

Abstract
Persistence of hepatitis B virus (HBV) covalently closed circular DNA (cccDNA) under current antiviral therapy is a major barrier to eradication of chronic hepatitis B (CHB). Curing CHB will require novel strategies for specific disruption of cccDNA. The clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system is a newly developed tool for site-specific cleavage of DNA targets directed by a synthetic guide RNA (gRNA) base-paired to the target DNA sequence. To examine whether this system can cleave HBV genomes, we designed eight gRNAs against HBV of genotype A. With the HBV-specific gRNAs, the CRISPR/Cas9 system significantly reduced the production of HBV core and surface proteins in Huh-7 cells transfected with an HBV-expression vector. Among eight screened gRNAs, two effective ones were identified. Interestingly, one gRNA targeting the conserved HBV sequence acted against different genotypes. Using a hydrodynamics-HBV persistence mouse model, we further demonstrated that this system could cleave the intrahepatic HBV genome-containing plasmid and facilitate its clearance in vivo, resulting in reduction of serum surface antigen levels. These data suggest that the CRISPR/Cas9 system could disrupt the HBV-expressing templates both in vitro and in vivo, indicating its potential in eradicating persistent HBV infection.
Introduction
Current antiviral treatment with nucleos(t)ide analogues (NAs) or interferon fails to cure chronic hepatitis B (CHB). Rebound viremia often occurs after the cessation of antiviral treatment. It is clear now that NA alone has little or no effect on the elimination of replicative templates of hepatitis B virus (HBV), the covalently closed circular DNA (cccDNA), despite its effective suppression of HBV replication by inhibiting viral reverse transcriptase.1,2 Moreover, HBV cccDNA exhibits staggering stability and declines slowly under antiviral therapy, thus representing a major barrier to viral eradication.3,4,5,6
Curing CHB requires elimination of persistent HBV cccDNA or infected hepatocytes, which is so far still an unattainable goal. Theoretically, combined with effective suppression of HBV replication by NAs, specific disruption of the HBV genome may result in eradication of HBV cccDNA, hence curing chronic HBV infection.7 Two genome editing technologies, zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), can cleave the host genomes in a site-specific fashion and have been under investigation for specific degradation of the HBV genomes in vitro and in vivo.8,9,10 Both of them are composed of protein-based programmable, sequence-specific DNA-binding modules linked to a nonspecific DNA cleavage domain.11,12,13
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) system is a novel genome editing tool derived from the adaptive immune system of bacteria and archaea in which it provides the host with resistance to invading foreign viruses or plasmids.14,15,16,17 The type II CRISPR/Cas system consists of two short RNAs, CRISPR RNAs (crRNAs) and trans-activating crRNAs (tracrRNAs), and the DNA endonuclease Cas9. The mature crRNA::tracrRNA complex directs Cas9 DNA endonuclease to the target DNA sequence called the protospacer on the target DNA next to the protospacer adjacent motif (PAM) for site-specific cleavage.18 This system is further simplified by fusing the crRNA and tracrRNA into a single chimeric guide RNA (gRNA). Compared with ZFNs and TALENs, the CRISPR/Cas system can be easily reprogrammed to cleave virtually any DNA sequence by simply redesigning the crRNAs or gRNAs. Recently, the CRISPR/Cas system has been successfully applied to human cells19,20,21,22 and several model organisms for genome editing.23,24,25,26 Here, we evaluated the potential of using the CRISPR/Cas9 system to disrupt the HBV genome in vitro and in vivo.
