

Abstract
Background: Cystinosis is an autosomal recessive disorder marked by intralysosomal cystine accumulation. Patients present with generalized proximal tubular dysfunction called renal Fanconi syndrome. Urinary carnitine loss results in plasma and muscle carnitine deficiency, but no clinical signs of carnitine deficiency have been described. Also, the optimal dose of carnitine supplementation is undefined. This study aimed to determine whether currently recommended carnitine doses result in adequate correction of plasma carnitine.
Methods: Five cystinosis patients with renal Fanconi syndrome, aged 2–18 years, were included. l-carnitine was prescribed 50 mg/kg/day since diagnosis: median 36 (range 18–207) months. Total and free plasma and urine carnitine and carnitine profiles were measured at study onset, after stopping l-carnitine for 3 months and 3 months after reintroducing l-carnitine 50 mg/kg/day.
Results: At study onset, plasma free carnitine was normal in all patients, total carnitine (1/5), acetylcarnitine (3/5), and several short- and medium-chain acylcarnitines ≤10 carbons (5/5) were increased indicating carnitine over-supplementation. Three months after cessation, carnitine profiles normalized and 3/5 patients showed plasma carnitine deficiency. Three months after reintroduction, plasma free carnitine normalized in all patients, however, carnitine profiles were disturbed in 4/5 patients. Urine free carnitine, acetylcarnitine, and acylcarnitines ≤10 carbons were increased in all patients independent of carnitine supplementation.
Conclusion: Administration of recommended doses l-carnitine (50 mg/kg/day) resulted in over-supplementation. Although the drug is considered to be rather safe, long-term effects of over-supplementation remain unknown warranting cautious use of high doses. Plasma carnitine profile might be used as a monitor, to prevent overdosing.
Background
Cystinosis is a rare, autosomal recessive disorder. It is caused by mutations in the CTNS gene (17p13) which encodes for the lysosomal cystine carrier cystinosin. Mutations in the CTNS gene lead to the intralysosomal accumulation of cystine (Town et al. 1998, Kalatzis et al. 2001). The most frequent infantile, nephropathic form manifests around the age of 3–6 months with renal Fanconi syndrome. This general proximal tubular damage is characterized by the enhanced excretion of various small molecules, including salts, glucose, amino acids, low-molecular weight proteins and carnitine. If untreated with the cystine-depleting agent cysteamine, cystinosis leads to end-stage renal failure around the age of 10 years. Besides the specific treatment with cysteamine, cystinosis patients with renal Fanconi syndrome are treated symptomatically by supplementation of solutes lost into urine, such as bicarbonate, potassium, phosphate, citrate, and in some cases l-carnitine (Gahl et al. 2002).
Carnitine is an important transport molecule of long-chain fatty acids (acyl groups) and acyl-Co-enzyme A esters (acyl-CoA) across the mitochondrial inner membrane. The latter are degraded inside the mitochondrium by the process of β-oxidation until acetyl-CoA is left, which is further channelled as a substrate for several enzymes. In most tissues, acetyl-CoA enters the Krebs cycle with subsequent ATP generation; in ketogenic tissues, such as liver, it can also be used for ketone synthesis during carbohydrate starvation. Toxic fatty acyl-CoA esters can bind carnitine to form an acylcarnitine, which can be transported out of the mitochondrion. The cytosolic acetylcarnitine can act as an “acetyl-CoA buffer” and re-enter the mitochondrion when intramitochondrial acetyl-CoA levels have decreased, while acylcarnitines can be exported out of the cell and excreted in the urine. Since acyl and acetyl-groups can be transferred between carnitine and CoA, carnitine does not only affect the intracellular and intramitachondrial concentrations of acyl-CoA and acetyl-CoA, but it also maintains the acyl-CoA/CoA ratio and protects the body from toxic fatty acids (Bartlett and Eaton 2004).
In cystinosis, decreased tubular absorption of free carnitine and acylcarnitines leads to a deficiency of free carnitine in both plasma and muscle (Bernardini et al. 1985, Steinmann et al. 1987). Carnitine has a molecular weight of 162 Da and is freely filtered by the glomerulus. It is subsequently reabsorbed for 99% in the proximal tubule by a sodium-dependent transporter called OCTN2 (Tamai et al. 1998). Recently, a second carnitine transporter was identified in mouse proximal tubular cells, called Oat9, which transports carnitine without the need for a sodium or H+ gradient (Tsuchida et al. 2010). The latter transporter has, however, not been demonstrated in humans thus far.
