

Abstract
Objective
To determine whether patient expectancy plays a role in observed placebo and nocebo effects in two clinical trials.
Method
Data were re-analyzed from two fluoxetine-discontinuation studies conducted from March 1990–September 1992 and May 1997–December 2002. Outpatients aged 18–65 years with DSM-III-R Major Depressive Disorder (MDD) responding to 12-week duration open treatment were randomized to continued fluoxetine or placebo for an additional year. Participants in one of the included studies received a fixed dose of fluoxetine 20mg daily, while the second study utilized flexible fluoxetine doses up to 60mg daily. Mixed effects longitudinal models determined whether the possible randomization to placebo at 12 weeks resulted in significant depressive symptom worsening across treatments. Correlations were computed between early symptom change (weeks 1–3 of open treatment) and post-randomization symptom change (weeks 13–16 following randomization).
Results
Participants continuing to receive fluoxetine and those switched to placebo had significantly higher mean HAM-D scores immediately post-randomization compared to the final weeks of open treatment (p < 0.001 for both fluoxetine and placebo-treated patients). In both studies, early HAM-D change was significantly correlated with post-randomization HAM-D change for patients receiving fluoxetine (r = −0.46, p < 0.001) as well as placebo (r = −0.48, p < 0.001).
Conclusions
The possibility of receiving placebo following 12 weeks of open fluoxetine was associated with significant symptom worsening in two large fluoxetine-discontinuation studies. Worsening depression scores following randomization were significantly associated with the degree of improvement participants experienced during weeks 1–3 of open treatment. These results suggest that treatment changes influence patients’ expectations of improvement, which in turn affect their depressive symptoms.
INTRODUCTION
The probability of receiving placebo as opposed to active medication influences treatment outcome in antidepressant clinical trials (1–4). Medication response rates are lowest in drug-placebo trials (51.7% response) and increase in drug-drug-placebo trials (57.7% response) and drug-drug trials (65.4% response) (5). Moreover, antidepressant trials comprising a greater number of active treatment arms have increased placebo response and decreased drug-placebo differences (6–7).
Such marked differences depending on trial design (i.e., placebo-controlled vs. active comparator) imply that a medication’s pharmacologic effect is only one contributor to symptom change. Non-pharmacologic mechanisms also contribute to outcome, including spontaneous improvement and worsening, a patient’s expectation of benefit from the treatment, therapeutic effects of the treatment situation, positive life events, and sources of measurement error and bias. The relative contributions of these non-pharmacologic factors may change across different treatment settings, resulting in different observed medication responses. While the pharmacologic effects of a medication can be estimated from the differential response between drug and placebo, elucidating the role of various sources of “placebo” effects is more complicated because studies have not been designed to isolate them.
Since clinical trial participants become aware of their probability of receiving active medication vs. placebo during the informed consent discussion, it has been suggested that patient expectancy may in some cases explain the relationship between study design and antidepressant response (8–9). The induction of positive expectancies about treatment outcome has been shown to significantly improve antidepressant response (10) and is hypothesized to be a primary mechanism of placebo effects in clinical trials (11). Conversely, information that generates negative expectancies may lead to worsening (i.e., nocebo effects) (12). Informing patients about possible side effects of drug administration has been shown to increase the occurrence of these side effects (13–14), and diminished medication effects are observed when delivered by neutral clinicians compared to positive clinicians (15).
To differentiate the contribution of patient expectancy from other factors that may influence antidepressant response, we re-analyzed data from two large, multicenter discontinuation trials treating participants having Major Depressive Disorder (MDD) with fluoxetine for 12 weeks followed by randomized continuation treatment with either fluoxetine or placebo (16–17). Reasoning that changes in depressive symptoms caused by patient expectancy would occur in the initial few weeks following a change in treatment (18), we evaluated symptom change: (1) at the initiation of open fluoxetine treatment and (2) at possible randomization to placebo at 12 weeks. We hypothesized that due to a decrease in patient expectancy, depression scores would significantly worsen in the 4 weeks following randomization for patients receiving continued fluoxetine as well as those switched to placebo. Furthermore, we predicted that individuals who experienced substantial improvement during the first three weeks of open treatment with fluoxetine would be the same individuals to experience significant worsening following the 12 week randomization time point (i.e., from weeks 13–16). We complemented our investigation of these primary hypotheses with follow-up analyses aimed at ruling out spontaneous improvement/worsening, positive life events, and rater bias as explanations of the observed patterns of symptom change.