Results
Coexpression of HBV-specific gRNAs and Cas9 suppressed the production of HBV proteins in vitro
To construct HBV-specific gRNAs, we searched for a potential 20-base target sequence on the genome of the genotype A HBV-expression vector pAAV/HBV1.2 with the initiating 5′G and the 3′-downstream PAM (GN19-NGG). A 5′G is required for transcription from the U6 promoter in the gRNA-expression cassette. We designed a panel of eight HBV-specific gRNAs targeting different regions of the HBV genome (Figure 1a). The sequences and locations of these HBV-specific gRNAs are detailed in Table 1. To examine the efficiency of each individual gRNA in suppressing HBV protein expression, we cotransfected the HBV-, individual gRNA-, and Cas9-expression vectors to Huh7 cells. By measuring the intracellular core (HBcAg) and surface antigen (HBsAg) expression levels, we found that the gRNAs P1, S1, PS2, and PS3 exhibited higher efficacy in suppressing the expression of HBV proteins by 70, 40, 30, and 20%, respectively (Figure 2a). To improve the efficiency of the CRISPR/Cas9 system, we further constructed a dual expression vector containing both the gRNA- and Cas9-expression cassettes. We chose the gRNAs P1, S1, and PS2 because of their higher suppressive ability. The gRNA XCp was also chosen because its target sequence is highly conserved in the HBV genomes of genotypes A, B, and C. Interestingly, all four dual expression vectors with the gRNAs P1, S1, XCp, and PS2 significantly suppressed the intracellular expression of surface proteins by 96, 90, 82, and 77%, respectively (Figure 2b). We also measured the HBsAg levels in the culture supernatant. The gRNAs P1, XCp, and PS2 suppressed the HBsAg levels by 64, 36, and 16%, respectively (Figure 2c). Surprisingly, although the gRNA S1 significantly suppressed the expression of intracellular HBsAg and HBcAg, it failed to reduce the extracellular HBsAg levels.
Figure 1.

Designs of gRNAs targeting (hepatitis B virus) HBV-specific sequences. (a) Illustration of the gRNA-targeted sequences located in the HBV genome. (b) Designs of the chimeric HBV-specific gRNAs.
Table 1. Sequences of the protospacer and PAM targeted by HBV-specific gRNAs in the HBV genome of the pAAV/HBV1.2 vector.

Figure 2.
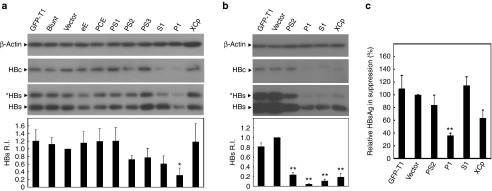
Examination of the efficacy of individual (hepatitis B virus) HBV-specific gRNAs in inhibition of HBV protein expression. The HBV-expression vector was cotransfected to Huh7 cells with (a) each individual gRNA-expression vector and the hCas9 vector or (b) each individual gRNA/Cas9 dual expression vector. The lysate was collected after 48 hours and the levels of intracellular HBcAg and HBsAg were analyzed by using Western blot analysis and quantification of the band intensity. (c) Levels of HBsAg in culture supernatant. The gRNA targeting GFP (GFP-T1), backbone plasmid for the construction of gRNA-expression cassette (blunt), and plasmid containing the U6 promoter/gRNA scaffold (vector) served as negative controls. The bands of *HBs and HBs represent glycosylated and nonglycosylated small surface antigens of HBV, respectively. The relative intensity of HBsAg was shown by quantification of both bands of *HBs and HBs normalized to that of Vector in the bottom panel. RI, relative intensity. The RI and percentages of HBsAg expression were calculated from the results of three independent experiments and presented as mean ± SEM. Statistical significance was computed by Student's t-test and indicated as asterisks (*P < 0.05, **P < 0.01).
We further examined the effects of the CRISPR/Cas9 system on the HBV replicative templates, including relaxed circular-DNA (rc-DNA) and cccDNA. In the cell-line culture system, the HBV-expressing plasmid only generates very few cccDNA, if not none, although it can produce progeny virions efficiently. In contrast, the duck hepatitis B virus (DHBV)-expressing plasmid efficiently produces its cccDNA, particularly in a mutant lacking the surface antigen.27 Therefore, we utilized the DHBV system and designed three DHBV-specific gRNAs T1, T7, and Tr6. 293T cells were cotransfected with the surface-deficient DHBV-expressing plasmid and individual gRNA/Cas9 dual expression vectors. Compared with the mock gRNA-transfection control, the DHBV-specific gRNAs slightly reduced the levels of DHBV-expressing plasmid, as demonstrated by Southern blot, but efficiently suppressed the levels of the rc-DNA and cccDNA (Supplementary Figure S1).