It has been suggested to administer oral l-carnitine in a daily amount of 50–100 mg/kg, resulting in normalization of plasma carnitine levels within days (Gahl et al. 1988). Muscle carnitine levels, however, may need years to normalize (Gahl et al. 1993). Since clinical signs of carnitine deficiency such as cardiomyopathy or metabolic encephalopathy have never been reported in cystinosis patients, it remains unclear whether there is an indication for carnitine supplementation in cystinosis patients, and if so, what would be the optimal dose regimen. Also, the effect of carnitine supplementation on the altered carnitine profile in cystinosis patients is unknown. Therefore, we aimed to study whether the currently recommended oral supplementation of 50 mg l-carnitine/kg/day leads to alterations in the carnitine profile in cystinosis patients with renal Fanconi syndrome.
Patients and Methods
Five cystinosis patients with renal Fanconi syndrome, aged 2–18 years old, were included in this study, two of them were male. No patient had a renal graft. Their clinical characteristics are shown in Table 1. Glomerular filtration rate (GFR) was calculated using the bedside Schwartz formula (Schwartz et al. 2009).
Table 1.
Clinical characteristics
| Patient | Sex | Age at diagnosis (months) | CTNS gene mutations | Age at onset study (years) | Glomerular filtration rate (mL/min/1.73 m2) |
|---|---|---|---|---|---|
| 1 | Female | 14 | 57 kb del | 11 | 21 |
| 57 kb del | |||||
| 2 | Female | 9 | 57 kb del | 2 | 75 |
| c.926 dup | |||||
| 3 | Male | 6 | 57 kb del | 18 | 52 |
| 57 kb del | |||||
| 4 | Female | 10 | 57 kb del | 4 | 87 |
| c.926 dup | |||||
| 5 | Male | 11 | c.681G>A | 3 | 123 |
| c.1015G>A |
All patients were treated with 50 mg/kg/day l-carnitine since they were diagnosed with cystinosis and had received l-carnitine supplements during at least 18 months prior to enrolment in this study. Blood and urine carnitine profiles were measured at the study onset (daily dose 50 mg/kg), after 3 months of cessation of l-carnitine supplements and 3 months after reintroduction of l-carnitine at a daily dose of 50 mg/kg. In patient 1, no urine sample could be obtained after 3 months without carnitine supplements. Free carnitine concentrations and a carnitine profile were measured in blood and urine. Total carnitine concentrations were determined only in plasma, not in urine. All samples were measured in the laboratory of Radboudumc (Nijmegen, The Netherlands). Normal values were provided by the laboratory, different reference ranges were used for different age groups (Mueller et al. 2003).
Concentrations of free carnitine and acylcarnitines were measured by tandem mass spectrometry, essentially as described before (Vreken et al. 1999). Total carnitine concentrations were calculated by summation of free and acylcarnitine concentrations.
Results
The diagnosis of cystinosis was confirmed by a molecular analysis of the CTNS gene showing a disease causing mutation on both alleles in all five patients (Table 1). GFR ranged between 21 and 123 mL/min/1.73 m2, all patients had full-blown renal Fanconi syndrome with pronounced aminoaciduria. None of the patients reported muscle weakness before or during the study.
At the onset of this study, all patients were supplemented with a daily l-carnitine dose of 50 mg/kg. Plasma free carnitine was normal in all patients, plasma total carnitine was elevated in patient 1 and plasma acetylcarnitine was elevated in patients 1, 2, and 4. Several short- and medium-chain acylcarnitines (up to 10 carbons) were increased in all patients on carnitine therapy (Fig. 1). Urine analysis showed increased excretions of free carnitine, acetylcarnitine, and acylcarnitines with a length of up to 10.
Fig. 1.
