

Abstract
Recent studies have suggested that the gut microbiome may be an important factor in the development of colorectal cancer (CRC). Abnormalities in the gut microbiome have been reported in patients with CRC; however, this microbial community has not been explored as a potential screen for early stage disease. We characterized the gut microbiome in patients from three clinical groups representing the stages of CRC development: health, adenoma, and carcinoma. Analysis of the gut microbiome from stool samples revealed both an enrichment and depletion of several bacterial populations associated with adenomas and carcinomas. Combined with known clinical risk factors of CRC (e.g. BMI, age, race), data from the gut microbiome significantly improved the ability to differentiate between healthy, adenoma, and carcinoma clinical groups relative to risk factors alone. Using Bayesian methods, we determined that using gut microbiome data as a screening tool improved the pre-test to post-test probability of adenoma over 50-fold. For example, the pre-test probability in a 65 year-old was 0.17% and, after using the microbiome data, this increased to 10.67% (1 in 9 chance of having an adenoma). Taken together the results of our study demonstrate the feasibility of using the composition of the gut microbiome to detect the presence of precancerous and cancerous lesions. Furthermore, these results support the need for more cross sectional studies with diverse populations and linkage to other stool markers, dietary data, and personal health information.
Introduction
Worldwide, colorectal cancer (CRC) is the third most commonly diagnosed malignancy and accounts for over a half million deaths annually (1). Development of CRC is a stepwise process by which localized precancerous adenomatous polyps (ademonas) develop in the colon and progress into invasive and metastatic cancerous tumors (carcinomas) overtime (2). Development of carcinomas is largely preventable if adenomas are detected and removed (3), with a CRC survival rate exceeding 90% if the diagnosis occurs while the disease is still localized. However, there is a dramatic decline in survival following invasion and metastasis (4). Thus, early detection at the adenoma stage of this disease has been critical for successful treatment and survival.
From 1975 to 2010, death rates from colorectal cancer have steadily decreased in the United States, with a 2.8% average annual decline (4). Screening with high sensitivity fecal occult blood testing (FOBT), sigmoidoscopy, and colonoscopy has improved survival rates and is recommended for adults 50 to 75 years of age (5). In particular, colonoscopies allow for full examination of the bowel with the opportunity for same-session colonic biopsies and removal of polyps. However, over 30% of adults in the US do not receive age and risk-appropriate screenings and surveys indicate that 50–60% of adults prefer non-invasive screening methods (6, 7). Lack of compliance with these recommendations may be due in part to the intrusiveness and uncomfortable nature of the colonoscopy procedure. Furthermore, the healthcare costs of screening for CRC by colonoscopy are considerable, ranging from $800 to $3160 per procedure in 2012, which was undergone by more than 48 million 50–75 year-old Americans (8, 9). Therefore, there is a need to develop cost-effective non-invasive screening methods to prioritize individuals for further evaluation by colonoscopy. One of the most commonly used non-invasive screening procedures is the guaiac fecal occult blood test (gFOBT), which detects blood in an individual’s feces (10). Occult blood in stool can indicate the presence of advanced adenomas and carcinomas in the colon, but can also indicate a wide variety of other disorders and factors that may lead to false positive tests (11). Although the specificity of the method ranges from 87–98% (10), the sensitivity can be as low as 9–12% (10). With repeated testing using multiple stool samples and regular screening intervals, sensitivity can be dramatically improved (3). Despite these limitations, gFOBT has been shown to reduce mortality from CRC by 15 to 33%, highlighting the effectiveness of non-invasive screening measures (12–14).
Approximately 70% of CRC cases develop spontaneously and are of unknown etiology (2). Factors associated with increased risk of CRC include diet, alcohol, and chronic inflammation of the gastrointestinal tract (15–17). Recently, there has been increasing appreciation for a largely understudied variable in CRC, the gut microbiome. This collection of symbiotic microorganisms inhabits the gastrointestinal tract and is associated with diseases such as obesity and inflammatory bowel disease (18, 19). In animal studies, evidence suggests that through interaction with the immune system, production of cancer-associated metabolites, and the release of genotoxic virulence factors, bacteria can directly contribute to the development of CRC (20–22). Furthermore, in human studies, patients with CRC have an abnormal gut microbiome structure when compared to healthy patients (23–25). Taken together, this suggests that the gut microbiome might be a candidate biomarker for early detection of CRC.
We hypothesized that using novel microbiome biomarkers of CRC in concert with known clinical risk factors could improve the ability to identify candidates for colonoscopy. We compared the microbiome of healthy individuals, persons with adenomas, and patients with colorectal carcinomas. We sequenced the V4 region of the 16S rRNA gene from the feces of each individual using the Illumina MiSeq sequencing platform. The resulting data were used to test our hypothesis that the incorporation of microbiome data would significantly improve the ability to distinguish among the three types of individuals, beyond clinical (demographic) data and FOBT results. This analysis demonstrates that the microbiome provides a powerful source of biomarkers for identifying individuals harboring adenomas and carcinomas.
Material and Methods
Study design and sample collection
As part of the National Cancer Institute-funded Early Detection Research Network (EDRN), the Great Lakes-New England Clinical Epidemiological Center (GLNE CEC) created a biorepository that included whole evacuated stool for studies on potential molecular markers for the detection of colonic precancerous and cancerous conditions and cancer risk assessment. This study was approved by the University of Michigan Institutional Review Board and all subjects provided informed consent. Eligible patients were 18 years of age or older, able to tolerate 58 ml of blood removal at two time points, willing to complete an gFOBT kit, able to provide informed consent, and had colonoscopy and histologically confirmed colonic disease status. Patients were excluded if known HIV or chronic viral hepatitis, known HNPCC or FAP, inflammatory bowel disease, any surgery, radiation or chemotherapy for their current colorectal cancer or colonic adenoma. Colonoscopies were performed and fecal samples were collected from subjects in 4 locations: Toronto (Ontario, Canada), Boston (Massachusetts, USA), Houston (Texas, USA), and Ann Arbor (Michigan, USA). Following endoscopic examination, patients without colonic abnormalities were designated as healthy. Examinations that revealed the presence of lesions resulted in a biopsy and subsequence diagnosis of adenoma or carcinoma. For each patient, clinical data were collected including demographic information and the results of the gFOBT (Table 1). There were no significant differences in age or current medication use among the three patient groups. However, among our samples, men, whites, and those with greater BMI were more likely to have colorectal cancer (Table 1).
