

Abstract
Spinocerebellar ataxia type 10 (SCA10) is an autosomal dominant neurodegenerative disorder manifested by ataxia with a variable presentation of epileptic seizures, which is caused by a large expansion of an intronic ATTCT pentanucleotide repeat in ATXN10 on 22q13.3. Herein, we report the first description of SCA10 in a Peruvian family, supporting the Amerindian origin of SCA10 and the Panamerican geographical distribution of the disease in North, Central and South America. Moreover, the presence of an interruption motif in the SCA10 expansion along with epileptic seizures in this family supports the correlation between the two, as seen in other families. Finally, this is the first SCA10 patient ever observed outside of America, specifically in Italy. Since this patient is a Peruvian immigrant of Amerindian ancestry, our case report highlights the growing need for awareness amongst clinicians of seemingly geographically restricted rare diseases.
Keywords: Spinocerebellar ataxia, SCA10, Epilepsy
Introduction
Spinocerebellar ataxia type 10 (SCA10; OMIM#603516) is a rare form of autosomal dominant cerebellar ataxia variably associated with epilepsy and other nervous system disorders, which is caused by a large expansion of an intronic ATTCT pentanucleotide repeat in ATXN10 on 22q13.3. Normal alleles contain from 9 to 32 repetitions [1], while affected people’s alleles contain up to 4500 repetitions [2]. SCA10 has been so far described only in patients of Amerindian ancestry in Latin America and among Sioux Indian descendants [3–5]. While clinical manifestations of families described in Brazil are those of a predominantly “pure” cerebellar syndrome, in the Mexican patients cerebellar ataxia is often accompanied by epilepsy and occasionally peripheral neuropathy [3,4]. Herein, we report the first description of SCA10 in a Peruvian family with a discussion of phenotypic and genotypic characteristics.
Case report
A 47-year-old Peruvian woman, of pure Amerindian ancestry, originated from the Paijan region of Northern coastal Peru (III-6, Fig. 1). She had immigrated to Italy in her mid-twenties. She was evaluated in our center for progressive gait unsteadiness with onset at 35 years of age and frequent falls over the last few years. At neurological examination she showed ataxic gait. She was still able to walk, but needed bilateral support. Cerebellar signs, including moderate scanning speech, intentional tremor, mild dysmetria and dysdiadochokinesia were also observed as well as bilateral fixation nystagmus and saccadic pursuit. She scored a 57 (out of 100) on the Scale for Assessment and Rating of Ataxia (SARA) [6]. No peripheral neuropathy, pyramidal, extrapyramidal signs nor mental retardation were detected. Brain MRI showed moderate pancerebellar atrophic changes (Fig. 2). During her hospitalization for rehabilitation purposes, brief episodes of sudden impairment of consciousness were noted. This prompted an EEG examination, which showed bilateral focal epileptiform discharges over the occipito-temporal areas. A diagnosis of complex partial seizures was made. Treatment with oral leviracetam, 500 mg b.i.d, resulted in good seizure control. Anti-cerebellar, and anti-GAD65, anti-gliadin, anti-tissue transglutaminase and anti-endomysial antibodies, thyroid function screening, serum vitamin E and alphafetoprotein were within normal values. The association of an idiopathic progressive cerebellar disease with epilepsy in a woman of Amerindian origin, suggested a possible diagnosis of SCA10. Indeed fluorescent repeat-primed PCR analysis showed a clear heterozygous ATTCT expansion in ATXN10 gene, well above normal range (>1000 repeats; N.V. <13) (Fig. 3). Testing for SCA1, SCA2, SCA3, SCA12 and FXN analyses gave entirely normal results. Southern blot analysis confirmed a heterozygous SCA10 repeat expansion of 1760 repeats. Upon further investigation, several other family members were reported as affected by a similar disorder (Fig. 1), with a clear autosomal dominant pattern of inheritance. The proband’s brother, III-2, was examined and found to have mild cerebellar axial ataxia with no apparent epileptic episodes. Southern blot analysis of his genomic DNA confirmed a heterozygous expansion of the SCA10 locus of 820 repeats. The proband’s two sons (IV-1 and IV-2), were found to be both clinically and genetically healthy. Haplotype analysis of single nucleotide polymorphisms (SNPs) surrounding their SCA10 expansion (data not shown) is identical to Brazilian, Mexican, Argentinean and Sioux SCA10 expansions [5,7,8] indicating a shared ancestral origin.
Figure 1.
