google Scholar]
- 17.Nuttall RK, Sampieri CL, Pennington CJ, Gill SE, Schultz GA, Edwards DR. Expression analysis of the entire MMP and TIMP gene families during mouse tissue development. FEBS Letters. 2004;563:129–134. doi: 10.1016/S0014-5793(04)00281-9. [DOI] [PubMed] [Google Scholar]
- 18.Hartenstein B, Dittrich BT, Stickens D, Heyer B, Vu TH, Teurich S, et al. Epidermal development and wound healing in matrix metalloproteinase 13-deficient mice. J Invest Dermatol. 2006;126:486–496. doi: 10.1038/sj.jid.5700084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfaffen S, Hemmerle T, Weber M, Neri D. Isolation and characterization of human monoclonal antibodies specific to MMP-1A, MMP-2 and MMP-3. Experimental Cell Research. 2010;316:836–847. doi: 10.1016/j.yexcr.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Balbín M, Fueyo A, Tester AM, Pendás AM, Pitiot AS, Astudillo A, et al. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35:252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- 21.Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, et al. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131:5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inada M, Wang Y, Byrne MH, Rahman MU, Miyaura C, Lopez-Otin C, et al. Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc Natl Acad Sci U S A. 2004;101:17192–17197. doi: 10.1073/pnas.0407788101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertram JS, Janik P. Establishment of a cloned line of Lewis Lung Carcinoma cells adapted to cell culture. Cancer Lett. 1980;11:63–73. doi: 10.1016/0304-3835(80)90130-5. [DOI] [PubMed] [Google Scholar]
- 24.Goerge T, Barg A, Schnaeker E-M, Poppelmann B, Shpacovitch V, Rattenholl A, et al. Tumor-derived matrix metalloproteinase-1 targets endothelial proteinase-activated receptor 1 promoting endothelial cell activation. Cancer Research. 2006;66:7766–7774. doi: 10.1158/0008-5472.CAN-05-3897. [DOI] [PubMed] [Google Scholar]
- 25.Agarwal A, Tressel SL, Kaimal R, Balla M, Lam FH, Covic L, et al. Identification of a metalloprotease-chemokine signaling system in the ovarian cancer microenvironment: implications for antiangiogenic therapy. Cancer Research. 2010;70:5880–5890. doi: 10.1158/0008-5472.CAN-09-4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blackburn JS, Brinckerhoff CE. Matrix metalloproteinase-1 and thrombin differentially activate gene expression in endothelial cells via PAR-1 and promote angiogenesis. Am J Pathol. 2008;173:1736–1746. doi: 10.2353/ajpath.2008.080512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agarwal A, Covic L, Sevigny LM, Kaneider NC, Lazarides K, Azabdaftari G, et al. Targeting a metalloprotease-PAR1 signaling system with cell-penetrating pepducins inhibits angiogenesis, ascites, and progression of ovarian cancer. Molecular Cancer Therapeutics. 2008;7:2746–2757. doi: 10.1158/1535-7163.MCT-08-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cisowski J, O’Callaghan K, Kuliopulos A, Yang J, Nguyen N, Deng Q, et al. Targeting protease-activated receptor-1 with cell-penetrating pepducins in lung cancer. Am J Pathol. 2011;179:513–523. doi: 10.1016/j.ajpath.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foley CJ, Luo C, O’Callaghan K, Hinds PW, Covic L, Kuliopulos A. Matrix metalloprotease-1a promotes tumorigenesis and metastasis. J Biol Chem. 2012;287:24330–24338. doi: 10.1074/jbc.M112.356303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jozic D, Bourenkov G, Lim N-H, Visse R, Nagase H, Bode W, et al. X-ray structure of human proMMP-1: new insights into procollagenase activation and collagen binding. J Biol Chem. 2005;280:9578–9585. doi: 10.1074/jbc.M411084200. [DOI] [PubMed] [Google Scholar]
- 31.Van Wart HE, Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci USA. 1990;87:5578–5582. doi: 10.1073/pnas.87.14.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen N, Kuliopulos A, Graham RA, Covic L. Tumor-derived Cyr61(CCN1) promotes stromal matrix metalloproteinase-1 production and protease-activated receptor 1-dependent migration of breast cancer cells. Cancer Research. 2006;66:2658–2665. doi: 10.1158/0008-5472.CAN-05-2082. [DOI] [PubMed] [Google Scholar]
- 33.Vizoso FJ, González LO, Corte MD, Rodríguez JC, Vázquez J, Lamelas ML, et al. Study of matrix metalloproteinases and their inhibitors in breast cancer. Br J Cancer. 2007;96:903–911. doi: 10.1038/sj.bjc.6603666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heppner KJ, Matrisian LM, Jensen RA, Rodgers WH. Expression of most matrix metalloprotease family members in breast cancer represents a tumor-induced host response. Am J Pathol. 1996;149:273–282. [PMC free article] [PubMed] [Google Scholar]
- 35.Griffin CT, Srinivasan Y, Zheng Y-W, Huang W, Coughlin SR. A Role for Thrombin Receptor Signaling in Endothelial Cells During Embryonic Development. Science. 2001;293:1666–1670. doi: 10.1126/science.1061259. [DOI] [PubMed] [Google Scholar]
- 36.Odake S, Morita Y, Morikawa T, Yoshida N, Hori H, Nagai Y. Inhibition of matrix metalloproteinases by peptidyl hydroxamic acids. Biochem Biophys Res Commun. 1994;199:1442–1446. doi: 10.1006/bbrc.1994.1392. [DOI] [PubMed] [Google Scholar]
- 37.Vanlaere I, Libert C. Matrix metalloproteinases as drug targets in infections caused by gram-negative bacteria and in septic shock. Clin Microbiol Rev. 2009;22:224–239. doi: 10.1128/CMR.00047-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gearing AJ, Beckett P, Christodoulou M, Churchill M, Clements J, Davidson AH, et al. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature. 1994;370:555–557. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]
- 39.Fowlkes JL, Enghild JJ, Suzuki K, Nagase H. Matrix metalloproteinases degrade insulin-like growth factor-binding protein-3 in dermal fibroblast cultures. J Biol Chem. 1994;269:25742–25746. [PubMed] [Google Scholar]
- 40.McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, et al. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem. 2001;276:43503–43508. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- 41.Ito A, Mukaiyama A, Itoh Y, Nagase H, Thogersen IB, Enghild JJ, et al. Degradation of interleukin 1beta by matrix metalloproteinases. J Biol Chem. 1996;271:14657–14660. doi: 10.1074/jbc.271.25.14657. [DOI] [PubMed] [Google Scholar]
- 42.Schönbeck U, Mach F, Libby P. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- 43.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.