Table 1.
Characteristics of subjects in each clinical group.
| Healthy | Adenoma | Cancer | P-value | |
|---|---|---|---|---|
| Age, years (mean, SD) | 55.3 (9.2) | 61.3 (11.1) | 59.4 (11.0) | 0.080 |
| Gender (n, %): | ||||
| Men | 11 (37%) | 18 (60%) | 21 (70%) | |
| Women | 19 (63%) | 12 (40%) | 9 (30%) | 0.029 |
| Race/Ethnicity: | ||||
| Non-Hispanic White | 21 (70%) | 27 (90%) | 28 (93%) | |
| Other | 9 (30%) | 3 (10%) | 2 (7%) | 0.026 |
| Body mass index (mean, SD) | 26.6 (5.2) | 27.4 (4.4) | 30.7 (7.2) | 0.022 |
| Current medication use (n, %) | 23 (77%) | 21 (70%) | 26 (87%) | 0.295 |
| Positive FOBT (n, %) | 0 (0%) | 4 (13%) | 22 (73%) | 0.001 |
All participants collected a whole evacuated stool in a hat with no preservatives after following the usual dietary and medication restrictions for 24 hours. Immediately after collection, the patient prepared a gFOBT six-panel kit (Sensa Hemocult II, Beckman-Coulter, Palo Alto, CA) from different areas of the stool. The whole stool was then packaged in an insulated box with ice packs and shipped to the processing center along with the gFOBT cards via next day delivery. Upon receipt the feces were stored at −80C. The gFOBT was processed and interpreted as soon as it arrived at the processing center. If any of the six wells were positive, the kit was recorded as positive for the participant. All participants had intact colonic lesions at time of stool collection. Study participants provided their stool sample between one and four weeks after their colonoscopy preparation. This period of time has previously been shown to be sufficient to allow the microbiome to recover (26). We were provided with 90 stool samples and linked data randomly chosen from disease groups of healthy (n=30), colonic adenoma (n=30), and colonic adenocarcinoma (n=30).
DNA extraction and 16S rRNA gene sequencing
Microbial genomic DNA was extracted using the PowerSoil-htp 96 Well Soil DNA isolation kit (Mo Bio Laboratories) using an EPMotion 5075 pipetting system. The V4 region of the 16S rRNA gene from each sample was amplified and sequenced using the Illumina MiSeq Personal Sequencing platform as described elsewhere (27). Sequences were curated as described previously using the mothur software package (28). Briefly, we reduced sequencing and PCR errors, aligned the resulting sequences to the SILVA 16S rRNA sequence database (29), and removed any chimeric sequences flagged by UCHIME (30). After curation, we obtained between 25,953 and 404,696 sequences per sample (median=95,464), with a median length of 253 bp. To limit effects of uneven sampling, we rarefied the dataset to 25,958 sequences per sample. Parallel sequencing of a mock community revealed an error rate of 0.03%. All fastq files and the MIMARKS spreadsheet are available at http://www.mothur.org/MicrobiomeBiomarkerCRC.
Gut Microbiome Biomarker Discovery Analysis
Sequences were clustered into operational taxonomic units (OTUs) at a 97% similarity cutoff and the relative abundance was calculated for OTUs in each sample. All sequences were classified using a naïve Bayesian classifier trained against the RDP training set (version 9; http://sourceforge.net/projects/rdp-classifier/) and OTUs were assigned a classification based on which taxonomy had the majority consensus of sequences within a given OTU (31). Differentially abundant OTUs were selected using the biomarker discovery algorithm, LEfSe (Linear discriminant analysis Effect Size) for each pairwise comparison of clinical groups (32) (Healthy vs. Adenoma, Healthy vs. Carcinoma, Adenoma vs. Carcinoma, Healthy vs. Colonic lesion). In short, LEfSe first uses a non-parametric factorial Kruskal-Wallis sum-rank test to identify differentially abundant OTUs. This is followed by a set of pairwise tests among clinical groups to ensure biological consistency using the Wilcoxon rank-sum test. Linear discriminant analysis (LDA) is then used to estimate the effect size of each differentially abundant OTU. We then ranked LEfSe statistics to assess greatest differences in microbial relative abundance across patient groups.