METHOD
Sources of data
Data from two clinical trials examining the efficacy of fluoxetine in preventing depression relapse during continuation/maintenance treatment were sequentially analyzed. Study 1 initially treated MDD patients with open fluoxetine 20 mg/d for 12 weeks, then randomized remitters to continued fluoxetine treatment vs. placebo substitution at one of three time points (see Figure 1) (16). Study 2 was designed as a replication study and utilized a similar design, with the primary exceptions that remitters to open-label fluoxetine were randomized 1:1 to continued fluoxetine or placebo (see Figure 1) and medication dose was titrated to 60 mg/d (17). Please see previous articles for full details (16–17, 19–21).
Figure 1.
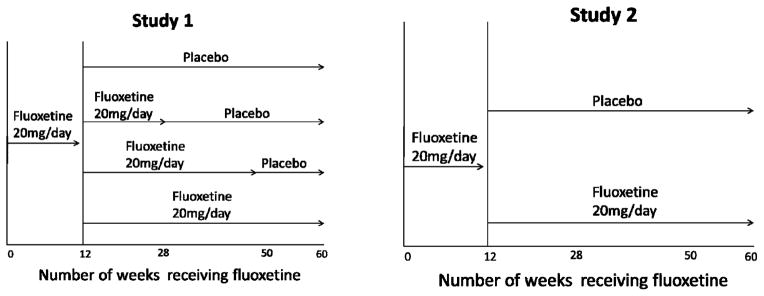
Design of two fluoxetine-discontinuation studies investigating depression relapse in subjects after 12 weeks of open treatment.
Subjects
Study 1 enrolled outpatients aged 18–65 years who met Diagnostic and Statistical Manual of Mental Disorders, Third Edition, Revised (DSM-III-R) (22) criteria for MDD for at least the 1 month preceding study participation. Subjects were also required to have a modified 17-item Hamilton Rating Scale for Depression (mHAM-D) (23) score ≥ 16 (described below). Exclusion criteria were acute, severe, or unstable medical problems, pregnancy or lactation, serious suicide risk, history of psychosis, mania, or organic mental disorder, substance use disorder within the past year, previous fluoxetine treatment for ≥ 3 months in a previous episode, or non-response to 8-weeks of fluoxetine treatment at a dose ≥ 20mg during the current episode. Study 2 used similar selection criteria, except no minimum mHAM-D score was required for study entry and subjects were excluded for substance use disorder within the previous 6 months (rather than 1 year).
Study Assessments
A modified form of the HAM-D was used in which hypersomnia and hyperphagia were substituted for insomnia, anorexia, and weight loss items in patients with reverse neurovegetative symptoms. Subjects in whom the modified mHAM-D was used for eligibility continued to use the reverse vegetative items throughout the study, whereas patients who presented with insomnia, anorexia, and weight loss used the standard neurovegetative items for the duration of the study.
Open-label Treatment with Fluoxetine
In Study 1, subjects whose depression persisted (i.e., mHAM-D remained ≥ 16) following a no-treatment observation week began 12 weeks of open-label fluoxetine 20mg/day. Remission at the end of the open-treatment period was defined as 3 consecutive weeks with both a mHAM-D ≤ 7 and failure to meet DSM-III-R criteria for MDD. Open treatment was similar in Study 2, with the exception that target fluoxetine doses were 10mg/day for the first week, 20 mg/day for weeks 2–4, 40mg/day for weeks 5–8, and 60mg/day for weeks 9–12 (increases only occurring if the patient had not remitted and tolerated the medication well). In Study 2, sustained remission was not required for patients to be randomized; instead, responders, defined as a Clinical Global Impressions—Severity (24) score of 1 or 2 at 12 weeks, moved into the continuation phase of the study.