Multiplex gRNAs exhibited stronger effects on suppressing the expression of HBV proteins
The CRISPR/Cas9 system has been used for multiplex genome cleavage.20 To examine the efficiency of multiplex genome cleavage in our experimental system, Huh7 cells were cotransfected with HBV-expression vector and the two most effective dual expression vectors, gRNAs P1 and XCp, alone or in combination. The results demonstrated that the combinatorial gRNAs P1 and XCp were more effective in suppressing intracellular HBsAg production than either gRNA alone, although the effect is only marginally significant (P = 0.078 for combination versus P1 and P = 0.085 for combination versus XCp) (Figure 3). It is likely because gRNA P1 or XCp alone was able to mediate efficient genome cleavage, thus limiting additional gain in genome cleavage from the addition of a second gRNA. We also conducted a T7 endonuclease 1 (T7E1) assay to determine the percentage of insertion/deletion (Indel) resulting from repaired double-strand breaks introduced by the CRISPR/Cas9 system. The results indicated that the occurrence of mutated HBV expression templates edited by gRNAs P1 and XCp alone was 13.6 and 9.3%, respectively. Besides, the percentage of Indel mediated by combinatorial gRNAs P1 and XCp was 25.6%, which was higher than that mediated by P1 or XCp alone. Interestingly, we also observed a shorter band (~580 bp) in the PCR product directly amplified from cells treated with combinatorial gRNAs P1 and XCp (Supplementary Figure S2). By sequencing, the band proved to be the cleaved DNA segment mediated by gRNAs P1 and XCp (data not shown). This indicates that this combination could result in an efficient dual cleavage and removal of a larger DNA fragment.
Figure 3.

Suppression of the (hepatitis B virus) HBV protein expression via the multiplex HBV-specific gRNA. The HBV-expression vector was cotransfected to Huh7 cells with the gRNA/Cas9 dual expression vectors. The lysate was collected after 48 hours and the levels of intracellular HBsAg were analyzed using Western blotting as shown by this representative figure. The band intensities were quantified by software ImageJ. The relative HBsAg intensity (HBs R.I.) in the bar graph of the bottom panel was the results of four independent experiments and presented as mean ± SEM. (b) The DNA extracted from the transfected Huh7 cells in panel (a) was analyzed by T7E1 assay for determining the percentage of Indel as shown in the bottom.
gRNA XCp targeting the conserved HBV sequence was effective for HBV genomes of different genotypes
We also examined the effects of the conserved gRNA XCp on the HBV genomes of genotypes B and C. Our data demonstrated that gRNA-XCp/Cas9 was equally effective in suppressing the viral protein expression of all three genotypes of HBV. In contrast, the most effective gRNA P1 against genotype A of HBV did not significantly suppress the viral protein expression of genotype B, although it significantly suppressed that of genotype C (Figure 4a). Nevertheless, the suppressive effect of gRNA P1 on viral protein expression of genotype C is much less than that of gRNA XCp (P < 0.05), whereas both gRNAs exhibited a similar inhibitory effect on genotype A. Analysis of the target sequences of gRNA P1 in the HBV genomes of these three genotypes revealed that the HBV genomes of genotypes B and C had a single-nucleotide polymorphism in the PAM, although they had the perfectly matched 20-bp target sequence (Figure 4b). In contrast, the target sequence and PAM of gRNA XCp are exactly the same in all three genotypes (Figure 4c). This result indicates the importance of PAM for genome cleavage by the CRISPR/Cas9 system, and also provides further evidence that the effects of gRNAs P1 and XCp are mediated by the CRISPR/Cas9 system.
Figure 4.

Effects of the gRNAs P1 and XCp on different genotypes of (hepatitis B virus) HBV. (a) HBV-expression vector was cotransfected to Huh7 cells with either the gRNA-P1/Cas9 or gRNA-XCp/Cas9 dual expression vector alone or in combination. The lysate was collected 48 hours post-transfection. The levels of intracellular HBsAg were determined by Western blot analysis. The band intensity was measured by software ImageJ and normalized to the intensity of the mock vector (Vector) for each HBV genotype. The HBs R.I. was calculated from the results of three independent experiments and presented as mean ± SEM. Statistical significance was calculated by the Student's t-test and indicated by asterisks (*P < 0.05, **P < 0.01). N.S. means no statistical significance. Sequence alignment of gRNAs P1 (b) and XCp (c) in HBV genotypes A, B, and C. The underlined nucleotides indicated the PAM motif for the recognition of Cas9.
