

Abstract
Unique tyrosine glycosylated amyloid-β (1–15) glycopeptides were synthesized with well-defined stereochemistry at the glycosidic linkages. Aided by these glycopeptides and tandem mass spectrometry analysis, the naturally existing amyloid-β glycopeptides, isolated from Alzheimer’s disease patients, were determined to contain an α-linked N-acetyl galactosamine at the modified tyrosine 10 residue. Glycosylation can significantly impact the property of amyloid-β as the glycopeptide has much lower affinity for Cu+ ions.
Alzheimer’s disease (AD) is the most common form of dementia, which is characterized by the formation of amyloid plaques composed of aggregates of amyloid-β (Aβ) polypeptides. AD is a serious concern for modern society as it is estimated that 5.4 million of Americans have AD with an approximate 200 billion dollars in cumulative cost in 2013.1 One of the major challenges in AD treatment is accurate diagnosis at an early stage. Aβ and tau proteins have been widely viewed as biomarkers for AD.2 However, they are of limited utility in diagnosis at the presymptomatic stage. There is an urgent need to identify AD biomarkers with higher sensitivity and specificity before cognitive decline appears.3 Recently, a series of novel Aβ glycopeptides, corresponding to the N-terminal fragments (Aβ 1–X, X ranging from residues 15 to 20) of Aβ, have been isolated from the cerebrospinal fluid of patients.4 It was found that these Aβ glycopeptides are present in AD patients at markedly higher levels compared to those from non-AD control patients.4 Moreover, cognitive impaired patients considered non-AD due to below threshold Aβ(1–42) levels actually exhibited similar Aβ glycopeptide profiles as AD patients, which may have indicated early AD development. Therefore, the Aβ glycopeptides can be potential new biomarkers to facilitate early AD diagnosis.
The structures of Aβ glycopeptides were analyzed via mass spectrometry (MS).4 While glycoproteins and glycopeptides with mammalian origin commonly bear carbohydrate chains on serines/threonines (O-linked glycans) or on asparagine residues (N-linked glycans),5,6 Aβ glycopeptides contain glycosylation on tyrosine 10 (Tyr10).4 Although tyrosine glycosylations have been found with glucose-α(1-O)Tyr or galactose-β(1-O)Tyr linkage,7–9 the carbohydrates on Aβ glycopeptides were determined to have a core N-acetyl hexosamine (HexNAc) moiety linked to the hydroxyl group of Tyr. The HexNAc was further extended with one hexose and two or three N-acetylneraminic acids (Neu5Ac). In analogy with other O-linked glycans, the core HexNAc was proposed to be N-acetyl galactosamine (GalNAc). An ambiguity in the structure is that the stereochemistry of the HexNAc-Tyr linkage could not be defined by the LC-MS/MS analysis alone. The uncertainty in structure has hindered understanding of its function as well as the efforts in generating glycopeptide specific monoclonal antibodies for its detection.
To enable structural determination, herein, we report the total synthesis of Aβ(1–15) glycopeptides bearing GalNAc on Tyr10 with well-defined steroechemistry, which joins the few examples of chemical synthesis of O-glycosyl tyrosine amino acid10–13 and glycopeptide14 reported to date. Aided by the synthetic Aβ(1–15) glycopeptides, the HexNAc in the natural glycopeptide from AD patients was determined to be α-linked GalNAc through MS. Glycosylation of Tyr10 can significantly change the properties of the Aβ peptide as evident from the much reduced affinity of the glycopeptide for Cu+ ion.