Randomization and Continuation Treatment
In Study 1, subjects whose depression met remission criteria after 12 weeks of open-label treatment were randomly assigned to 1 of 4 groups: (1) placebo for 50 weeks, (2) continued fluoxetine for 14 weeks followed by placebo for 36 weeks, (3) continued fluoxetine for 38 weeks followed by placebo for 12 weeks, or (4) continued fluoxetine for 50 weeks (see Figure 1). In Study 2, responders to open-label fluoxetine were randomized to 52 weeks treatment with placebo or continued fluoxetine at the dose to which they had remitted (see Figure 1). Subjects who met criteria for MDD for 2 consecutive weeks or who had mHAM-D ≥ 14 for 3 consecutive weeks were considered to have relapsed and removed from the study. Fluoxetine discontinuation was associated with a significantly increased risk of depression relapse at endpoint in both trials (Study 1: hazard ratio for relapse 2.22, 95% CI 1.62 – 3.05; Study 2: hazard ratio for relapse 1.73, 95% CI 1.20–2.51). In Study 1, 42% of fluoxetine-treated patients compared to 19% of placebo-treated patients remained in remission by 62 week follow up, while in Study 2, 54.1% of participants in the fluoxetine group and 28.0% of those in the placebo group remained well at 52 week follow-up.
Data Analysis
Since the primary goals of this analysis were to investigate post-randomization changes in mHAM-D scores and the correlation of these scores with early improvement, analyses were focused on data from the open-label treatment period (Weeks 1–12) and the first 4 weeks following randomization (Weeks 13–16). Analyses for Study 1 and Study 2 were identical.
To determine whether mHAM-D scores significantly increased following randomization, a longitudinal mixed effects model was fit to the repeated mHAM-D scores over time within patients. This model included a categorical week indicator, a group indicator which was nonzero in weeks 13–16 for those individuals randomized to placebo, and an interaction of group with weeks 13–16 to allow for different means post-randomization in the two groups (25). A random intercept was included to account for correlation within individuals over time, and estimates were obtained by restricted maximum likelihood. Weekly estimated means and associated 95% confidence intervals were calculated to facilitate interpretation.
Next, to test whether early improvement during open-label treatment with fluoxetine (i.e., mHAM-D decreases during weeks 1–3) predicts significant symptom worsening following randomization, we fit a piecewise linear longitudinal mixed effects model to the repeated mHAM-D scores with change points allowed at weeks 3 and 10 as well as random intercepts and slopes for individuals. Week 10 rather than Week 12 was selected to more conservatively identify change following randomization, since mHAM-D scores at weeks 11–12 may have been subject to rater bias (i.e., the desire to have patients meet remission criteria and be randomized at Week 12). This modeling produced individualized linear trajectories (slopes) between each knot based on best linear unbiased predictors (BLUPs) (26). “Early mHAM-D Change” (a subject’s modeled mHAM-D at week 3 minus mHAM-D modeled at week 0) was tested for correlation with “Post-randomization mHAM-D Change” (a subject’s modeled mHAM-D at week 16 minus modeled mHAM-D at week 10). Additionally, the percent variability in Post-randomization mHAM-D Change explained by Early mHAM-D Change and randomization group, respectively, were assessed using regression. Finally, average trajectories associated with individuals demonstrating very high and very low changes in their mHAM-D post-randomization were estimated.
Additional Analyses
In contrast to patient expectancy, fluctuation in a patient’s natural course of illness is likely to occur randomly throughout treatment. To test whether some individuals randomly improved or worsened (as opposed to change being linked specifically to study events), we identified subjects having low mHAM-D (<=7) in weeks 4–6 and examined their mean mHAM-D scores in weeks 8–10. A finding of no change during this interval in this highly selected group would suggest post-randomization changes are indeed specific to the randomization time-point and not due to random fluctuation.
To determine whether early improvement and post-randomization symptom increase might be caused by rater bias (i.e., baseline score inflation or pre-randomization score deflation), we inspected the distributions of mHAM-D scores of included subjects in each study at baseline and just prior to the week 12 randomization. Peaks in the distributions at mHAM-D score thresholds would indicate possible rater bias, while a normal distribution of scores would make rater bias less likely.