The HBV-specific CRISPR/Cas9 system facilitated the clearance of intrahepatic HBV templates in vivo
The HBV hydrodynamics-mouse model is a well-established model of HBV persistence.28,29 Therefore, we further examined the efficacy of the CRISPR/Cas9 system in the cleavage of HBV genome-containing plasmids in vivo by using this model. The HBV-expression vector and the CRISPR/Cas9 dual expression vectors were coinjected into the tail veins of C57BL/6 mice by hydrodynamics. We found that serum HBsAg levels were significantly lower in mice receiving HBV-specific gRNAs P1 and XCp on day 2 postinjection (Figure 5a). On day 6, serum HBsAg levels were significantly reduced only in gRNA-P1-treated mice (Figure 5a). In addition, using Southern blot analysis, we found that the levels of intrahepatic HBV-expressing vectors, an indicator for persistent HBV genomes, were significantly reduced in those mice receiving HBV-specific gRNAs P1 and XCp (Figure 5b). To confirm that this CRISPR/Cas9 system specifically cleaves the target sequences, we performed the T7E1 assay, which showed target-specific mutagenesis in mice receiving the gRNA-P1 or -XCp/Cas9 dual expression vector, but not in control mice (Figure 5c). According to the T7E1 assay, the targeted disruption occurred in 5 and 4% of intrahepatic HBV genomes in mice receiving gRNAs P1 and XCp, respectively. This result is in contrast to the efficient reduction in the levels of intrahepatic HBV-expressing vectors on Southern blot analysis. We further carried out the clonal sequencing of intrahepatic HBV DNA extracted from mice receiving gRNA P1 to demonstrate the disruption of the HBV genome by the CRISPR/Cas9 system. We analyzed 18 clones in total. Five clones (27.8%) with Indel were noted, suggesting that nonhomologous end joining (NHEJ) repair has occurred at the cleaved target sequence of gRNA P1 (Supplementary Figure S3). Taken together, our results indicated that the CRISPR/Cas9 system cleaved the HBV genome-expressing template and facilitated its clearance.
Figure 5.

In vivo destruction of intrahepatic (hepatitis B virus) HBV genomes by the HBV-specific CRISPR/Cas9 system. Sera from mice receiving pAAV/HBV 1.2 together with the gRNA/Cas9 dull expression plasmid, vector (n = 4), gRNA P1 (n = 4), or gRNA XCp (n = 4), were collected after hydrodynamic injection. (a) Levels of HBsAg in sera were measured at days 2 and 7 posthydrodynamic injection as shown in the upper and bottom panel, respectively. The results were presented as individual samples with mean ± SEM. Statistical significance was calculated by the Student's t-test and indicated by asterisks (*P < 0.05, **P < 0.01). (b) Analysis of intrahepatic HBV DNA by Southern blotting. 0.1 ng of pAAV/HBV 1.2 plasmid was loaded as a positive control. Two samples from each group were analyzed in this experiment. The band intensity was determined by software ImageJ. The numbers in the bottom indicate the relative intensities of the HBV expression plasmids in each sample. (c) Results of T7E1 assay. The percentage of mismatched sequences from two of the gRNA-P1- or gRNA-XCp-treated mice (Indel %) was determined by T7E1 assay. The numbers in the bottom indicate the percentage of Indel.