To access the Aβ glycopeptide, we began with the preparation of the glycosylated Tyr building block from the D-galactosamine hydrochloride 1. As stereochemistry at the glycosyl linkage is not known, we aimed for both α and β anomers. To facilitate the formation of the α glycosyl linkage, the amine moiety of galactosamine 1 was converted to azide through the copper catalyzed diazo transfer reaction (Scheme 1a).15 Global acetylation followed by selective removal of the anomeric acetate (Ac) and addition of trichloroacetonitrile led to the trichloroacetimidate donor 2. Trimethylsilyl triflate (TMSOTf) promoted glycosylation of Fmoc protected tyrosine 3 by donor 2 forming the glycosylated tyrosine, which was then reduced with simultaneous acetylation leading to 4 as a single stereoisomeric product in 56% yield for the two steps. NMR analysis of 4 (3JH1–H2 = 3.0 Hz) suggested that the newly formed glycosidic linkage was α. Catalytic hydrogenolysis of 4 generated tyrosine carboxylic acid 5, which was ready for glycopeptide synthesis.
Scheme 1.
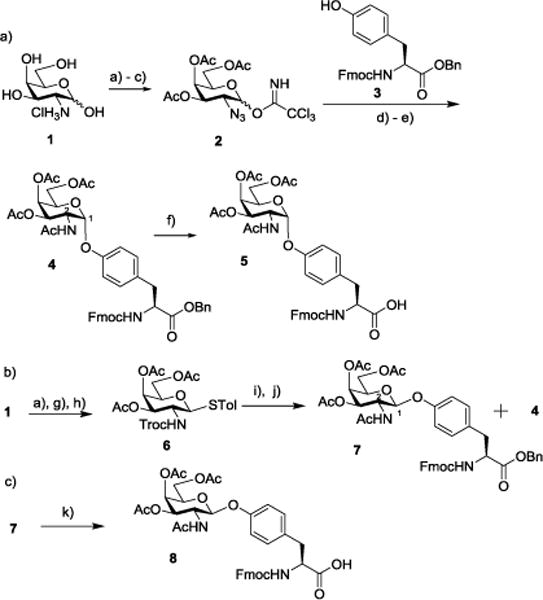
Reagents and conditions: (a) i. triflic azide, K2CO3, CuSO4, H2O/Toluene/MeOH, r.t.; ii. Ac2O, pyridine, DMAP, 0°C to r.t., 71% for 2 steps; (b) hydrazine acetate, DMF, r.t., 82%; (c) trichloroacetonitrile, K2CO3, DCM, r.t., 75%; (d) 3, TMSOTf, 4Å MS, DCM/Et2O (1:1), −30°C (α only); e) Zn, Ac2O, AcOH, THF, r.t., 56% for 2 steps; (f) H2, Pd/C, AcOH, DCM/MeOH, r.t., 94%. (g) i. H2, Pd/C, p-toluenesulfonic acid monohydrate, THF, r.t., ii. 2,2,2-trichloroethyl chloroformate, TEA, THF, 0°C, 80% for 2 steps; (h) p-toluenethiol, BF3• Et2O, DCM, r.t., 85%; (i) NIS, trifluoromethanesulfonic acid, 3, 4Å MS, DCM, −20°C; (j) Zn, Ac2O, AcOH, THF, r.t., 52% for 2 steps (7: 26%; 4: 26%); (k) H2, Pd/C, AcOH, DCM/MeOH, r.t., 90%.
In order to form β-linked glycosyl tyrosine, trichloroethyl carbamate (Troc) protected galactosamine thioglycosyl donor 6 was prepared from D-galactosamine hydrochloride 1 (Scheme 1b). With N-iodosuccinimide (NIS) and trifluoromethane sulfonic acid as the promoters, donor 6 was coupled with tyrosine acceptor 3, which was followed by reduction of 2-N-trichlorethoxycarbonyl (Troc) group and acetylation leading to two anomeric isomers 7 and 4 in 1:1 ratio. 7 and 4 were separated by silica gel chromatography and 7 was determined to be the β anomer (3JH1–H2 = 8.5 Hz) by 1H-NMR. The formation of a large quantity of 4 in the presence of 2-N-Troc moiety capable of neighboring group participation in donor 6 was presumably due to the higher thermodynamic stability of 4 resulting from the anomeric effect. 7 was subjected to catalytic hydrogenolysis yielding the β-linked tyrosine carboxylic acid 8 (Scheme 1c).