RESULTS
Characteristics of Randomized Patients and Response to Treatment
In Study 1, 395 patients remitted during open-label fluoxetine (51.0% of subjects treated openly) and were randomized in the continuation phase (299 received fluoxetine, while 96 were switched to placebo). In Study 2, 278 of the 570 subjects (48.7%) who began open-label treatment were randomized (139 to continued fluoxetine and 139 to placebo). Clinical and demographic characteristics of randomized subjects are presented in Table 1. Subjects continued on fluoxetine treatment did not differ from those randomized to placebo on any of the characteristics.
Table 1.
Characteristics of randomized subjects in two fluoxetine-discontinuation studies.
| Study 1 | Study 2 | |||
|---|---|---|---|---|
| Characteristic | Fluoxetine (N=299) | Placebo (N=96) | Fluoxetine (N=134) | Placebo (N=135) |
| Age | 39.6 ± 10.2 | 40.0 ± 10.5 | 39.8 ± 11.3 | 38.5 ± 11.1 |
| % Male | 34.4 | 20.8 | 50.0 | 31.7 |
| Baseline mHAM-D | 20.7 ± 3.5 | 21.5 ± 3.7 | 17.7 ± 5.1 | 17.2 ± 4.5 |
| mHAM-D at randomization | 2.9 ± 2.2 | 2.7 ± 2.3 | 4.8 ± 3.0 | 5.3 ± 3.5 |
Note: Age and gender data were only available for a subset of subjects in Study 2, N=82 in each group. mHAM-D data reflect the entire randomized sample.
Post-Randomization mHAM-D Change
Individual trajectories of mHAM-D scores over the first 16 weeks of Study 1 and Study 2 are shown in Figure 2. In both studies, individuals remaining on fluoxetine had significantly higher mean mHAM-D scores post-randomization (i.e., weeks 13–16) than immediately prior to randomization (i.e., weeks 10–12). Average mHAM-D scores increased in Study 1 (N=299) from 3.3 pre-randomization to 5.0 post-randomization (t = 7.9, p<0.0001) and in Study 2 (N=139) from 5.5 pre-randomization to 6.6 post-randomization (t = 3.7, p=0.0002). Moreover, in Study 1 individuals substituted to placebo (n=96) showed a significantly larger worsening than those remaining on fluoxetine during weeks 13–16 (2.6 additional mHAM-D points, t = 6.5, p< 0.0001), while individuals randomized to placebo in Study 2 (n=139) worsened no more on average than the group that remained on fluoxetine during weeks 13–16 (0.3 additional mHAM-D points, t = 0.79, p = 0.38).
Figure 2.

Individual and group trajectories of mHAM-D change during the first 16 weeks of treatment in Study 1 and 2.
Further analyses suggested the changes identified during weeks 1–3 and 13–16 were not random, as subjects with low mHAM-D scores during weeks 4–6 did not experience significant symptom increases during weeks 8–10 (Study 1, mean mHAM-D 3.50 during weeks 4–6 vs. 3.45 during weeks 8–10, t = 0.25, p=0.80; Study 2, mean mHAM-D 3.27 during weeks 4–6 vs. 3.47 during weeks 8–10, t = −0.66, p = 0.51). Histograms depicting distributions of mHAM-D scores at baseline and weeks 10–12 revealed no evidence of significant rater bias at study entry or in the peri-randomization period.
Correlation Between Early mHAM-D Change and Post-Randomization mHAM-D Change
Figure 3 plots Post-randomization mHAM-D Change (i.e., change from week 10–16) as a function of Early mHAM-D Change (i.e., change from week 1–3) for Studies 1 and 2. In Study 1, the correlation of Early mHAM-D Change with Post-Randomization mHAM-D Change was −0.46 (p< 0.0001) for subjects receiving continued fluoxetine and −0.47 (p <0.0001) for those switched to placebo. Each additional point of mHAM-D improvement in the first 3 weeks of the study was associated with a 0.57 point worsening post-randomization (t = −10.03, p<.0001), while controlling for randomization to placebo or fluoxetine. Early mHAM-D Change predicted 23.8% of the variability in Post-randomization mHAM-D Change, while treatment condition explained only 4.3% of the variability in Post-randomization mHAM-D Change.
Figure 3.

Scatter plots of Post-randomization mHAM-D Change vs. Early mHAM-D Change for Study 1 and 2.