Data Analyses
Analyses of patient-level characteristics across the three clinical groups utilized Pearson chi-square test for categorical data and one-way ANOVA for continuous variables. Clinical variables evaluated were age, gender, race/ethnicity, body mass index (BMI, kg/m2), and current medications. One missing value for BMI was imputed. Logit models were generated using both clinical and microbiome data as independent variables to contrast differences across disease groups (i.e., healthy versus adenomas; healthy versus cancer; adenomas versus cancer). OTUs demonstrating the highest LDAs were entered into a logit model and their ability to discriminate group classification was evaluated using area under the receiver operator characteristic (ROC) curve. We used a maximum of 6 OTUs for each model to avoid potentially over-fitting the model. It is important to note that in the first phase of the data analyses, the greatest ranked differences in OTUs (represented by the LEfSe statistic) were used to select the OTUs, not through multiple hypothesis testing. Differences between nested logit models were compared using the test for the equality of ROC areas (33). Data were available on gFOBT status and therefore, this was entered as an independent variable when comparing adenoma versus carcinoma. While we considered possible options for validation, both cross-validation and bootstrapping have been shown to be unreliable in small samples (34). However, Bayesian intervals have been recommended for analyses of cross-classification in small samples and therefore, we calculated 95% Bayesian intervals for the Youden’s J statistic (maximum percentage correctly classified) in the final microbiome models (34). It is important to note that, in our cross-classification, there was no knowledge of types of micro-organisms present in the feces at the time of determination of lesions (normal, adenoma, carcinoma). Therefore, our cross-classification variables are assumed to be independent in this regard (blinded assessment) and fulfill underlying assumptions of testing. We tested using an experiment wide error rate (i.e. α) of 0.05 and performed 2-tailed tests. Analyses were conducted in Stata/MP 13.1.
We used Bayesian methods to estimate the probability of adenoma based on relative abundance data taken from the gut microbiome (35). Since CRC screening involves detection of early stages of disease, data from the model differentiating adenoma from healthy colons formed the basis of a preliminary screening test. Sensitivity, specificity and positive likelihood ratios were calculated based on our study results, with failure to detect any appreciable level of any of these 5 OTUs (0 relative abundance) indicating possible pathology (i.e., positive test). Since the false positive rate of this test was 0%, we applied a continuity correction of 0.1 to each cell and calculated the likelihood ratio of a positive test and the 95% confidence intervals using Jeffreys’ Bayesian credible interval (36). The likelihood ratio was then applied to the pre-test probability of CRC based on national Surveillance, Epidemiology and End Results (SEER) data, years 2000–2010 (4).
Results
Comparison of healthy and adenoma clinical groups
We utilized logit regression models to differentiate between patients in the healthy and adenoma clinical groups. Preliminary models were generated using age, gender, race/ethnicity, BMI, and medication use as independent variables. For these subjects, both age and race were significantly associated with the presence of adenomas (AUC=0.713; 95% CI: 0.580–0.845; p=0.009). There were also differences in the gut microbiome between individuals with and without adenomas. Relative to healthy subjects, subjects with adenomas had higher relative abundances of OTUs affiliated with the Ruminococcaceae (OTU 21), Clostridium (OTU 60), Pseudomonas (OTU 3322), and Porphyromonadaceae (OTUs 1901 and 1903); they had lower relative abundances of OTUs affiliated with the Bacteroides (OTUs 1889 and 1913), Lachnospiraceae (OTU 36), Clostridiales (OTU 38), and Clostridium (OTUs 20, 97, 99) (Supplementary Fig. 1). The model that yielded the greatest differentiation between adenoma and healthy groups included age, race, and 5 OTUs (OTUs 38, 99, 136, 1889, 1913) (Figure 1A). The addition of these 5 OTUs significantly improved the predictive ability of the model beyond that of age and race only (AUC=0.896; 95% CI: 0.816–0.976; p=0.002) (Figure 1B). Youden’s J statistic fell at a sensitivity of 90% and specificity of 80% in this model, yielding a 4.5 fold increase in post-test to pre-test probability of detecting adenoma (95% 3.3, 6.0 fold).
Figure 1. Microbial biomarkers improve accuracy of predictive models for healthy and adenoma clinical groups.
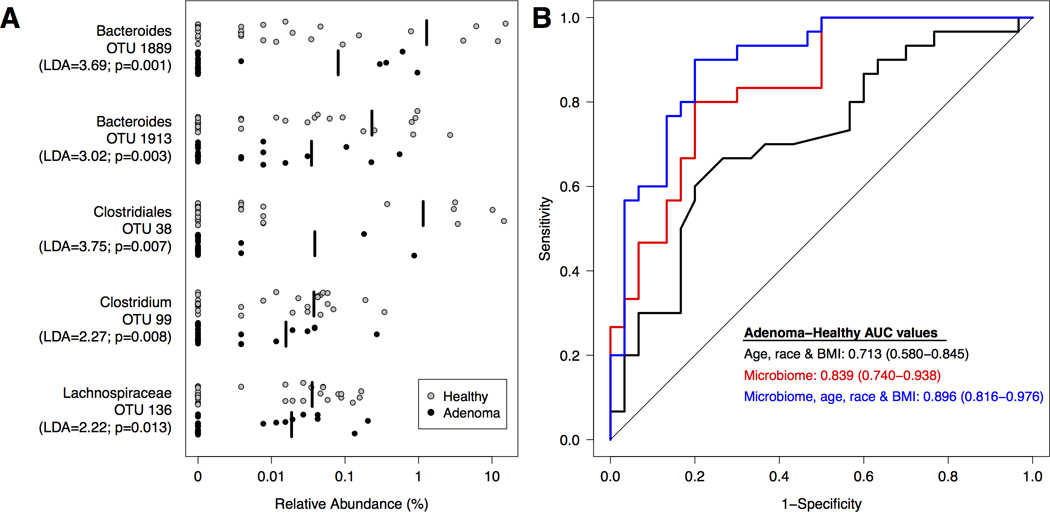
A. Relative abundance of differentially abundant OTUs for all healthy (n=30; grey) and adenoma (n=30; black) subjects. The mean relative abundance is represented for each clinical group by a vertical black line. B. ROC curves for microbial biomarkers alone, clinical data alone, and microbial biomarkers with clinical data. The straight line represents the null model.