With the glycosyl tyrosines 5 and 8 in hand, glycopeptide syntheses were performed using the solid phase method on 2-chlorotrityl resin (Scheme 2). Iterative peptide coupling using Fmoc protected amino acid building blocks including the glycosylated tyrosines followed by Fmoc deprotection produced glycopeptides, which were cleaved off the resin using a cocktail of trifluoroacetic acid (TFA), water and triisopropylsilane (TIPS). Treatment of the resulting peptides with base removed all O-acetates on GalNAc and the N-terminal Fmoc. Subsequent HPLC purification generated pure Aβ(1–15) glycopeptides 9 and 10 containing α and β linked GalNAc respectively. In a similar manner, the unglycosylated Aβ(1–15) 11 was synthesized.
Scheme 2.
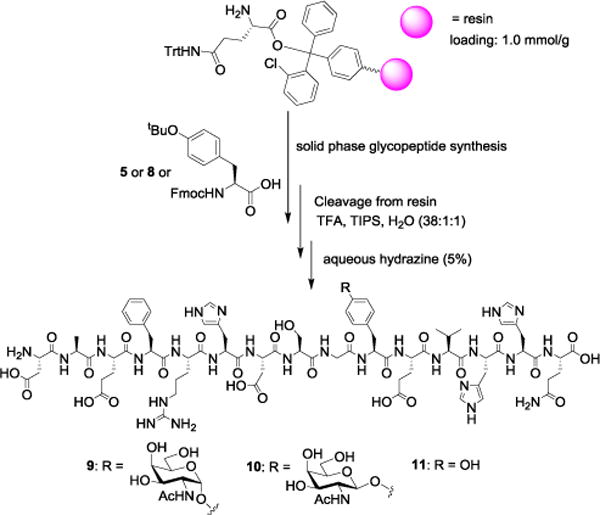
Synthesis of Aβ glycopeptides 9 (3%), 10 (5%) and peptide 11 (9%).
The synthetic glycopeptides provide valuable standards to establish MS methods for stereochemical assignment. The Aβ(1–15) glycopeptides 9 and 10 were analyzed with nano-flow liquid chromatography electrospray ionization tandem mass spectrometry (LC-MS/MS or MS2) using a hybrid linear ion trap-Fourier transform ion cyclotron resonance mass spectrometer. Collision induced dissociation (CID) was utilized to achieve glycosidic and partial peptide fragmentation and electron capture dissociation (ECD) was utilized for determining the amino acid position of the glycan. The CID-MS/MS (MS2) spectra were collected in the linear ion trap (Fig. 1A for glycopeptide 9 and Fig. 1B for glycopeptide 10) resulting from the [M+4H]4+ precursors (Fig. S1). The theoretical mass (monoisotopic) for the [M+4H]4+ precursor of compounds 9 and 10 is m/z 508.2213. The measured masses for compound 9 and compound 10 were m/z 508.2207 (1.14 ppm off) and 508.2210 (0.63 ppm off) respectively. Both compounds lost GalNAc+H+ from the [M+3H]3+ peptide ion (m/z 609.6). Although qualitatively the spectra were similar, quantitative analysis revealed key differences distinguishing the two structures. For 9, the [M+4H]4+ peptide ion (m/z 457.7), resulting from neutral loss of GalNAc, had lower intensity than a number of GalNAc substituted b- and y-ions (e.g., [y14+GalNAc]2+ at m/z 479.8) (Fig. 1A). The reverse was observed for 10 where the [M+4H]4+ ion was more prominent compared to the [y14+GalNAc]2+ ion (Fig. 1B). The ratios for the m/z 457.7 and 479.8 ions were found to be 0.5 ± 0.1 and 2.6 ± 0.7 (m ± SD) for 9 and 10 respectively. Thus, GalNAc was more easily cleaved from the Tyr residue during CID when it was attached to the peptide in β-configuration (glycopeptide 10) compared to the α-configuration (glycopeptide 9). This is most likely due to the higher stability of the glycosidic linkage in 9 as a result of the anomeric effect. Therefore, MS can be used to reliably differentiate these two isomeric glycopeptides.