In Study 2, the correlation of Early mHAM-D Change with Post-randomization mHAM-D Change was −0.48 (p < 0.0001) for fluoxetine and −0.43 (p < 0.0001) for placebo (see Figure 3). Each additional point of mHAM-D improvement in the first 3 weeks resulted in a 0.46 point worsening post-randomization mHAM-D (t = −8.44, p< 0.0001. Early mHAM-D Change predicted 20.8% of the variability in post-randomization mHAM-D Change, while treatment explained 0.2% of the variability in Post-randomization mHAM-D Change.
Figure 4 presents the average trajectories for individuals on the extreme ends of early improvement in mHAM-D scores and post-randomization worsening. These curves demonstrate in a different way the pattern already described, such that individuals with the steepest improvement during the first 3 weeks of open treatment are the patients with the greatest mHAM-D worsening following randomization at week 12. Subjects who continued to improve post-randomization tended to be individuals who only showed mild improvement during the first 3 weeks of open treatment with fluoxetine.
Figure 4. Average trajectories for individuals who remained on fluoxetine and had the highest 10% (i.e., most change) and lowest 10% (i.e., least change) symptom differences post-randomization.
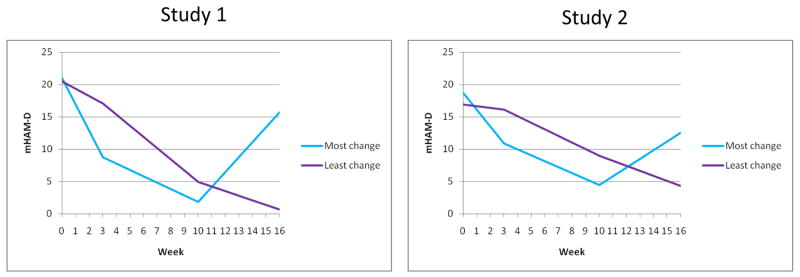
Curves are based on average mHAM-D trajectories across 29 (most change) and 30 (least change) individuals in Study 1 and 14 (most change) and 13 (least change) individuals in Study 2.
DISCUSSION
The reported analyses support the hypothesis that symptom increases and decreases immediately following treatment changes are affected by patient expectancy. Some depressed patients appear more prone to the effects of expectancy than others, since there were high correlations between likelihood of symptom change at each of two treatment changes. Strikingly, the post-randomization increase in mHAM-D scores occurred irrespective of treatment assignment, as worsening mHAM-D score following randomization was significantly associated with the degree of mHAM-D improvement during weeks 1–3 of open treatment in patients continuing to receive medication as well as those switched to placebo. Analyses of Study 2 paralleled those of Study 1, demonstrating a remarkable consistency in findings across the two patient samples.
One explanation of these results is that treatment changes (e.g., institution of a new treatment or discontinuation of an established treatment) influence patients’ expectations of improvement, which in turn affect their depressive symptoms. In the studies examined, patients were informed at the start of open label treatment that they would receive a medication known to be effective in the treatment of depression. This knowledge likely instills a positive expectancy of improvement that may ameliorate the symptoms of depression. At week 12, participants are aware that they may be randomized to placebo, which may decrease their expectancy of continued improvement (i.e., decreased placebo effect) or increase their expectation of worsening (i.e., increased nocebo effect). Such expectancy effects in continuation studies of antidepressants have been found by Zimmerman et al (2007), who compared relapse rates to antidepressants and placebo in studies using a placebo-substitution (i.e., open acute treatment with active medication followed by randomization to continued medication or placebo) vs. extension designs (i.e., responders to double-blind acute treatment with medication or placebo continue taking what they responded to in continuation phase) (27). Overall relapse rates were reported to be lower in extension studies, likely due to a greater expectation of continued positive response in these studies where patients are aware they will continue taking the agent that made them better.