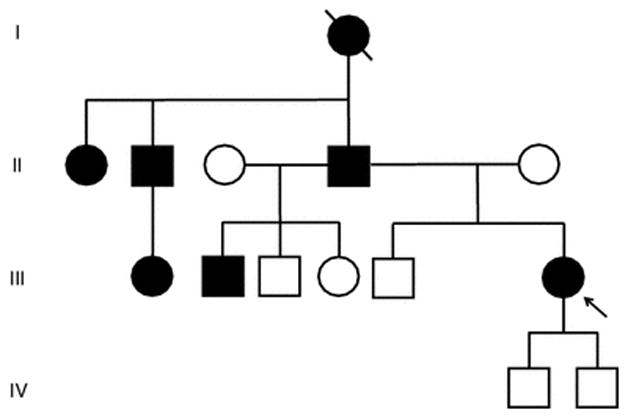
Family tree showing autosomal dominant pattern of inheritance. Proband (III-6) and her half-brother (III-2) have been genotyped and found to carry an expanded ATTCT in the SCA10 gene, 1,760 and 820 repeats, respectively. The proband’s two healthy sons have been found not to carry the expanded allele.
Figure 2.
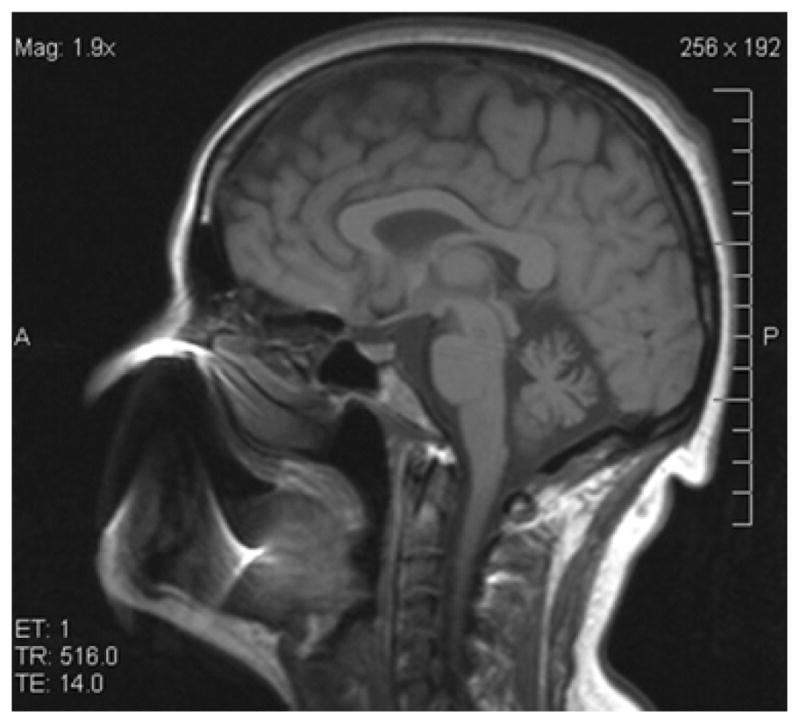
T1-weighted sagittal brain MRI showing cerebellar atrophy in patient III-6.
Figure 3.
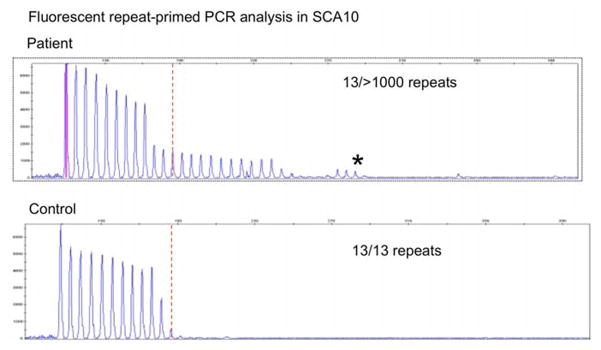
Fluorescent triplet repeat-primed PCR (TP-PCR) analysis of the ATXN10 gene, showing an expanded ATTCT pentanucleotide repeat in the proband. The interruptions in the series of peaks at the high molecular weight products indicate that interruption motifs are present in SCA10 expansion.
Discussion
SCA10 is the second most common autosomal dominant cerebellar ataxia in Brazil (after SCA 3) and in Mexico (after SCA 2) [3]. The ATTCT expansion in ATXN10 is hypothesized to have an ancestral, common origin in Amerindian populations [5,7]. We report the first SCA10 family occurring in Peru, which fits the Panamerican geographical distribution of the disease in North, Central and South America and supports the Amerindian origin of SCA10. The vast majority of Brazilian SCA10 patients develop pure cerebellar ataxia and have a very low incidence of epileptic seizures [4,9] while Mexican SCA10 patients are more likely to develop variable extra-cerebellar symptoms including epilepsy, psychiatric disorders, polyneuropathy and soft pyramidal signs [10]. Indeed, epileptic seizures are found in the vast majority (60%) of Mexican patients [9]. Our proband (III-6) shows a phenotype of completely pure cerebellar ataxia, similar to that of the Brazilian patients, and also has epileptic seizures, as found in Mexican patients.