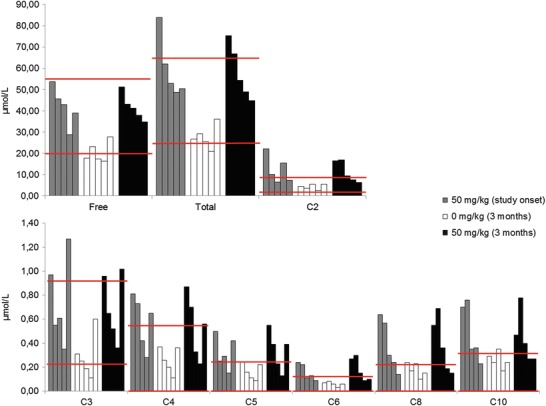
Carnitine profile in plasma. Red lines indicate normal range. Each bar represents an individual patient
After stopping l-carnitine supplementation during 3 months, blood and urine carnitine-ester profiles normalised in all patients. However, free carnitine was decreased in three patients (patients 1, 3, and 4) and total carnitine was decreased in one patient (patient 4).
Three months after reintroduction of l-carnitine at a daily dose of 50 mg/kg, free plasma carnitine normalised in plasma while it remained above normal values in urine in all patients; plasma total carnitine was above normal values in patients 1 and 2. Plasma acetylcarnitine was above normal values in patients 1, 2, and 3, while it was above normal values in urine in all patients. Several short-chain acylcarnitines with a length of up to 10 carbons were above normal values in 4/5 patients in both blood and urine, patient 4 was the only one who had a normal plasma carnitine profile.
Discussion
In this study, we analyzed plasma and urine carnitine levels in children with Fanconi syndrome due to cystinosis, both on and off l-carnitine supplementation.
Since children with renal Fanconi syndrome may have a high urinary loss of both free carnitine and carnitine-esters (Bernardini et al. 1985, Steinmann et al. 1987), it is recommended to treat them with l-carnitine. In the USA a daily dose of 100 mg/kg l-carnitine is advised (Gahl et al. 1988), while in Europe a daily dose of 50 mg/kg l-carnitine is mostly prescribed. We found alterations in plasma and urine carnitine profiles in all five cystinosis patients with renal Fanconi syndrome supplemented with 50 mg/kg l-carnitine during at least 18 months and 3 months after reintroducing l-carnitine 50 mg/kg, after 3 months cessation of l-carnitine supplementation. These alterations include increased levels of total carnitine, acetylcarnitine, and several short- and medium-chain acylcarnitine with a length up to 10 carbons in plasma and urine. On the other hand, plasma free carnitine (patients 1, 3, and 4) and plasma total carnitine (patient 4) declined below normal values when l-carnitine supplementation was stopped during 3 months.
The increased urinary (acyl)carnitine excretion in these patients can be explained by the fact that they all suffered from renal Fanconi syndrome (Bernardini et al. 1985, Steinmann et al. 1987), since 99% of filtered carnitine is reabsorbed in the proximal tubule by OCTN2 transporter (Tamai et al. 1998). Strikingly, the patient with the lowest GFR had the highest plasma total, free, and acetylcarnitine levels. One could hypothesize that a lower GFR is accompanied by lower urine carnitine losses. However, it is impossible to draw this conclusion in only one patient. Also, in previous studies in cystinosis patients, no correlation was found between serum creatinine (as a measure of GFR) and plasma carnitine levels (Bernardini et al. 1985, Gahl et al. 1988).
The increased (acyl)carnitine levels in plasma are more peculiar. Since these patients excrete high amounts of (acyl)carnitines in their urine, it could be expected to find corresponding decreased (acyl)carnitine profiles in patients’ plasma. However, the plasma carnitine profile in patients under supplementation in our study mimics plasma carnitine profiles in healthy subjects who receive l-carnitine supplements. In a study conducted by Hongu et al. 6 healthy women received 680 mg l-carnitine l-tartrate per day and were compared to 7 healthy woman receiving placebo and 6 healthy woman receiving 940 mg choline bitartrate per day. After 7 days they increased plasma and urine levels of total carnitine, free carnitine, acid-soluble acylcarnitine, and acetylcarnitine were found in the group receiving l-carnitine supplementation, while acid-insoluble acylcarnitine was normal in both plasma and urine (Hongu and Sachan 2003). Thus, increased levels of plasma total carnitine, acetylcarnitine, and short- and medium-chain acylcarnitines in our patients most likely reflect a state of over-supplementation.