We also explored the possibility that some patients experience random mood fluctuations independent of anticipated treatment change. This appeared unlikely to explain the observed patterns, since we found no significant symptom increases or decreases at arbitrary time points not associated with changes in treatment. We also considered regression to the mean and rater bias as alternative explanations of the observed patterns of symptom change. Inflation of baseline scores in order to meet the minimum cut-off for initial enrollment might be expected to cause decreased scores during weeks 1–3 (28–30). Similarly, mHAM-D scores might be artificially decreased to facilitate randomization at week 12, resulting in increased scores post-randomization once the mHAM-D score restriction is released. Again, there was no evidence that score inflation or deflation by raters contributed to the observed pattern of results, since no clustering of mHAM-D scores near the cut-off points was observed. To further mitigate the effects of rater bias on the present analyses, we analyzed week 10 in addition to week 12 as the change point in our linear models of mHAM-D scores following randomization. Similar results were obtained across these analyses, again suggesting minimal contribution of rater bias.
Developing methods of prospectively identifying participants likely to experience expectancy effects may facilitate efforts to minimize placebo response in clinical trials, thereby making it easier to detect a signal of efficacy for a putative antidepressant over placebo. Methods of predicting expectancy effects may also allow patients to be targeted in clinical treatment with interventions designed to increase patient expectancy and improve treatment outcomes. While simplistic attempts to identify these individuals (i.e., single-blind placebo lead in periods) have generally failed to influence placebo response, these data suggest that more sophisticated methods of predicting expectancy effects should be studied (31).
Finally, several limitations must be considered when interpreting the findings presented. Most importantly, the studies analyzed did not measure expectancy or attempt to assess expectation effects, so it is an inference that expectancy caused the observed patterns of symptom change. The role of patient expectancy in antidepressant outcome must be prospectively tested in randomized controlled trials which manipulate expectation to make firmer conclusions about its causative role. Another limitation may have been confounding expectancy effects with clinical worsening caused by the loss of therapeutic effects of fluoxetine. However, fluoxetine discontinuation would be expected to have a delayed onset owing to the long half-life of this medication (32). Fluoxetine discontinuation also would not explain the worsening observed in the patients receiving continued fluoxetine, the magnitude of which was identical to that observed in patients switched to placebo at 12 weeks.
In summary, analyses of depression scores from two large clinical studies were consistent with the hypothesis that patient expectancy contributes significantly to symptom change in the first weeks following change in treatment and occurs independent of the pharmacologic effects of treatment. Following an acute period of open treatment, patients who are aware they may be randomized to receive placebo experience significant worsening in depressive symptoms, even if they actually continue taking medication. Depressed patients who experience substantial early improvement (presumably due to positive expectancies instilled by knowing effective treatment has begun) are likely to experience a substantial worsening when their expectancies are diminished. Thus, patient expectancy of improvement or worsening should be considered when interpreting the results of both acute and discontinuation antidepressant studies. Optimizing expectancy may be explored as a useful therapeutic technique in the clinical treatment of patients with depression.
Acknowledgments
Within the past 3 years, Dr. Stewart reports receiving honoraria from Sanofi-Aventis and Merck as well as participating on a Bristol-Myers Squibb advisory board. Support for the design and conduct of the study was provided by a National Institute of Mental Health grant K23 MH085236 (BRR). Dr. Rutherford had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. This paper has not been previously presented.
Footnotes
Disclosures: Dr. Rutherford, Mr. Glass, and Dr. Wall have no disclosures to report.
Contributor Information
Bret R Rutherford, Email: brr8@columbia.edu, Columbia University College of Physicians and Surgeons, New York State Psychiatric Institute, 1051 Riverside Drive, Box 98, New York, NY 10032, 212 543 5746 (telephone), 212 543 6100 (fax).
Melanie M. Wall, Columbia University College of Physicians and Surgeons, New York State Psychiatric Institute.
Andrew Glass, Columbia University College of Physicians and Surgeons, New York State Psychiatric Institute.
Jonathan W. Stewart, Columbia University College of Physicians and Surgeons, New York State Psychiatric Institute.