Comparison of healthy and carcinoma clinical groups
Next, we generated logit models using clinical and microbiome data to differentiate between patients in the healthy and carcinoma groups. Age, race, and BMI were predictive of carcinomas (AUC=0.798; 95% CI: 0.686–0.910; p<0.001). We observed that relative to healthy subjects, subjects with carcinomas had higher abundances of OTUs associated with Fusobacterium (OTU 2458), Porphyromonas (OTU 1905), Lachnospiraceae (OTUs 31, 59, 32, 116, 85), and Enterobacteriaceae (OTU 2479); they had lower relative abundances of OTUs affiliated with the Bacteroides (OTU 1889), Lachnospiraceae (OTUs 23, 30, 253, 136), and Clostridiales (OTU 42) (Supplementary Figure 2). To test the hypothesis that the gut microbiome could improve our ability to predict the presence of carcinomas, we added these OTUs to the logit model we generated based on the subjects’ age, race, and BMI (Figure 2B). The model with the greatest discriminatory ability included age, race, BMI and 6 OTUs (OTUs 136, 1901, 1905, 1913, 2479, 2458; Figure 2A). This model significantly improved the ability to distinguish between healthy and carcinoma compared to the model containing age, race and BMI only (AUC=0.922; 95% CI: 0.858–0.986; p=0.012; Figure 2B). Youden’s J statistic occurred at a sensitivity of 90% and a specificity of 83.3% in the full model, yielding a 5.4 fold increase in post-test to pre-test probability of detecting carcinoma (95% 4.1, 7.0 fold).
Figure 2. Microbial biomarkers improve accuracy of predictive models for healthy and carcinoma clinical groups.
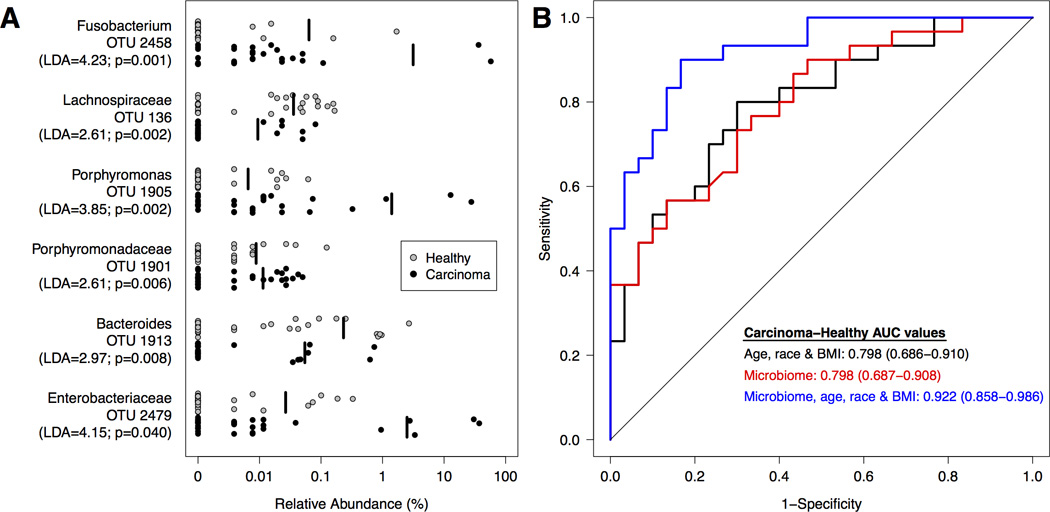
A. Relative abundance of differentially abundant OTUs for all healthy (n=30; grey) and carcinoma (n=30; black) subjects. The mean relative abundance is represented for each clinical group by a vertical black line. B. ROC curves for microbial biomarkers alone, clinical data alone, and microbial biomarkers with clinical data. The straight line represents the null model.
Comparison of healthy individuals to those with colonic lesions
Next, we explored the ability of the gut microbiome to differentiate between healthy subjects and those with either adenoma or carcinomas. Thus, we combined the clinical and microbiome data from adenoma and carcinoma subjects to create a combined colonic lesion group. We then generated a logit model to differentiate between healthy subjects and the colonic lesion group. Clinical variables that were predictive of colonic lesion were age, gender, and race (AUC=0.754; 95% CI: 0.648–0.859) (Figure 3). To test the hypothesis that the gut microbiome could improve our ability to predict the presence of colonic lesions regardless of stage, we added 6 OTUs (OTU 136, 253, 1889, 1897, 1913, 2891) (Supplementary Fig. 3) to this logit model. Age, gender, race, and these 6 OTUs significantly improved the ability to distinguish between the healthy and colonic lesion combined groups (AUC=0.936; 95% CI: 0.887–0.985; p<0.0001) (Figure 3).
Figure 3. Microbial biomarkers improve accuracy of predictive models for differentiating between healthy subjects and those with colonic lesions.
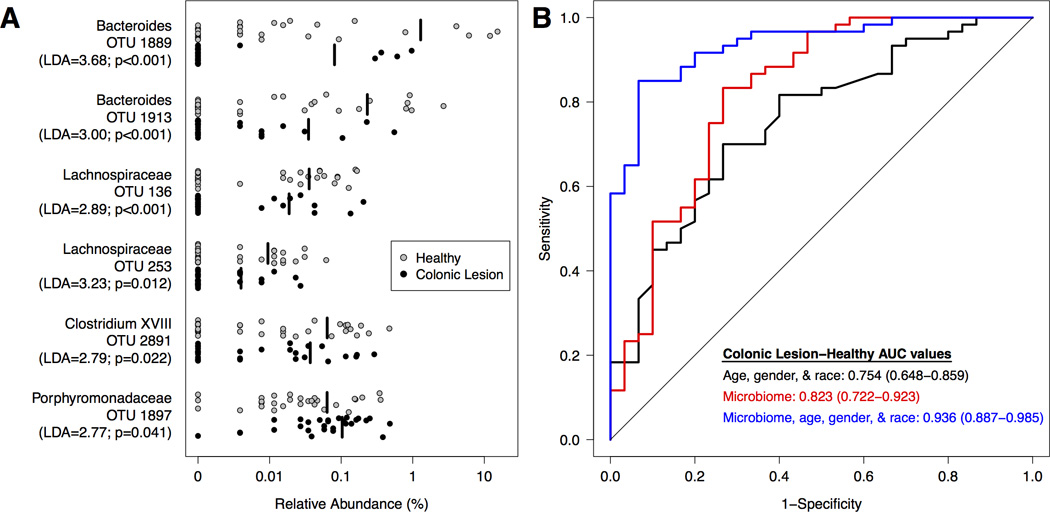
Adenoma and carcinoma subjects were combined into a single clinical group (Lesions; n=60). A. Relative abundance of differentially abundant OTUs for healthy (n=30; grey) subjects and those with lesions (n=60; black). The mean relative abundance is represented for each clinical group by a vertical black line. B. ROC curves for microbial biomarkers alone, clinical data alone, and microbial biomarkers with clinical data. The straight line represents the null model.