RP-PCR analysis indicates that the proband and her brother have interruption motifs present in their SCA10 expansions. This is indicated by interruptions in the series of peaks at the high molecular weight products (see Figure 3). This phenomenon in the RP-PCR pattern has been seen for other patients with confirmed interruptions, especially those of the “ATCCT-positive” type of interruptions which are highly correlated with epileptic seizures [8–11]. The presence of an interruption motif of an unidentified type along with epileptic seizures in this patient supports the correlation between the two seen in other families [8].
Comparing the paternally inherited SCA10 expansion size between the proband and her brother indicates a high degree of instability in this family. Prior work has shown that paternally inherited, interrupted SCA10 expansions are highly unstable with propensity towards shrinkage in intergenerational transmission [11]. However, we can only infer intergenerational instability from the disparity in the expansion sizes of the siblings as we do not have genomic DNA from the father. Yet, these finding fit with prior studies of SCA10 families and suggests that, both in terms of clinical and molecular features, the SCA10 expansion in this family behaves similar previously described paternally inherited SCA10 expansions alleles that harbor “ATCCT-type” interruption motifs. Though we do note that the interruption motif is unidentified in this family.
Finally, this is the first SCA10 patient ever observed outside of America, specifically in Italy. Although this patient is a Peruvian immigrant of Amerindian ancestry, our case report highlights the growing need for awareness amongst clinicians of seemingly geographically restricted rare diseases. Therefore, SCA10 should be considered in patients with suitable genetic background, especially if cerebellar ataxia is associated with epilepsy.
METHODS
Patient Samples
Following informed, written consent, blood was drawn from the patient. High molecular weight genomic DNA was extracted from leucocytes following standard procedures.
SCA10 RP-PCR analysis
SCA10 ATTCT repeat in intron 9 of the ATXN10 gene was determined by fluorescent triplet repeat-primed PCR (RP-PCR) analysis [12,13], using three primers including one fluorescent-dye-conjugated forward primer (SCA10-5′-6-FAM-cagatggcagaatgataaactcaa), a first reverse primer TP-R1 (5′-tacgcatcccagtttgagacgagaatagaatagaatagaatagaat) and a second reverse TP-R2 anchor tail (5′-agaatagaatagaatagaatagaat). TP-PCR was carried out as follows: 50ng of genomic DNA, 0.8 μM of locus-specific (forward) and 0.08 μM of TP-R1 primer and 0.8 μM of the TP-R2 primer, 200 μmol/L dNTPs, 1.5 mmol/L MgCl2, and 1U of TaqGold in 1X specific buffer (LifeTechnologies). Amplified fragments were separated in 3130xl Genetic Analyzer and the SCA10 expansion was identified by the presence of a smeared ladder of peaks with a characteristic 5-bp periodicity, using GeneMapper v4.0 (LifeTechnologies).
Southern blot analysis
Southern blot analysis was performed as described [2]. Briefly, 10 micrograms of high molecular weight genomic DNA was digested with EcoRI overnight, separated on a 0.8% agarose gel and transferred to a nitrocellulose membrane. The membrane was subsequently probed with a 32P-labeled, 800-bp PCR fragment located directly upstream of the SCA10 expansion. SCA10 expansions were sized using molecular weight standards in the gel.
SCA10 haplotype analysis
SCA10 haplotype analysis was performed using single nucleotide polymorphisms (SNPs) rs5764850 and rs72556348. These SNPs flank the SCA10 expansion have been used to define a C (rs5764850)-expansion-G (rs72556348)-G-C SCA10 haplotype [7] and is found is all described tested SCA10 families to date [8]. PCR primers, amplification conditions and RFLP restriction digests were performed as described [8].
Figure 4.
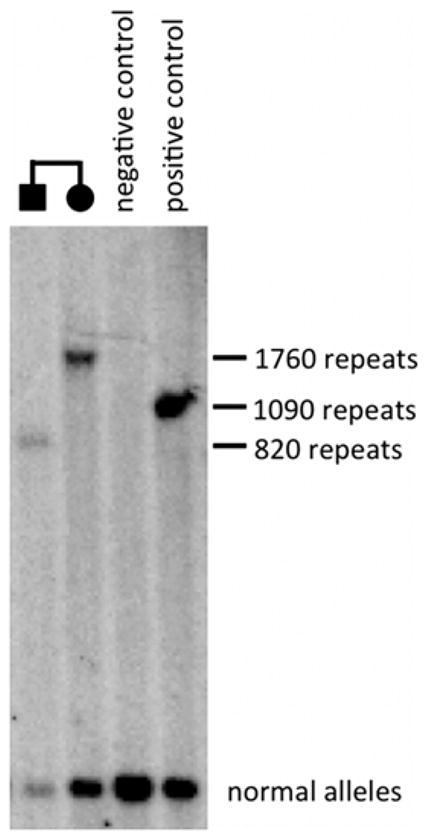
Southern blot analysis showing a heterozygous SCA10 repeat expansion of 1,760 repeats in proband (III-6) and of 820 repeats in her less-affected brother (III-2).