References
- 1.Rutherford BR, Roose SP. A Model of Placebo Effects in Antidepressant Clinical Trials. Am J Psychiatry. 2013;170:723–733. doi: 10.1176/appi.ajp.2012.12040474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roose SP, Schatzberg AF. The Efficacy of Antidepressants in the Treatment of Late-Life Depression. J Clin Psychopharm. 2005;25(Suppl 1):S1–7. doi: 10.1097/01.jcp.0000162807.84570.6b. [DOI] [PubMed] [Google Scholar]
- 3.Rutherford BR, Sneed JR, Roose SP. Does Study Design Affect Outcome? The Effects of Placebo Control and Treatment Duration in Antidepressant Trials. Psychother Psychosom. 2009;78:172–181. doi: 10.1159/000209348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sneed JR, Rutherford BR, Rindskopf D, Roose SP. Design makes a difference: a meta-analysis of antidepressant response rates in placebo-controlled versus comparator trials in late-life depression. Am J Geri Psychiatry. 2008;16:65–73. doi: 10.1097/JGP.0b013e3181256b1d. [DOI] [PubMed] [Google Scholar]
- 5.Sinyor M, Levitt AJ, Cheung AH, Schaffer A, Kiss A, Dowlati Y, Lanctot KL. Does inclusion of a placebo arm influence response to active antidepressant treatment in randomized controlled trials? results from pooled and meta-analyses. J Clin Psychiatry. 2010;71:270–279. doi: 10.4088/JCP.08r04516blu. [DOI] [PubMed] [Google Scholar]
- 6.Khan A, Kolts RL, Thase ME, Krishnan RR, Brown W. Research Design Features and Patient Characteristics Associated with the Outcome of Antidepressant Clinical Trials. Am J Psychiatry. 2004;161:2045–2049. doi: 10.1176/appi.ajp.161.11.2045. [DOI] [PubMed] [Google Scholar]
- 7.Papakostas GI, Fava M. Does the probability of receiving placebo influence clinical trial outcome? A meta-regression of double-blind, randomized clinical trials in MDD. Eur Neuropsychopharm. 2009;19:34–40. doi: 10.1016/j.euroneuro.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 8.Rutherford BR, Wager TD, Roose SP. Expectancy Effects in the Treatment of Depression: A Review of Experimental Methodology, Effects on Patient Outcome, and Neural Mechanisms. Current Reviews in Psychiatry. 2010;6:1–10. doi: 10.2174/157340010790596571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaudiano BA, Hughes JA, Miller IW. Patients’ treatment expectancies in clinical trials of antidepressants versus psychotherapy for depression: a study using hypothetical vignettes. Compr Psychiatry. 2013;1:28–33. doi: 10.1016/j.comppsych.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rutherford BR, Marcus SM, Wang P, et al. A randomized, prospective pilot study of patient expectancy and antidepressant outcome. Psychol Med. 2013;43:975–982. doi: 10.1017/S0033291712001882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stewart-Williams S, Podd J. The placebo effect: dissolving the expectancy versus conditioning debate. Psychol Bull. 2004;30:324–340. doi: 10.1037/0033-2909.130.2.324. [DOI] [PubMed] [Google Scholar]
- 12.Barsky AJ, Aintfort R, Rogers MP, Borus JF. Nonspecific Medication Side Effects and the Nocebo Phenomenon. JAMA. 2002;287:622–627. doi: 10.1001/jama.287.5.622. [DOI] [PubMed] [Google Scholar]
- 13.Nestoriuc Y, Orav EJ, Liang MH, Horne R, Barsky AJ. Prediction of nonspecific side effects in rheumatoid arthritis patients by beliefs about medicines. Arthritis Care Res. 2010;62:791–799. doi: 10.1002/acr.20160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers MG, Cairns JA, Singer J. The consent form as a possible cause of side effects. Clin Pharmacol Ther. 1997;42:250–253. doi: 10.1038/clpt.1987.142. [DOI] [PubMed] [Google Scholar]