Discussion
The targeted genome editing tools have the potential for elimination of persistent HBV genomes.8,9,10 Both ZFNs and TALENs utilize protein-based programmable, sequence-specific DNA-binding modules, whose construction is usually time-consuming and labor-intensive. In contrast, the CRISPR/Cas9 system only requires one to design the 20-base sequence-matching gRNAs. Previous studies reported that HBV-specific TALENs have a mutagenesis rate ranging between 30 and 70%.9,10 Our study demonstrated that the CRISPR/Cas9 system was as effective as TALENs in mediating gene disruption in vitro, although its mutagenesis rate is lower (~5% by T7E1 and ~27% by clonal sequencing) in vivo. Additionally, simultaneous cleavage of multiple sites can be achieved by using multiplex gRNAs that could increase the efficiency for disrupting HBV genomes and reduce the emergence of viral mutations escaping from cleavage. As demonstrated here, the use of combinatorial gRNAs P1 and XCp exhibited slightly higher efficiency in reducing the HBV protein production than use of either individual gRNA alone and generated a larger fragment of cleaved DNA, exemplifying the advantage of utilizing multiplex gRNAs. Using the HBV hydrodynamics-mouse model, we showed that the dual gRNA/Cas9 expression vector could significantly reduce the serum levels of HBsAg in vivo. Finally, we demonstrated by Southern blot analysis that the HBV-specific gRNAs/Cas9 reduced the intrahepatic HBV templates.
Although the CRISPR/Cas9 system is a promising strategy to eradicate the persistent HBV genome, there are several remaining issues that need to be addressed for further application. First, the efficiency of this system can be improved. In our hydrodynamics system, serum HBsAg was reduced, but still persisted following CRISPR/Cas9 treatment. Theoretically, any residual HBV genome can potentially lead to rebound viremia and repopulation of HBV in the liver after cessation of the antiviral therapy. To overcome this obstacle, an efficient delivery system with a high vector to target cell ratio, such as adenoviral or adeno-associated viral vectors, would be required to carry the CRISPR/Cas9 system.7 When used in combination with the antiviral therapy for effective suppression of viral replication, repeated treatment with the CRISPR/Cas9 system may eventually lead to eradication of persistent HBV genomes. Another concern for the CRISPR/Cas system is its off-target effect. A previous study showed that a 13-bp “seed” region located at the 3′-end of the protospacer sequence is critical for its site-specific cleavage, indicating that Cas9 can tolerate up to seven mismatches at the 5′-end of the 20-base protospacer sequence.18 Although no apparent toxicity was observed in our in vitro and in vivo experiments, the off-target effects of our gRNAs cannot be excluded. Several groups have characterized the off-target effects of the CRISPR/Cas9 system.30,31 Therefore, designing an efficient gRNA requires selection of specific target sequences with low off-target activity.20 Alternatively, this issue may be solved by using a Cas9 nickase, which cleaves only complementary strands, combined with paired gRNAs targeting different strands of the DNA double helix to generate targeted DNA double-stranded breaks with high specificity.30,32,33 It remains to be seen whether the Cas9 nickase with paired gRNAs is able to eliminate intrahepatic cccDNA efficiently. Another concern is the integrated HBV genomes, which are commonly detected in CHB patients, and can also be targeted by the CRISPR/Cas9 system. Whether the cleavage of integrated HBV genomes can affect the stability of host genomes should be carefully evaluated.
One limitation of this study is the use of the HBV hydrodynamics-mouse model, which is not a model of HBV infection and no cccDNA is detectable. In CHB patients, up to 50 copies of cccDNA exist in each infected hepatocyte, and some of them are epigenetically modified.34 It remains unclear whether episomal cccDNA can be disrupted with high efficiency by the CRISPR/Cas9 system. Interestingly, a very recent study reported that the CRSIPR/Cas9 system could cleave the large-genome DNA viruses, adenovirus and herpes simplex virus, with high efficiency.35 Besides, the CRISPR/Cas9 system was also utilized to disrupt the latent integrated HIV provirus.36 These suggested that this system effectively works on both the integrated and extrachromosomal viral DNA genomes. Nevertheless, animal models that harbor the bona fide cccDNAs, including the human–mouse hepatocyte chimeric model or the natural woodchuck hepatitis virus (WHV)-infected woodchuck model, are still required for further validation of our results.
In conclusion, as a proof of concept, we have shown the utility of the CRISPR/Cas9 system in disrupting intrahepatic HBV templates without apparent toxicity. Combined with effective NAs in suppression of HBV replication, the robust and easy-to-design CRISPR/Cas9 system may exhibit the potential for eradication of persistent HBV DNA in CHB patients, raising the hope for curing chronic HBV infection.