Comparison of adenoma and carcinoma clinical groups
Finally, we generated logit models using clinical and microbiome data to differentiate between patients in the adenoma and carcinoma groups. A patient’s BMI was the only clinical variable that discriminated between the adenoma and carcinoma clinical groups (AUC=0.658; 95% CI: 0.518–0.799; p=0.023). When examining populations within the gut microbiome, relative to subjects with adenomas, those with carcinomas harbored higher relative abundances of OTUs that affiliated with the Fusobacterium (OTU 2458), Bacteroides (OTU 1882), Phascolarctobacterium (OTU 2395), and Porphyromonas (OTU 1905). In contrast, OTUs affiliated with Blautia (OTU 9), Ruminococcus (OTU 16), Clostridium (OTUs 60 and 93), Lachnospiraceae (OTU 12 and 23) were more abundant in subjects with adenomas (Supplementary Figure 4). Next, we constructed a logit model to differentiate between the adenoma and carcinoma clinical groups using BMI with microbiome data. The model that provided the greatest differentiation between carcinoma and adenoma included BMI and 4 OTUs (OTUs 1905, 2395, 2458, 3235; Figure 4A). This model provided significantly greater discrimination than BMI alone (AUC=0.963; 95% CI: 0.921–1.00; (p<0.001; Figure 4B). Examination of the relative abundance of OTUs associated with the Fusobacterium genera revealed no significant associations between Fusobacterium and the stage or location of carcinomas.
Figure 4. Microbial biomarkers improve accuracy of predictive models for adenoma and carcinoma clinical groups.
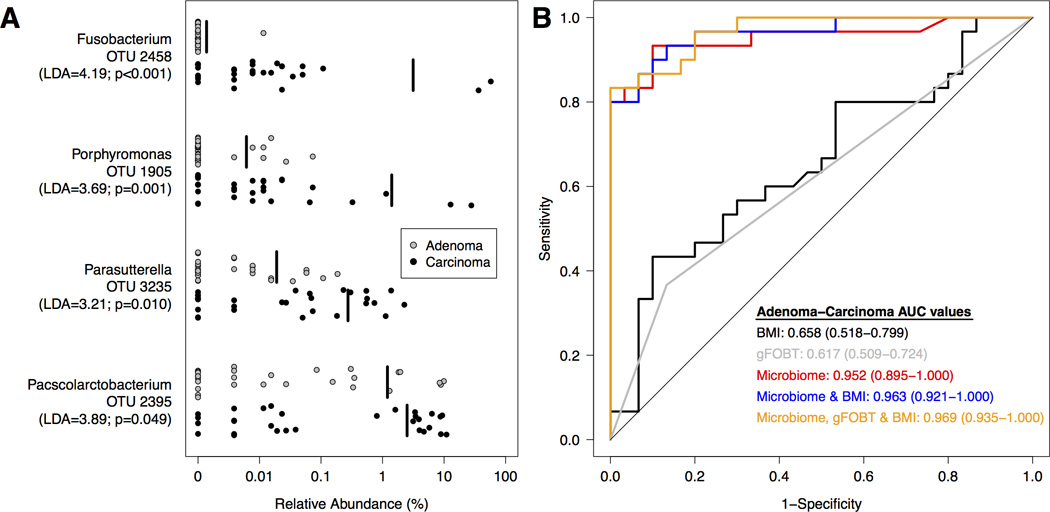
A. Relative abundance of differentially abundant OTUs for adenoma (n=30; grey) and carcinoma (n=30; black) subjects. The mean relative abundance is represented for each clinical group by a vertical black line. B. ROC curves for microbial biomarkers alone, clinical data alone, FOBT alone, microbial biomarkers with clinical data, and microbial biomarkers with FOBT and clinical data. For each comparison, the straight line represents the null model.
Complementing gFOBT test with microbiome-based models
Because gFOBT is the most common, non-invasive screening tool for CRC, we evaluated whether the microbiome-based models could be improved by including gFOBT results. The gFOBT test had 100% specificity in our study when comparing healthy individuals to those with colonic lesions. That is, patients without colonic lesions tested negative on the gFOBT. In an analysis comparing adenoma and carcinoma groups, the odds ratio for gFOBT was 3.76 (95% CI 1.04–13.65) when entered as a single explanatory variable, with AUC=0.617. In contrast, the microbiome data alone yielded an AUC of 0.952. The model combining BMI, gFOBT, and the microbiome data (OTUs 1905, 2395, 2458, 3235) provided excellent discriminatory ability (AUC=0.969; 95% CI: 0.935–1.000; Figure 4B).