- 15.Uhlenhuth EH, Rickels K, Fisher S, Park LC, Lipman RS, Mock J. Drug, doctor’s verbal attitude, and clinic setting in the symptomatic response to pharmacotherapy. Psychopharmacologia. 1966;9:392–418. doi: 10.1007/BF00406450. [DOI] [PubMed] [Google Scholar]
- 16.Stewart JW, Quitkin FM, McGrath PJ, et al. Use of Pattern Analysis to Predict Differential Relapse of Remitted Patients with Major Depression During 1 Year of Treatment with Fluoxetine or Placebo. Arch Gen Psychiatry. 1998;55:334–343. doi: 10.1001/archpsyc.55.4.334. [DOI] [PubMed] [Google Scholar]
- 17.McGrath PJ, Stewart JW, Petkova E, et al. Predictors of Relapse During Fluoxetine Continuation or Maintenance Treatment of Major Depression. J Clin Psychiatry. 2000;61:518–524. doi: 10.4088/jcp.v61n0710. [DOI] [PubMed] [Google Scholar]
- 18.Quitkin FM, Rabkin JG, Ross D. Identification of true drug response to antidepressants: use of pattern analysis. Arch Gen Psychiatry. 1984;41:782–786. doi: 10.1001/archpsyc.1984.01790190056007. [DOI] [PubMed] [Google Scholar]
- 19.McGrath PJ, Stewart JW, Quitkin FW, et al. Predictors of Relapse in a Prospective Study of Fluoxetine Treatment of Major Depression. Am J Psychiatry. 2006;163:1542–1548. doi: 10.1176/ajp.2006.163.9.1542. [DOI] [PubMed] [Google Scholar]
- 20.Amsterdam JD, Fawcett J, Quitkin FM, et al. Fluoxetine and Norfluoxetine Plasma Concentrations in Major Depression: A Multicenter Study. Am J Psychiatry. 1997;154:963–969. doi: 10.1176/ajp.154.7.963. [DOI] [PubMed] [Google Scholar]
- 21.Quitkin FM, Petkova E, McGrath PJ, et al. When Should a Trial of Fluoxetine for Major Depression Be Declared Failed? Am J Psychiatry. 2003;160:734–740. doi: 10.1176/appi.ajp.160.4.734. [DOI] [PubMed] [Google Scholar]
- 22.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Third Edition, Revised. Washington, DC: American Psychiatric Association; 1987. [Google Scholar]
- 23.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guy W. New Clinical Drug Evaluation Unit (ECDEU) Assessment Manual for Psychopharmacology. Vol. 1976. Rockville, MD: National Institute of Mental Health; 1976. Clinical Global Impressions; pp. 218–222. [Google Scholar]
- 25.Diggle P, Heagerty P, Liang KY, Zeger S. Analysis of Longitudinal Data. Oxford, UK: Oxford University Press; 2002. [Google Scholar]
- 26.Harville DA. Variance Component Estimation. JASA. 1977;72:320–340. [Google Scholar]
- 27.Zimmerman M, Posternal MA, Ruggero CJ. Impact of study design on the results of continuation studies of antidepressants. J Clin Psychopharm. 2007;27:177–181. doi: 10.1097/JCP.0b013e31803308e1. [DOI] [PubMed] [Google Scholar]
- 28.Rief W, Nestoriuc Y, Weiss S, Welzel E, Barsky AJ, Hofmann SG. Meta-analysis of the placebo response in antidepressant trials. J Affect Disord. 2009;118:1–8. doi: 10.1016/j.jad.2009.01.029. [DOI] [PubMed] [Google Scholar]
- 29.Mundt JC, Greist JH, Jefferson JW, Katzelnik DJ, DeBrota DJ, Chappell PB, Modell JG. Is It Easier to Find What You Are Looking for if You Think You Know What It Looks Like? J Clin Psychopharm. 2007;27:121–125. doi: 10.1097/JCP.0b013e3180387820. [DOI] [PubMed] [Google Scholar]
- 30.Landin R, DeBrtoa DJ, DeVries TA, Potter WZ, Demitrack The Impact of restrictive Entry Criterion During the Placebo Lead-In Period. Biometrics. 2000;56:271–278. doi: 10.1111/j.0006-341x.2000.00271.x. [DOI] [PubMed] [Google Scholar]
- 31.Trivedi M, Rush J. Does a Placebo Run-In or a Placebo Treatment Cell Affect the Efficacy of Antidepressant Medications? Neuropsychopharmacology. 1995;11:33–43. doi: 10.1038/npp.1994.63. [DOI] [PubMed] [Google Scholar]
- 32.Rosenbaum JF, Fava M, Hoog SL, Ascroft RC, Krebs WB. Selective serotonin reuptake inhibitor discontinuation syndrome: a randomized clinical trial. Biol Psychiatry. 1998;44:77–87. doi: 10.1016/s0006-3223(98)00126-7. [DOI] [PubMed] [Google Scholar]