It remains unclear whether over-supplementation with l-carnitine can be harmful. l-carnitine is considered to be a rather safe drug, with the only reported side effects being a fishy body odor, diarrhea, and nausea. Doses up to 2,000 mg/day are considered safe, the available data regarding doses above 2,000 mg/day are not sufficient for a confident conclusion on long-term safety (Hathcock and Shao 2006). However, in very-long-chain acyl-CoA dehydrogenase deficiency (VLCADD), controversy exists regarding the l-carnitine supplementation. It does not prevent decreased tissue carnitine levels and the accumulation of certain acylcarnitines can have toxic effects (Spiekerkoetter and Wood 2010).
Hypothetically, l-carnitine could be carcinogenic since it can be metabolized into trimethylamine and trimethylamine-N-oxide, both of which can be further metabolized into the potential carcinogenic compound N-nitrosodimethylamine (Bain et al. 2005). However, at this moment there is no evidence that l-carnitine might indeed be carcinogenic in vivo in doses used in this study. In addition, dietary l-carnitine can be metabolized into trimethylamine-N-oxide by gut bacteria, which is proatherogenic, and chronic dietary l-carnitine supplementation in mice can cause atherosclerosis (Koeth et al. 2013). Most of the cystinosis patients develop ESRD requiring renal replacement therapy associated with increased risk of cardiovascular disease and malignancy due to the use of immunosuppressive treatment. However, most cystinosis patients with renal graft are not receiving carnitine treatment, since renal Fanconi syndrome becomes much less pronounced after renal transplantation. Whether carnitine overdose in pre-transplant cystinosis patients can enhance the risk of malignancy and/or cardiovascular disease in the post-transplant period is impossible to estimate.
The rationale to prescribe high doses of l-carnitine of up to 100 mg/kg/day originates from the fact that muscle carnitine levels take years to normalize (Gahl et al. 1993). The clinical consequences of low muscle carnitine levels in cystinosis patients, however, remain unknown. Clinical symptoms of carnitine deficiency are highly variable and may include metabolic encephalopathy and cardiomyopathy, while other patients remain asymptomatic (Breningstall 1990). Myopathy, which can be another symptom of carnitine deficiency (Breningstall 1990), is frequently reported in cystinosis patients. However, myopathy in cystinosis patients generally develops after 10 years of age and progresses after renal transplantation (Gahl et al. 2002), when secondary carnitine deficiency is no longer present (Gahl et al. 2007). Therefore, the myopathy observed in older cystinosis patients is most likely a trait of the disease and not linked to excess urinary carnitine excretion. Noteworthy, rat studies have shown that the induction of carnitine deficiency does not alter exercise capacity despite decreased muscle carnitine levels (Simi et al. 1990).
As for the clinical relevance of carnitine supplementation in cystinosis patients, controversy exists regarding the recommended dose for l-carnitine administration. Plasma carnitine profiling could be a useful tool to determine the optimum l-carnitine dose in individual patients. The goal of l-carnitine supplementation should be to normalize total carnitine, free carnitine, and short-chain acylcarnitines. If plasma carnitine profiling is not easily accessible, we suggest using plasma free carnitine, total carnitine, and acetylcarnitine levels to monitor l-carnitine supplementation.
In conclusion, we demonstrate that the current dosing schemes of carnitine result in over-supplementation. Plasma carnitine profiling can be a useful tool to monitor carnitine doses in cystinosis patients under carnitine therapy.
Synopsis
The indications, optimal dose regimen, and efficacy of l-carnitine supplementation in cystinosis are unknown; however, current dosing schemes result in over-supplementation.
Compliance with Ethics Guidelines
Conflict of Interest
All the authors of this chapter declare that there are no conflicts of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2005. Informed consent was obtained from all patients for being included in the study.
Details of the Contributions of Individual Authors
Martine Besouw planned the study, gathered data, and wrote the manuscript.
David Cassiman interpreted data and corrected the manuscript.
Elisabeth Cornelissen conducted the study and corrected the manuscript.
Leo Kluijtmans interpreted data and corrected the manuscript.
Lambertus van den Heuvel interpreted data and corrected the manuscript.
Elena Levtchenko supervised the study, planned the study, and corrected the manuscript.