Fig. 1.
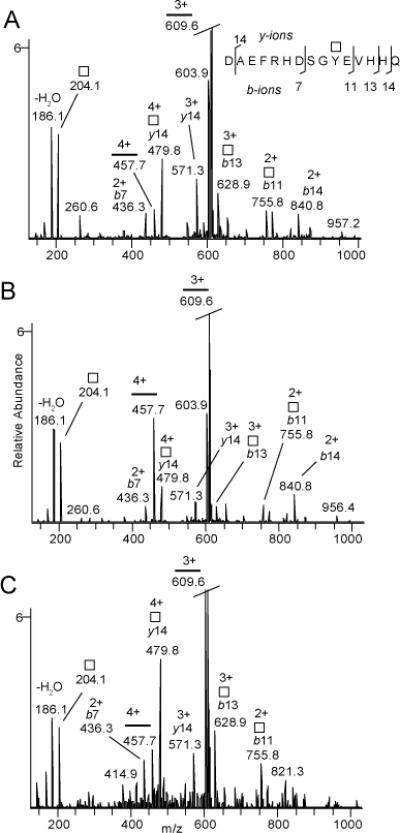
CID-MS2 of Aβ(1–15) glycopeptides 9 (A) and 10 (B); and CID-MS3 of HexNAc substituted Aβ(1–15) from human cerebrospinal fluid (C). The spectra are intensity zooms of the corresponding CID-MS2 and CID-MS3 spectra (See Figs. S1 and S2). Square symbolizes free or glycopeptide linked HexNAc and horisontal line the peptide 1DAEFRHDSGYEVHHQ15.
Native Aβ(1–15) glycopeptide isolated from human cerebrospinal fluid (CSF) was examined next by MS. Aβ peptides and glycopeptides were immunopurified using 6E10 antibody and any terminal Neu5Ac residues were selectively hydrolyzed using 0.1 M formic acid at 353 K for 30 minutes resulting in Aβ glycopeptides enriched in de-sialylated HexHexNAc (Hex denotes a hexose).16 The [M+4H]4+ precursor of Aβ(1–15)+HexHexNAc was observed at m/z 548.7350 (Fig. S2A) and CID-MS2 resulted in abundant neutral loss of Hex into Aβ(1–15)+HexNAc at m/z 508.4 (Fig. S2B) isomeric to 9 and 10. Further consecutive fragmentation (CID-MS3) led to a prominent loss of HexNAc+H+ and a spectrum closely matching that of the CID-MS2 of 9 (Fig. 1C). Specifically, CID-MS3 spectra of the native glycopeptide possessed a more abundant [y14+GalNAc]2+ (m/z 479.8) and a less abundant [M+4H]4+ peptide ion (m/z 457.7) with the intensity ratio of 0.4 ± 0.2 for the peak at m/z 457.7 over that with m/z 479.8. Thus, we conclude that the native Aβ(1–15) glycopeptides from CSF are composed of an α-linked HexNAc moiety, most likely an α-GalNAc moiety. An electron capture dissociation (ECD) MS spectrum of the [M+4H]4+ ion for compound 9 also confirmed the peptide sequence and pinpointed that the GalNAc residue was attached to Tyr10 (Fig. S3).