Materials and methods
Plasmids. The human codon-optimized Cas9 (hCas9) expression vector, GFP-T1 gRNA, and gRNA cloning vectors were obtained from Addgene (Cambridge, MA) plasmid 41815, 41819, and 41824, respectively.19 The HBV-expression vector pAAV/HBV1.2 (genotype A) has been described previously.28 The full genomes of HBV genotypes B and C obtained from CHB patients were also constructed as previously described.28 Plasmids were prepared by using the Geneaid plasmid purification kit (Taoyuan, Taiwan) or the Qiagen endofree plasmid kit (Hilden, Germany).
Design and cloning of HBV-specific gRNA. The candidate 20-base protospacer sequence was derived from the HBV genome sequence of pAAV/HBV1.2 (genotype A), containing the initiating 5′G and the downstream 3′PAM with GG dinucleotide (GN19-NGG). A total of eight gRNAs were designed based on this criterion (Table 1). The 5′ 20-base nucleotides were cloned to the gRNA vector using the Gibson assembly kit (New England BioLabs, Ipswich, MA) by following the manufacturer's protocol to construct HBV-specific gRNA-expression vectors. The fragment containing the U6 promoter and HBV-specific gRNA was PCR amplified from the individual gRNA-expression vector and was subsequently inserted into the hCas9 expression vector by using the MfeI restriction sites to construct the HBV-specific gRNA/hCas9 dual expression vector.
Transfection of cell lines. Huh7 cells were maintained in Dulbecco's modified Eagle medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum at 37 °C and 5% CO2. A total of 4 × 105 Huh7 cells per well were seeded in a six-well plate 24 hours prior to transfection. The HBV-specific gRNA/hCas9 expression vectors or HBV-specific gRNA/hCas9 dual expression vectors were cotransfected with the HBV-expression vector to Huh7 cells by using lipofectamine 2000 or lipofectamine 3000 (Life Technologies) according to the manual of the manufacturer.
Detection of extracellular and intracellular HBV proteins. At 48 hours post-transfection, culture supernatant was collected and the level of HBs antigen was determined by Abbott Architect i1000 (Abbott Park, IL). Cells were washed with phosphate-buffered saline (PBS) and lyzed with RIPA buffer (50 mmol/l Tris pH 7.5, 150 mmol/l NaCl, 10 mmol/l EDTA, 1% NP-40, 0.1% SDS) containing protease inhibitor cocktails (Roche Diagnostics, Mannheim, Germany), followed by 14,000 rpm for 30 minutes at 4 °C. Cell lysates were subjected to SDS–15% polyacrylamide gel electrophoresis (PAGE), followed by Western blot analysis using primary antibody (anti-β-actin, anti-HBs antigen, or anti-HBc antigen), secondary antibody and being detected by Millipore Immobilon Western Chemiluminescent HRP Substrate (Billerica, MA). The band intensity was determined by ImageJ software (version 10.2, Bethesda, MD).
Hydrodynamic injection of the HBV-expression and CRISPR/Cas9-expression vectors in mice. The protocol for the generation of the HBV persistent mouse model by hydrodynamic injection (HDI) has been described.28,29 Briefly, 6 μg pAAV/HBV1.2 and 10 μg gRNA/Cas9 dual expression vector were injected into the tail veins of mice (C57BL/6 at the age of 6–8 wk) in a volume of PBS equivalent to 8% of the mouse body weight within 5 seconds. Sera were sampled at different time points. The concentration of HBs antigen was determined by AXSYM systems (Abbott Park, IL) and indicated the persistence of HBV. All mouse care and experiments were performed with the approval of the Institutional Animal Care and Use Committee at the National Taiwan University College of Medicine.