Application of Microbiome Results to Population Data
To further test the capacity of the gut microbiome as a CRC screening candidate, we extracted data from Surveillance, Epidemiology and End Results (SEER) for age-specific incidence rates of CRC in the United States. Since likely candidates for CRC screening would target identification of early stage disease (adenoma), we designed a preliminary screening test based on the 5 OTUs (OTUs 38, 99, 136, 1889, 1913), which were enriched in healthy subjects compared to patients with adenomas. Persons who had any detectable levels (Relative abundance > 0) of these 5 OTUs were more likely to have healthy colons and constituted a negative test. Using a Bayesian model, we calculated the positive likelihood ratio for this preliminary screening test and applied it to population probabilities of CRC for each age group (Table 2). The likelihood ratio of this test was 71 (95% CI: 64.78, 77.22) (sensitivity=23.3% [7/30], specificity=100% [30/30]). As can be seen in Table 2, individuals who are 65 years of age had a pre-test probability of CRC of 0.17% based on nationwide SEER data. When we applied the OTU test to this age group, the probability of adenoma was 10.67% after knowing the microbiome data (1 in 9 chance of having an adenoma). For people 50 years of age, the results suggest a one in 26 chance of having an adenoma with a positive OTU test, and for adults 80 years of age; a positive OTU test yielded a 1 in 5 chance of having an adenoma.
Table 2.
Post-test probability of microbiome-based adenoma screen.
| Age at diagnosis (years) |
Incidence Rate (per 100,000 people)a |
Pre-test probability |
Pre-test odds |
Post-test oddsb |
Post-test probability |
95% confidence interval for post-test probability |
|---|---|---|---|---|---|---|
| 35–39 | 8.2 | 0.0001 | 0.000082 | 0.0058 | 0.0058 | 0.0045–0.0074 |
| 40–44 | 15.8 | 0.0002 | 0.000158 | 0.0112 | 0.0111 | 0.0092–0.0133 |
| 45–49 | 29.1 | 0.0003 | 0.000291 | 0.0207 | 0.0203 | 0.0177–0.0232 |
| 50–54 | 55.8 | 0.0006 | 0.000558 | 0.0396 | 0.0381 | 0.0345–0.0420 |
| 55–59 | 77.0 | 0.0008 | 0.000771 | 0.0547 | 0.0519 | 0.0477–0.0564 |
| 60–64 | 112.0 | 0.0011 | 0.001122 | 0.0796 | 0.0738 | 0.0688–0.0790 |
| 65–69 | 168.0 | 0.0017 | 0.001683 | 0.1195 | 0.1067 | 0.1008–0.1129 |
| 70–74 | 223.4 | 0.0022 | 0.002239 | 0.1590 | 0.1372 | 0.1306–0.1440 |
| 75–79 | 283.3 | 0.0028 | 0.002841 | 0.2017 | 0.1678 | 0.1606–0.1752 |
| 80–84 | 337.1 | 0.0034 | 0.003382 | 0.2401 | 0.1936 | 0.1859–0.2014 |
| 85+ | 376.4 | 0.0038 | 0.003778 | 0.2682 | 0.2115 | 0.2036–0.2196 |
Based on Surveillance, Epidemiology and End Results data, Years 2000–2010.
Likelihood Ratio of a positive test = 71.
For comparison purposes, we assessed the pre-to-post-test probabilities of detecting adenoma based on the gFOBT results in this sample. The likelihood ratio of a positive gFOBT was 41 (95% CI: 34.75 – 47.25), which was lower than the likelihood ratio of a positive microbiome test (i.e., LR+=71). For a person who is 65 years of age with a positive gFOBT, the post-test probability of adenoma was 6.46%, indicating a 1 in 15 chance of having an adenoma. This contrasts with the 10.67% probability of adenoma (1 in 9 chance) using a positive microbiome test in the same 65-year old. While both tests had good specificity in this sample, the sensitivity of the microbiome test was greater than the sensitivity of the gFOBT.
Discussion
Our results suggest that relative abundance data from the human gut microbiome differentiates individuals with healthy colons from those with adenomas and carcinomas. Most importantly, there was a significant difference in the gut microbiome of people with colonic adenomas compared to those with healthy colons. This has considerable importance in secondary prevention because screening for early stage colorectal cancer hinges on the ability to detect early pathologic changes. In this regard, we found that failure to detect at least 1 of the 5 OTUs served as a signal of the presence of adenoma. The probability of having an adenoma rose over 50-fold with this added information regarding microbiome. Taken with the existing literature regarding the importance of the gut microbiome in health and disease, our study further suggests that the microbiome may play a crucial role in the etiology of colorectal cancer.
A strength of our study design was that we collected samples from three clinical groups that represented the multistage progression in CRC (healthy, adenoma, and carcinoma). This allowed us to identify a panel of bacterial populations that could indicate both the progression from healthy tissue to adenoma and the progression from adenoma to carcinoma. Interestingly, when we looked at each patient, we rarely observed significant enrichment of every bacterial population among the OTUs incorporated in the logit models. For example, 11 of the 30 carcinoma patients had no detectable levels of Fusobacterium. However using the relative abundance data for the remaining panel of microbial biomarkers, such as Porphyromonas, Bacteroides, and Enterobacteriaceae, we were able to accurately classify these subjects. This strongly suggests that there may be multiple underlying mechanisms by which the microbiome is involved in CRC and that CRC is likely a polymicrobial disease.