Footnotes
Competing interests: None declared
Contributor Information
M. Besouw, Email: martine.besouw@uzleuven.be
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Bain MA, Fornasini G, Evans AM. Trimethylamine: metabolic, pharmacokinetic and safety aspects. Curr Drug Metab. 2005;6:227–240. doi: 10.2174/1389200054021807. [DOI] [PubMed] [Google Scholar]
- Bartlett K, Eaton S. Mitochondrial beta-oxidation. Eur J Biochem. 2004;271:462–469. doi: 10.1046/j.1432-1033.2003.03947.x. [DOI] [PubMed] [Google Scholar]
- Bernardini I, Rizzo WB, Dalakas M, Bernar J, Gahl WA. Plasma and muscle free carnitine deficiency due to renal Fanconi syndrome. J Clin Invest. 1985;75:1124–1130. doi: 10.1172/JCI111806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breningstall GN. Carnitine deficiency syndromes. Pediatr Neurol. 1990;6:75–81. doi: 10.1016/0887-8994(90)90037-2. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Bernardini I, Dalakas M, et al. Oral carnitine therapy in children with cystinosis and renal Fanconi syndrome. J Clin Invest. 1988;81:549–560. doi: 10.1172/JCI113353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl WA, Bernardini I, Dalakas MC, et al. Muscle carnitine repletion by long-term carnitine supplementation in nephropathic cystinosis. Pediatr Res. 1993;34:115–119. doi: 10.1203/00006450-199308000-00001. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002;347:111–121. doi: 10.1056/NEJMra020552. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Balog JZ, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med. 2007;147:242–250. doi: 10.7326/0003-4819-147-4-200708210-00006. [DOI] [PubMed] [Google Scholar]
- Hathcock JN, Shao A. Risk assessment for carnitine. Regul Toxicol Pharmacol. 2006;46:23–28. doi: 10.1016/j.yrtph.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Hongu N, Sachan DS. Carnitine and choline supplementation with exercise alter carnitine profiles, biochemical markers of fat metabolism and serum leptin concentration in healthy women. J Nutr. 2003;133:84–89. doi: 10.1093/jn/133.1.84. [DOI] [PubMed] [Google Scholar]
- Kalatzis V, Cherqui S, Antignac C, Gasnier B. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. 2001;20:5940–5949. doi: 10.1093/emboj/20.21.5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller P, Schulze A, Schindler I, Ethofer T, Buehrdel P, Ceglarek U. Validation of an ESI-MS/MS screening method for acylcarnitine profiling in urine specimens of neonates, children, adolescents and adults. Clin Chim Acta. 2003;327:47–57. doi: 10.1016/S0009-8981(02)00327-3. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Muñoz A, Schneider MF, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol. 2009;20:629–637. doi: 10.1681/ASN.2008030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simi B, Mayet MH, Sempore B, Favier RJ. Large variations in skeletal muscle carnitine level fail to modify energy metabolism in exercising rats. Comp Biochem Physiol A Physiol. 1990;97:543–549. doi: 10.1016/0300-9629(90)90125-C. [DOI] [PubMed] [Google Scholar]
- Spiekerkoetter U, Wood PA. Mitochondrial fatty acid oxidation disorders: pathophysiological studies in mouse models. J Inherit Metab Dis. 2010;33:539–546. doi: 10.1007/s10545-010-9121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann B, Bachmann C, Colombo JP, Gitzelmann R. The renal handling of carnitine in patients with selective tubulopathy and with Fanconi syndrome. Pediatr Res. 1987;21:201–204. doi: 10.1203/00006450-198702000-00018. [DOI] [PubMed] [Google Scholar]
- Tamai I, Ohashi R, Nezu J, et al. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem. 1998;273:20378–20382. doi: 10.1074/jbc.273.32.20378. [DOI] [PubMed] [Google Scholar]
- Town M, Jean G, Cherqui S, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18:319–324. doi: 10.1038/ng0498-319. [DOI] [PubMed] [Google Scholar]
- Tsuchida H, Anzai N, Shin HJ, et al. Identification of a novel organic anion transporter mediating carnitine transport in mouse liver and kidney. Cell Physiol Biochem. 2010;25:511–522. doi: 10.1159/000303060. [DOI] [PubMed] [Google Scholar]
- Vreken P, Van Lint AE, Bootsma AH, Overmars H, Wanders RJ, Van Gennip AH. Quantitative plasma acylcarnitine analysis using electrospray tandem mass spectrometry for the diagnosis of organic acidaemias and fatty acid oxidation defects. J Inherit Metab Dis. 1999;22:302–306. doi: 10.1023/A:1005587617745. [DOI] [PubMed] [Google Scholar]