Extraction of intrahepatic HBV DNA. DNA was extracted from mice liver by the modified Hirt method as previously described.37 Briefly, a liver specimen from treated mice was lyzed in Hirt's lysis buffer (10 mmol/l Tris–HCl pH = 7.6, 10 mmol/l EDTA pH = 8.0, 0.7% SDS). After adding 5 mol/l NaCl, the sample was incubated at 4 °C overnight and centrifuged at 14,000 rpm for 30 minutes at 4 °C. The supernatant was collected and treated with 150 U/ml protease K for 2 hours at 37°C. The DNA was extracted twice with saturated phenol and once with phenol:chloroform. The extracted DNA was mixed with twice the volume of 100% ethanol at −80 °C overnight and subsequently precipitated by 14,000 rpm centrifugation for 20 minutes at 4 °C. The DNA pellet was washed with 70% ethanol and then resolved in 20 μl double-distilled H2O.
Southern blotting analysis of intrahepatic HBV DNA. Thirty μg of DNA extracted from the liver of HDI-treated mice was subjected to run 1% agarose gel electrophoresis at 40 V overnight. The agarose gel containing the DNA samples was treated with 0.25 N HCl for 15 minutes once, 0.5 N NaOH for 15 minutes twice, 0.5X TAE buffer for 10 minutes twice, and 0.25X TAE for 10 minutes once and then transferred to a nylon membrane. After UV cross-linking (UV stratalinker, Agilent Technologies, Santa Clara, CA), the membrane was hybridized with a DIG-labeled DNA probe covering 403 bp of X gene (Roche Applied Science, Mannheim, Germany). The signals were detected by exposure to films.
T7E1 assay. To demonstrate CRISPR/Cas9-mediated cleavage, we applied T7E1 (New England Biolabs) to digest imperfectly matched DNA after hybridization of DNA mixture derived from the intrahepatic DNA of HDI-treated mice. PCR products were amplified by HBV-Pol622-A (5′-AGAAAACTTCCTGTTAACAGGC-3′') and BglII-B (5′-CACTAGGGGTTCCTAGATCT-3′) and subjected to T7E1 assay as previously described.38 The band intensities were determined by software ImageJ (version 10.2) and analyzed by the method described previously.39
Statistical analysis. An unpaired, one-tailed Student's t-test was used to compute the difference between two independent groups.
SUPPLEMENTARY MATERIAL Figure S1. In vitro modification of the duck hepatitis B virus (DHBV) expression plasmid by the CRISPR/Cas9 system. Figure S2. Large deletion of the hepatitis B virus (HBV) expression plasmid by the combinatorial use of gRNAs P1 and XCp. Figure S3. Nucleotide alignment of clonal sequencing results.
Acknowledgments
We thank George Church for donating plasmids (hCas9, GFP-T1 gRNA, and gRNA cloning vectors) to Addgene. We also thank Mi-Hua Tao from Academia Sinica, Taiwan, for technical consultation. We thank the staff of Core Labs, Department of Medical Research, National Taiwan University Hospital for technical support of sequencing. This work was supported by a grant from the National Taiwan University Hospital (NTUH. 103-P01).
The authors have no conflict of interest to disclose.
Supplementary Material
In vitro modification of the duck hepatitis B virus (DHBV) expression plasmid by the CRISPR/Cas9 system.
Large deletion of the hepatitis B virus (HBV) expression plasmid by the combinatorial use of gRNAs P1 and XCp.
Nucleotide alignment of clonal sequencing results.