Our findings are supported by previous evidence. Three research groups reported that Fusobacterium spp. were enriched on the surface of tumors compared to adjacent healthy tissue (22, 37, 38). Building upon these clinical studies, animal and tissue culture-based studies have provided evidence that Fusobacterium may contribute to tumor multiplicity through the recruitment of immune cells to tumors (22, 37). These mechanistic studies agree with our findings that Fusobacterium may be a marker for the presence of tumors. In addition, enterotoxigenic Bacteroides fragilis (ETBF), a pathogenic variant of a common commensal, has been shown to directly influence the development of CRC in murine genetic models through the production of a metalloprotease toxin (39). In our samples, subjects with carcinomas showed an increase in the relative abundance of one Bacteroides population (OTU 1882) compared to subjects with adenomas. However, PCR-based screens for the toxin producing genes did not reveal the presence of ETBF. Additionally, we observed a significant decrease in the relative abundance of Bacteroides populations (OTUs 1889 and 1913) associated with the advancement of tumorigenesis. Finally, a polyketide synthetase operon from E. coli, was shown to influence the progression of tumors using a murine model of inflammation-derived tumorigenesis (21, 23). Although we did see an enrichment for non-E. coli Enterobacteriaceae in the carcinoma subjects relative to the healthy subjects, we were unable to detect significant differences in the relative abundance of E. coli across the three clinical groups.
It is tempting to speculate on the enrichment of Fusobacterium and Porphyromonas spp. in subjects with CRC. Both of these bacterial taxa are common commensals of the mouth and a wealth of literature has linked them to chronic inflammation and periodontal disease (40, 41). The mouth is a reservoir for these pathogens, allowing for colonization of the gastrointestinal tract under abnormal environmental conditions. During colorectal carcinogenesis, dramatic physiological changes occur in the microenvironment of colonic lesions (42). Tumor-associated fluxes in nutrients and shifts in inflammatory mediators may favor colonization by opportunistic pathogens such as Fusobacterium and Porphyromonas. As demonstrated by Kostic and colleagues, colonization by such pathogens can support the development and progression of CRC (22, 37). We were unable to detect a significant association between either population and carcinoma severity or location. Additional studies are needed to examine how and at what stage these bacterial populations are affecting the development of CRC and how they may be linked to the oral microbiome and related to oral disease.
As highlighted above, there is a clear association with the enrichment of pathogenic bacterial populations and colon tumorigenesis; however, in the present study we emphasize that the depletion of potentially protective bacteria likely plays a similar role CRC pathology. We identified several bacterial populations that were significantly depleted in CRC. Individuals with both adenomas and carcinomas showed a dramatic loss in OTUs associated with the genera Clostridium and Bacteroides, and the family Lachnospiraceae (43–45). Each of these bacterial taxa are well known producers of short chain fatty acids (SCFAs) in the colon. SCFAs are important microbial metabolites that supply nutrients to colonocytes and help maintain epithelial health and homeostasis. Specifically the SCFA, butyrate, has been shown to have substantial anti-tumorigenenic properties including the ability to inhibit tumor cell proliferation, initiate apoptosis in tumor cells (46), and mediate T-regulatory cell homeostasis (44). Loss of these important bacterial populations in concert with an enrichment of pathogenic populations likely plays a synergistic role in potentiating tumorigenesis.
Although our results are important, there are limitations to the investigation. A larger, more diverse sample of individuals is needed to augment and validate our findings. Furthermore, although our study clearly demonstrates the viability of using the gut microbiome as a biomarker for CRC, we cannot assess the bacterial populations’ role in causation or the mechanisms by which these populations affect the development and progression of CRC. Regardless, the feasibility, lack of invasive procedures, ability to be complement existing screening methods (e.g. gFOBT), and the strength of signal seen in this study support the further investigation and application of microbial biomarkers from stool as a method for CRC screening.
Supplementary Material
Acknowledgements
We thank the Great Lakes-New England Early Detection Research Network for providing the fecal samples that were used in this study. The analysis described in this study was supported by grants from the National Institutes of Health to PDS (R01HG005975 and P30DK034933) and MTR (5U01CA86400).
Footnotes
Conflicts of interest: The authors have no conflicts to declare.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA: a cancer journal for clinicians. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Fearon ER. Molecular genetics of colorectal cancer. Annual review of pathology. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 3.Levin B, Lieberman DA, McFarland B, Smith RA, Brooks D, et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA: a cancer journal for clinicians. 2008;58:130–160. doi: 10.3322/CA.2007.0018. [DOI] [PubMed] [Google Scholar]
- 4.Surveillance, Epidemiology, and End Results (SEER) National Cancer Institute, DCCPS, Surveillance Research Program, Surveillance Systems Branch; Program Research Data (1973–2010) released April 2013 based on the November 2012 submission ed: [Google Scholar]
- 5.Whitlock EP, Lin JS, Liles E, Beil TL, Fu R. Screening for colorectal cancer: a targeted, updated systematic review for the U.S. Preventive Services Task Force. Annals of internal medicine. 2008;149:638–658. doi: 10.7326/0003-4819-149-9-200811040-00245. [DOI] [PubMed] [Google Scholar]
- 6.Benson AB., 3rd Epidemiology, disease progression, and economic burden of colorectal cancer. Journal of managed care pharmacy : JMCP. 2007;13:S5–S18. doi: 10.18553/jmcp.2007.13.s6-c.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ling BS, Moskowitz MA, Wachs D, Pearson B, Schroy PC. Attitudes toward colorectal cancer screening tests. Journal of general internal medicine. 2001;16:822–830. doi: 10.1111/j.1525-1497.2001.10337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joseph DA, King JB, Miller JW, Richardson LC, Centers for Disease C, et al. Prevalence of colorectal cancer screening among adults--Behavioral Risk Factor Surveillance System, United States, 2010. MMWR Morbidity and mortality weekly report. 2012;61(Suppl):51–56. [PubMed] [Google Scholar]