Aβ peptides can bind with metal ions,17–19 which can facilitate nucleation contributing to plaque formation. At the same time, as ions such as free Cu+ are highly toxic to cells, it has been proposed that Aβ may play a neuroprotective role by scavenging metal ions.20 Thus, the Cu+ binding abilities of Aβ may influence plaque biology. This prompted us to analyze the affinity of glycopeptide 9 with Cu+ ion through a competitive binding assay.21 Cu(NO3)2 was reduced by sodium ascorbate to Cu+, which formed an orange complex with disodium bathocuproinedisulfonic acid (BC) with an absorbance maximum at 483 nm. When increasing amounts of Aβ glycopeptide 9 were added to a solution of BC-Cu complex, they competed with BC for Cu+ binding thus serially reducing the absorbance at 483 nm (Fig. 2). Based on absorbance changes, the dissociation constant of [Cu+ −9] complex was calculated to be 1.69 ± 0.84×10−14 M. In comparison, the dissociation constant of the unglycosylated Aβ peptide 11 with Cu+ was measured to be 2.72 ± 1.26×10−15 M, which was similar to the reported value of Aβ (1–16) binding with Cu+.21 Aβ peptide binds Cu+ in a linear bis-His geometry,20 thus most likely the Cu+ ion is ligated to His13 and His14 in unglycosylated Aβ peptide 11. The presence of a bulky GalNAc on Tyr10 in glycopeptide 9 could possibly sterically hinder Cu+ binding. The impact of reduced Cu+ affinity of the glycopeptide on plaque formation and toxicity will need to be established in vivo in the future.
Fig. 2.
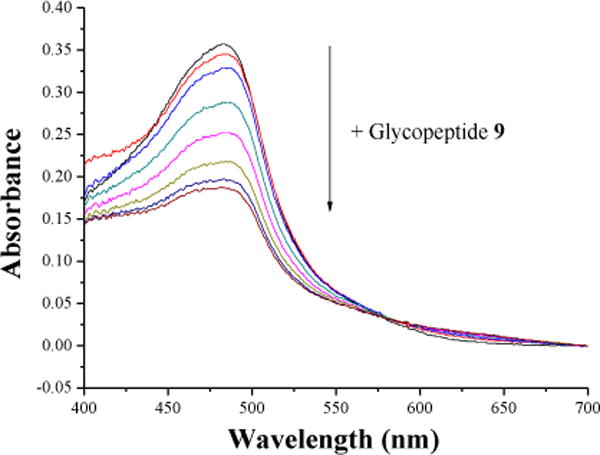
Competitive chelation of BC and Aβ glycopeptide 9 with Cu+. Increasing concentrations of glycopeptide 9 disrupted [CuBC2]3− complex resulting in reduction of the absorbance at 483 nm.
In conclusion, we developed viable synthetic routes to the first synthesis of Tyr O-glycosylated Aβ peptides. Aided by these well-defined synthetic samples and tandem MS analysis, we identified that Tyr10 and O-glycan were most likely linked through an α-GalNAc linkage in natural Aβ glycopeptide fragments isolated from AD patients. Glycosylation could significantly impact the property of the glycopeptide such as interactions with Cu+ ion. The determination of the glycopeptide structure can enable monoclonal antibody generation study and lay the groundwork towards further understanding of their roles as biomarkers.
Supplementary Material
Acknowledgments
This work was supported by Michigan State University, the National Science Foundation (CHE 1111550) and the National Institute of General Medical Sciences, NIH (R01GM072667) and the Swedish Research council (8266).
Footnotes
Electronic Supplementary Information (ESI) available: Synthesis procedures, characterization data for all new compounds and supplementary figures.
Contributor Information
Göran Larson, Email: goran.larson@clinchem.gu.se.
Xuefei Huang, Email: xuefei@chemistry.msu.edu.