Acknowledgments
We would like to acknowledge Massimiliano DiMicco, MD, for his brilliant clinical insight. The study was partly supported by NIH grant NS083564 (TA).
Footnotes
Conflicts of interest: On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical standard: This study has been approved by the appropriate ethics committee and has therefore been performed in the accordance with the ethical standards laid down in the 1964 Declaration of Helsinki.
References
- 1.Wang JL, Wu YQ, Lei LF, Shen L, Jiang H, Zhou YF, Yi JP, Zhou J, Yan XX, Pan Q, Xia K, Tang BS. Polynucleotide repeat expansion of nine spinocerebellar ataxia subtypes and dentatorubral-pallidoluysian atrophy in healthy Chinese Han population. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2010;27(5):501–505. doi: 10.3760/cma.j.issn.1003-9406.2010.05.006. (Article in Chinese) [DOI] [PubMed] [Google Scholar]
- 2.Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26:191, e4. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- 3.Teive HA, Munhoz RP, Arruda WO, Raskin S, Werneck LC, Ashizawa T. Spinocerebellar ataxia type 10: a review. Parkinsonism Relat Disord. 2011;17:655–661. doi: 10.1016/j.parkreldis.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Teive HA, Roa BB, Raskin S, Fang P, Arruda WO, Neto YC, Gao R, Werneck LC, Ashizawa T. Clinical phenotype of Brazilian families with spinocerebellar ataxia 10. Neurology. 2004 Oct 26;63(8):1509–1512. doi: 10.1212/01.wnl.0000142109.62056.57. [DOI] [PubMed] [Google Scholar]
- 5.Bushara K, Bower M, Liu J, McFarland KN, Landrian I, Hutter D, Teive HA, Rasmussen A, Mulligan CJ, Ashizawa T. Expansion of the Spinocerebellar Ataxia Type 10 (SCA10) Repeat in a Patient with Sioux Native American Ancestry. PLoS One. 2013 Nov 20;8(11):e81342. doi: 10.1371/journal.pone.0081342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitz-Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66:1717–1720. doi: 10.1212/01.wnl.0000219042.60538.92. [DOI] [PubMed] [Google Scholar]
- 7.Almeida T, Alonso I, Martins S, Ramos EM, Azevedo L, Ohno K, et al. Ancestral origin of the ATTCT repeat expansion in spinocerebellar ataxia type 10 (SCA10) Plos One. 2009;4:e4553. doi: 10.1371/journal.pone.0004553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McFarland KN, Liu J, Landrian I, Zeng D, Raskin S, Moscovich M, Gatto EM, Ochoa A, Teive HA, Rasmussen A, Ashizawa T. Repeat interruptions in spinocerebellar ataxia type 10 expansions are strongly associated with epileptic seizures. Neurogenetics. 2014;15(1):59–64. doi: 10.1007/s10048-013-0385-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teive HA, Munhoz RP, Raskin S, Arruda WO, de Paola L, Werneck LC, Ashizawa T. Spinocerebellar ataxia type 10: Frequency of epilepsy in a large sample of Brazilian patients. Mov Disord. 2010 Dec 15;25(16):2875–2878. doi: 10.1002/mds.23324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rasmussen A, Matsuura T, Ruano L, Yescas P, Ochoa A, Ashizawa T, Alonso E. Clinical and genetic analysis of four Mexican families with spinocerebellar ataxia type 10. Ann Neurol. 2001;50(2):234–239. doi: 10.1002/ana.1081. [DOI] [PubMed] [Google Scholar]
- 11.McFarland KN, Liu J, Landrian I, Gao R, Sarkar PS, Raskin S, Moscovich M, Gatto EM, Teive HA, Ochoa A, Rasmussen A, Ashizawa T. Paradoxical effects of repeat interruptions on spinocerebellar ataxia type 10 expansions and repeat instability. Eur J Hum Genet. 2013;21(11):1272–1276. doi: 10.1038/ejhg.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warner JP, Barron LH, Goudie D, Kelly K, Dow D, Fitzpatrick DR, Brock DJ. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J Med Genet. 1996;33(12):1022–1026. doi: 10.1136/jmg.33.12.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cagnoli C, Michielotto C, Matsuura T, Ashizawa T, Margolis RL, Holmes SE, Gellera C, Migone N, Brusco A. Detection of large pathogenic expansions in FRDA1, SCA10, and SCA12 genes using a simple fluorescent repeat-primed PCR assay. J Mol Diagn. 2004;6(2):96–100. doi: 10.1016/S1525-1578(10)60496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]