- 9.Howden L, Meyer J. Age and sex composition, 2010. 2010 Census Briefs: US Department of Commerce, US Census Bureau. 2012 [Google Scholar]
- 10.Collins JF, Lieberman DA, Durbin TE, Weiss DG Veterans Affairs Cooperative Study G. Accuracy of screening for fecal occult blood on a single stool sample obtained by digital rectal examination: a comparison with recommended sampling practice. Annals of internal medicine. 2005;142:81–85. doi: 10.7326/0003-4819-142-2-200501180-00006. [DOI] [PubMed] [Google Scholar]
- 11.Young GP, St John DJ, Winawer SJ, Rozen P, Who, et al. Choice of fecal occult blood tests for colorectal cancer screening: recommendations based on performance characteristics in population studies: a WHO (World Health Organization) and OMED (World Organization for Digestive Endoscopy) report. The American journal of gastroenterology. 2002;97:2499–2507. doi: 10.1111/j.1572-0241.2002.06046.x. [DOI] [PubMed] [Google Scholar]
- 12.Hardcastle JD, Chamberlain JO, Robinson MH, Moss SM, Amar SS, et al. Randomised controlled trial of faecal-occult-blood screening for colorectal cancer. Lancet. 1996;348:1472–1477. doi: 10.1016/S0140-6736(96)03386-7. [DOI] [PubMed] [Google Scholar]
- 13.Mandel JS, Church TR, Bond JH, Ederer F, Geisser MS, et al. The effect of fecal occult-blood screening on the incidence of colorectal cancer. The New England journal of medicine. 2000;343:1603–1607. doi: 10.1056/NEJM200011303432203. [DOI] [PubMed] [Google Scholar]
- 14.Mandel JS, Church TR, Ederer F, Bond JH. Colorectal cancer mortality: effectiveness of biennial screening for fecal occult blood. Journal of the National Cancer Institute. 1999;91:434–437. doi: 10.1093/jnci/91.5.434. [DOI] [PubMed] [Google Scholar]
- 15.Chambers WM, Warren BF, Jewell DP, Mortensen NJ. Cancer surveillance in ulcerative colitis. The British journal of surgery. 2005;92:928–936. doi: 10.1002/bjs.5106. [DOI] [PubMed] [Google Scholar]
- 16.Huxley RR, Ansary-Moghaddam A, Clifton P, Czernichow S, Parr CL, et al. The impact of dietary and lifestyle risk factors on risk of colorectal cancer: a quantitative overview of the epidemiological evidence. International journal of cancer Journal international du cancer. 2009;125:171–180. doi: 10.1002/ijc.24343. [DOI] [PubMed] [Google Scholar]
- 17.Larsson SC, Rafter J, Holmberg L, Bergkvist L, Wolk A. Red meat consumption and risk of cancers of the proximal colon, distal colon and rectum: the Swedish Mammography Cohort. International journal of cancer Journal international du cancer. 2005;113:829–834. doi: 10.1002/ijc.20658. [DOI] [PubMed] [Google Scholar]
- 18.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 19.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, et al. Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, et al. The gut microbiome modulates colon tumorigenesis. mBio. 2013;4 doi: 10.1128/mBio.00692-13. e00692-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell host & microbe. 2013;14:207–215. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6:e16393. doi: 10.1371/journal.pone.0016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang T, Cai G, Qiu Y, Fei N, Zhang M, et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. The ISME journal. 2012;6:320–329. doi: 10.1038/ismej.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, et al. Human gut microbiome and risk for colorectal cancer. Journal of the National Cancer Institute. 2013;105:1907–1911. doi: 10.1093/jnci/djt300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Brien CL, Allison GE, Grimpen F, Pavli P. Impact of colonoscopy bowel preparation on intestinal microbiota. PLoS ONE. 2013;8:e62815. doi: 10.1371/journal.pone.0062815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Applied and environmental microbiology. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic acids research. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and environmental microbiology. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, et al. Metagenomic biomarker discovery and explanation. Genome biology. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- 34.Isaksson A, Wallman M, Goransson H, Gustafsson MG. Cross-validation and bootstrapping are unreliable in small sample classification. Pattern Recogn Lett. 2008;29:1960–1965. [Google Scholar]
- 35.Linnet K. A review on the methodology for assessing diagnostic tests. Clinical chemistry. 1988;34:1379–1386. [PubMed] [Google Scholar]
- 36.Jeffreys H. An invariant form for the prior probability in estimation problems. Proceedings of the Royal Society of London Series A, Mathematical and physical sciences. 1946;186:453–461. doi: 10.1098/rspa.1946.0056. [DOI] [PubMed] [Google Scholar]
- 37.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell host & microbe. 2013;14:195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome research. 2011 doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sears CL, Islam S, Saha A, Arjumand M, Alam NH, et al. Association of enterotoxigenic Bacteroides fragilis infection with inflammatory diarrhea. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2008;47:797–803. doi: 10.1086/591130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Signat B, Roques C, Poulet P, Duffaut D. Fusobacterium nucleatum in periodontal health and disease. Current issues in molecular biology. 2011;13:25–36. [PubMed] [Google Scholar]
- 41.Deshpande RG, Khan M, Genco CA. Invasion strategies of the oral pathogen porphyromonas gingivalis: implications for cardiovascular disease. Invasion & metastasis. 1998;18:57–69. doi: 10.1159/000024499. [DOI] [PubMed] [Google Scholar]
- 42.Peddareddigari VG, Wang D, Dubois RN. The tumor microenvironment in colorectal carcinogenesis. Cancer microenvironment : official journal of the International Cancer Microenvironment Society. 2010;3:149–166. doi: 10.1007/s12307-010-0038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruemmele FM, Schwartz S, Seidman EG, Dionne S, Levy E, et al. Butyrate induced Caco-2 cell apoptosis is mediated via the mitochondrial pathway. Gut. 2003;52:94–100. doi: 10.1136/gut.52.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.