Notes and References
- 1.Alzheimer’s Association 2013 Alzheimer’s disease facts and figures. 2013 doi: 10.1016/j.jalz.2013.02.003. http://www.alz.org/downloads/facts_figures_2013.pdf. [DOI] [PubMed]
- 2.Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, DeKosky ST, Gauthier S, Selkoe D, Bateman R, Cappa S, Crutch S, Engelborghs S, Frisoni GB, Fox NC, Galasko D, Habert MO, Jicha GA, Nordberg A, Pasquier F, Rabinovici G, Robert P, Rowe C, Salloway S, Sarazin M, Epelbaum S, de Souza LC, Vellas B, Visser PJ, Schneider L, Stern Y, Scheltens P, Cummings JL. Lancet Neurol. 2014;13:614–629. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- 3.Blennow K, Hampel H, Weiner M, Zetterberg H. Nat Rev Neurol. 2010;6:131–144. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 4.Halim A, Brinkmalm G, Rüetschi U, Westman-Brinkmalm A, Portelius E, Zetterberg H, Blennow K, Larson G, Nilsson J. Proc Natl Acad Sci USA. 2011;108:11848–11853. doi: 10.1073/pnas.1102664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grogan MJ, Pratt MR, Marcaurelle LA, Bertozzi CR. Annu Rev Biochem. 2002;71:593–634. doi: 10.1146/annurev.biochem.71.110601.135334. [DOI] [PubMed] [Google Scholar]
- 6.Dwek RA. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 7.Zarschler K, Janesch B, Pabst M, Altmann F, Messner P, Schäffer C. Glycobiology. 2010;20:787–798. doi: 10.1093/glycob/cwq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Messner P, Christian R, Neuninger C, Schulz G. J Bacteriol. 1995;177:2188–2193. doi: 10.1128/jb.177.8.2188-2193.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alonso MD, Lomako J, Lomako WM, Whelan WJ. FEBS Lett. 1994;342:38–42. doi: 10.1016/0014-5793(94)80580-6. [DOI] [PubMed] [Google Scholar]
- 10.Fahmi NE, Dedkova L, Wang B, Golovine S, Hecht SM. J Am Chem Soc. 2007;129:3586–3597. doi: 10.1021/ja067466n. [DOI] [PubMed] [Google Scholar]
- 11.Khodair AI, Winterfeld GA, Schmidt RR. Eur J Org Chem. 2003:1847–1852. [Google Scholar]
- 12.Salvador LA, Elofsson M, Kihlberg J. Tetrahedron. 1995;51:5643–5656. [Google Scholar]
- 13.Vargas-Berenguel A, Meldal M, Paulsen H, Jensen KJ, Bock K. J Chem Soc, Perkin Trans 1. 1994:3287–3294. [Google Scholar]
- 14.Jansson AM, Jensen KJ, Meldal M, Lomako J, Lomako WM, Olsen CE, Bock K. J Chem Soc, Perkin Trans 1. 1996:1001–1006. [Google Scholar]
- 15.Alper PB, Hung SC, Wong CH. Tetrahedron Lett. 1996;37:6029–6032. [Google Scholar]
- 16.Nilsson J, Rüetschi U, Halim A, Hesse C, Carlsohn E, Brinkmalm G, Larson G. Nat Methods. 2009;6:809–811. doi: 10.1038/nmeth.1392. [DOI] [PubMed] [Google Scholar]
- 17.Savelieff MG, Lee S, Liu Y, Lim MH. ACS Chem Biol. 2013;8:856–865. doi: 10.1021/cb400080f. [DOI] [PubMed] [Google Scholar]
- 18.Faller P, Hureau C, Berthoumieu O. Inorg Chem. 2013;52:12193–12206. doi: 10.1021/ic4003059. [DOI] [PubMed] [Google Scholar]
- 19.Kepp KP. Chem Rev. 2012;112:5193–5239. doi: 10.1021/cr300009x. [DOI] [PubMed] [Google Scholar]
- 20.Shearer J, Szalai VA. J Am Chem Soc. 2008;130:17826–17835. doi: 10.1021/ja805940m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feaga HA, Maduka RC, Foster MN, Szalai VA. Inorg Chem. 2011;50:1614–1618. doi: 10.1021/ic100967s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.