References
- Dandri M, Burda MR, Will H, Petersen J. Increased hepatocyte turnover and inhibition of woodchuck hepatitis B virus replication by adefovir in vitro do not lead to reduction of the closed circular DNA. Hepatology. 2000;32:139–146. doi: 10.1053/jhep.2000.8701. [DOI] [PubMed] [Google Scholar]
- Moraleda G, Saputelli J, Aldrich CE, Averett D, Condreay L, Mason WS. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J Virol. 1997;71:9392–9399. doi: 10.1128/jvi.71.12.9392-9399.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiang M, Rooney JF, Toole JJ, Gibbs CS. Biphasic clearance kinetics of hepatitis B virus from patients during adefovir dipivoxil therapy. Hepatology. 1999;29:1863–1869. doi: 10.1002/hep.510290626. [DOI] [PubMed] [Google Scholar]
- Werle–Lapostolle B, Bowden S, Locarnini S, Wursthorn K, Petersen J, Lau G, et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Caruntu FA, Molagic V. CccDNA persistence during natural evolution of chronic VHB infection. Rom J Gastroenterol. 2005;14:373–377. [PubMed] [Google Scholar]
- Sung JJ, Wong ML, Bowden S, Liew CT, Hui AY, Wong VW, et al. Intrahepatic hepatitis B virus covalently closed circular DNA can be a predictor of sustained response to therapy. Gastroenterology. 2005;128:1890–1897. doi: 10.1053/j.gastro.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Schiffer JT, Swan DA, Stone D, Jerome KR. Predictors of hepatitis B cure using gene therapy to deliver DNA cleavage enzymes: a mathematical modeling approach. PLoS Comput Biol. 2013;9:e1003131. doi: 10.1371/journal.pcbi.1003131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick TJ, Keck K, Bradshaw S, Jamieson AC, McCaffrey AP. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol Ther. 2010;18:947–954. doi: 10.1038/mt.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom K, Ely A, Mussolino C, Cathomen T, Arbuthnot P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol Ther. 2013;21:1889–1897. doi: 10.1038/mt.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang W, Lin J, Wang F, Wu M, Chen C, et al. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol Ther. 2014;22:303–311. doi: 10.1038/mt.2013.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG, 2nd, et al. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–118. doi: 10.1038/nature11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Xiao L, Li AS, Zhang X, Sirois P, Zhang J, et al. Biological and biomedical applications of engineered nucleases. Mol Biotechnol. 2013;55:54–62. doi: 10.1007/s12033-012-9613-9. [DOI] [PubMed] [Google Scholar]
- Deveau H, Garneau JE, Moineau S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol. 2010;64:475–493. doi: 10.1146/annurev.micro.112408.134123. [DOI] [PubMed] [Google Scholar]
- Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. 2013;10:741–743. doi: 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katic I, Großhans H. Targeted heritable mutation and gene conversion by Cas9-CRISPR in Caenorhabditis elegans. Genetics. 2013;195:1173–1176. doi: 10.1534/genetics.113.155754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köck J, Rösler C, Zhang JJ, Blum HE, Nassal M, Thoma C. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 2010;6:e1001082. doi: 10.1371/journal.ppat.1001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LR, Wu HL, Chen PJ, Chen DS. An immunocompetent mouse model for the tolerance of human chronic hepatitis B virus infection. Proc Natl Acad Sci USA. 2006;103:17862–17867. doi: 10.1073/pnas.0608578103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wei H, Wei H, Gao Y, Xu L, Yin W, et al. Blocking the natural killer cell inhibitory receptor NKG2A increases activity of human natural killer cells and clears hepatitis B virus infection in mice. Gastroenterology. 2013;144:392–401. doi: 10.1053/j.gastro.2012.10.039. [DOI] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levrero M, Pollicino T, Petersen J, Belloni L, Raimondo G, Dandri M. Control of cccDNA function in hepatitis B virus infection. J Hepatol. 2009;51:581–592. doi: 10.1016/j.jhep.2009.05.022. [DOI] [PubMed] [Google Scholar]
- Bi Y, Sun L, Gao D, Ding C, Li Z, Li Y, et al. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog. 2014;10:e1004090. doi: 10.1371/journal.ppat.1004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebina H, Misawa N, Kanemura Y, Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. 2013;3:2510. doi: 10.1038/srep02510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Zhu C, Gupta A, Hall VL, Rayla AL, Christensen RG, Dake B, et al. Using defined finger-finger interfaces as units of assembly for constructing zinc-finger nucleases. Nucleic Acids Res. 2013;41:2455–2465. doi: 10.1093/nar/gks1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, Rebar EJ. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- Ono Y, Onda H, Sasada R, Igarashi K, Sugino Y, Nishioka K. The complete nucleotide sequences of the cloned hepatitis B virus DNA; subtype adr and adw. Nucleic Acids Res. 1983;11:1747–1757. doi: 10.1093/nar/11.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In vitro modification of the duck hepatitis B virus (DHBV) expression plasmid by the CRISPR/Cas9 system.
Large deletion of the hepatitis B virus (HBV) expression plasmid by the combinatorial use of gRNAs P1 and XCp.
Nucleotide alignment of clonal sequencing results.